

Abstract
Amyloid-β (Aβ) peptides and other amyloidogenic proteins can form a wide range of soluble oligomers of varied morphologies at the early aggregation stage, and some of these oligomers are biologically relevant to the pathogenesis of Alzheimer's disease. Spherical micelle-like oligomers have been often observed for many different types of amyloids. Here, we report a hybrid computational approach to systematically construct, search, optimize, and rank soluble micelle-like Aβ25-35 structures with different side-chain packings at the atomic level. Simulations reveal for the first time, to our knowledge, that two Aβ micelles with antiparallel peptide organization and distinct surface hydrophobicity display high structural stability. Stable micelles experience a slow secondary structural transition from turn to α-helix. Energetic analysis coupled with computational mutagenesis reveals that van der Waals and solvation energies play a more pronounced role in stabilizing the micelles, whereas the electrostatic energies present a stable but minor energetic contribution to peptide assemblies. Modeled Aβ micelles with shapes and dimensions similar to those of experimentally derived spherical structures also provide detailed information about the roles of structural dynamics and transition in the formation of amyloid fibrils. The strong binding affinity of our micelles to antibodies implies that micelles may be a biologically relevant species.
Introduction
The β-amyloid (Aβ), with 39–42 amino acids, is a major component of neurotic plaques in Alzheimer's disease (AD), and its aggregation from unstructured monomers to β-structured oligomers, elongated protofibrils, and entwined mature fibrils is believed to play a critical role in the pathogenesis of AD (1,2). Increasing evidence from numerous structural determinations and biophysical experiments (3,4) suggests that soluble and highly ordered Aβ oligomers are likely to be the primary neurotoxic species pathologically linked to AD. Moreover, these oligomers generally display distinct structural morphologies (5–8) as a result of structural differences in peptide size, packing, and sequence, as well as different environmental conditions. Polymorphism appears to be a hallmark of amyloid structures, and different morphologies have been related to various assembly pathways to amyloid fibrils, driven by various intermolecular interactions (9–13).
Aβ micelle-like oligomers have been observed by atomic force microscopy (AFM) and electron microscopy (EM) (5,14,15), with sizes ranging from 3 nm to 35 nm depending on the experimental conditions and amino acid sequences used (14,16). Different sizes of spherical aggregates appear to be the precursor for fibril nucleation at the very early stage of aggregation process (17,18), representing different assembly stages or paths to mature fibrils. It has been suggested that interactions of Aβ micelles with other monomers or oligomers can perturb the micelle state and lead to a structural transition toward other oligomers with a distinct morphology (19,20). Of importance, it has been reported that a variety of antibodies (e.g., A11, IgG, 6E10, and IAPP) can specifically recognize micelle-like oligomers formed by different amyloidogenic proteins/peptides (16,21,22), implying that micelle-like oligomers are more likely to be on-pathway products of fibrillogenesis (18). Furthermore, studies on the interactions of Aβ spherical particles with the lipid bilayer (23,24) have shown that spherical Aβ oligomers can directly reduce the energy barrier for ions to pass through the membrane without the formation of amyloid-pore structures in the cell membrane. Despite their pathological importance, however, atomic structures of Aβ-micelles are still not available, primarily because of their small size, short lifetime, and low concentration.
Aβ25-35 is an intermembrane domain of APP, and is thought to be the bioactive and toxic fragment of Aβ because Aβ25-35 can form fibrils very quickly in senile plaques and impair hippocampal neurons in human brains in a manner similar to that of full-length Aβ40/42 (25). Aβ25-35 is often selected as a model system for studying cellular dysfunction, aggregation, and maladaptive response (26). The short, 11-residue sequence is more convenient for experimental synthesis (1,27) and computational simulation (28,29). Both experimental and computational studies have depicted many important structural and biological characteristics of Aβ25-35 aggregates. Ma and Nussinov (29) performed molecular-dynamics (MD) simulations to study the free-energy landscape of Aβ25-35 monomers in correlation with their conformational change. They found that both the α-helix and the β-strand can contribute to the formation of amyloid intermediates. using replica exchange MD simulations, Wei and Shea (28) revealed that the Aβ25-35 monomer adopts a helical structure in apolar organic solvent, and presents a collapsed coil or, to a lesser extent, a β-hairpin conformation in pure water. Of importance, Kaminsky et al. (30) reported that Aβ25-35 can infuse into the lateral ventricles of the brain and increase the p53 level in nuclei from the neocortex, suggesting that Aβ25-35 can affect nuclear enzyme systems.
In this work, we developed a computational strategy that includes conformational searches, structural optimization, potential energy evaluations, and all-atom MD to predict and model atomic structures of Aβ25-35 micelles at a very early stage of the aggregation process. First, an in-house-developed peptide packing program, coupled with an implicit generalized Born with a simple switching function (GBSW) approach (31), is used to systematically build, search, and optimize the molecular structure of Aβ-micelles based on the packing energy. The four lowest-energy micelle-like structures of ∼50 Å in diameter, consisting of 62 Aβ25-35 peptides, are selected from 2016 candidates by optimizing interpeptide organizations (i.e., parallel and antiparallel) and surface hydrophobicity (N-terminus versus C-terminus) and by further refining the interpetide displacements and intrapeptide self-rotation (Fig. 1). These four preselected micelle-like structures are subsequently subjected to explicit-solvent MD simulations to examine their structural stability. Simulations show that the micelles with antiparallel interpeptide organization display higher structural stability than those with parallel organization, resulting from the optimization of geometrical side-chain matches among peptides, surface hydrophobicity, and electrostatic interactions. A comparative energy analysis shows that peptide-peptide van der Waals (VDW) interactions and peptide-water solvation interactions are dominant forces for associating peptides and facilitating secondary structure conversion from turn to helix and, to a lesser extent, β-strand. Molecular docking of stable micelles with antiamyloid antibodies displays a high binding affinity, implying that these micelles may be a biologically relevant species.
Figure 1.
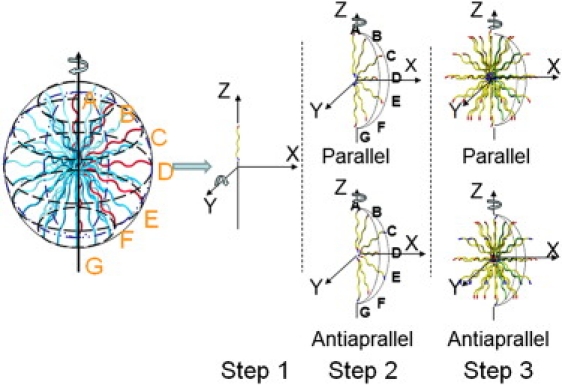
Three-step strategy for constructing Aβ25-35 micelles with parallel and antiparallel peptide orientations. Step 1: A single peptide is aligned to the z axis. Step 2: The peptide is replicated and rotated along the y axis at every 30° to form a semicircle by peptides A–G. The relative orientation (parallel versus antiparallel) and displacement between adjacent peptides are adjusted within the semicircle. Step 3: Peptides B–F (but not A and G) are rotated and copied along the z axis at every 30° to form a micelle consisting of 62 peptides.
Materials and Methods
Model construction of Aβ25-35 micelles
The initial coordinate for monomeric Aβ25-35 was generated by averaging 20 NMR-based structures (PDB code: 1QWP, GSNKGAIIGLM) (32) in a hexafluoroisopropanol/water (80:20) system. This was used as a building block for micelle construction because Aβ monomers are generally more structurally stable and less aggregated in a hexafluoroisopropanol solution. Aβ25-35 monomer exhibits a well-ordered α-helical structure (residues 28–34) with a large nonpolar C-terminus and relatively small polar N-terminus (Fig. S1 in the Supporting Material) possessing a cone shape. The N- and C-termini were blocked by NH3+ and COO− groups, respectively.
In building a compact Aβ micelle, we considered the initial relative orientation and position between peptides to be critical factors affecting the final packing energy and structural stability of the micelles. The interpeptide orientation was chosen to be in either a parallel or an antiparallel orientation, and the interpeptide position was used to crossly stagger adjacent side chains. The parallel organization employed two different micelle models: one with a large nonpolar C-terminus exposed to the solvent, and one with a small polar N-terminus exposed to the solvent. The antiparallel organization consisted of one micelle structure with 26 peptides exposing the N-terminus to the solvent and 36 peptides exposing the C-terminus to the solvent, and versa vice for the other structure. Thus, based on interpeptide orientation and position, four basic models were constructed that possessed distinct parallel/antiparallel organizations and surface hydrophobicity quantified by the ratio of the number of N-/C-termini exposed on the outer spherical surface.
Fig. 1 shows the three-step procedure involved in building a micelle. First, a single Aβ25-35 peptide is aligned to the z axis at a minimal distance of ∼4 Å from the origin of the Cartesian coordinate. Second, the peptide is replicated and rotated along the y axis at every 30° to form a semicircle of seven peptides (peptides A–G) with the same parallel orientation in the xz plane. Peptides B, D, and F are rotated an additional 15° along the z axis so that peptides A, C, E, and G, and peptides B, D, and F are located in different planes. For the antiparallel packing, peptides A, C, E, and G are reversed to impose an opposite orientation relative to peptides B, D, and F. Finally, peptides B–F (but not A and G) are rotated and copied along the z axis at every 30° to form a micelle consisting of different circle layers, i.e., B, C, D, E, and F layers. Each layer consists of 12 peptides, excluding A and G layers, leading to total of 62 peptides in the micelle with initial diameter of ∼50 Å. Four micelle structures were subjected to coarse structure optimization by using energy minimization with the GBSW implicit solvent model (31).
For each coarsely-optimized micelle, we further refined the structure by adjusting the peptide self-rotating angle (Φ) along the helical axis and peptide displacement between different layers (λ), i.e., each peptide was rotated along its helical axis at every 15° to avoid side-chain clash while all neighboring peptides were alternatively shifted along the helical axis from −1.2 to 2.8 Å at every 0.2 Å (i.e., peptides A, C, E, and G were moved with respect to peptides B, D, and F along the opposite direction). The structure-refinement procedure generated 504 distinct structures for each coarsely-optimized micelle. A total of 2016 wild-type (WT) micelles were energy-minimized by using 300 steps of steepest descent with backbone constraint, followed by 200 steps of conjugate gradient and 300 steps of steepest descent minimization without position constraints in the presence of the GBSW implicit solvent (31). The four lowest-energy micelles, one from each category (i.e., parallel organization with all N-termini exposed to the solvent (P_N), parallel organization with all C-termini exposed to the solvent (P_C), antiparallel organization with dominant N-termini exposed to the solvent (AP_N), and antiparallel organization with dominant C-termini exposed to the solvent (AP_C)) were selected and subjected to explicit-solvent MD simulation (Table S1) to examine their structural and energetic aspects at the early stage of the aggregation process.
Results and Discussion
Conformational search for stable Aβ25-35 micelles
The Aβ25-35 peptide displays a typical cone shape with a small hydrophilic N-terminus and a large hydrophobic C-terminus. Based on four different categories of constructed micelles, the P_N micelles represent a structural organization with favorable interface hydrophobicity and unfavorable geometrical packing because all small, hydrophilic N-termini are exposed to the bulk solvent. Conversely, the P_C micelles represent an opposite structural organization with unfavorable hydrophobic C-termini exposed to the solvent, but favorable geometrical packing. For the antiparallel peptide orientation, the AP_N and AP_C micelles compromise the packing in geometry and surface hydrophobicity. We ensure that each micelle category contains 504 different packings by varying the self-rotating angle (Φ) every 15° and the interlayer displacement (λ) every 0.2 Å along the peptide helical axis. Fig. S2 shows the packing-energy landscape, calculated by the GBSW method, as a function of peptide self-rotation and interlayer displacement. Overall, the P_C micelles display the most favorable average conformational energy (−1.7386×103 kcal/mol) for 504 candidates, and the P_N micelles show the least favorable average conformational energy (1.8587×104 kcal/mol). In similarity to the antiparallel peptide organization, the averaged conformational energy is lower for the AP_C micelles (4.8967×103 kcal/mol) compared to the AP_N micelles (7.9246×103 kcal/mol). This indicates that the geometrical peptide packing makes a more dominant contribution to the packing energy.
During the Φ and λ variations, two discrete but similar energy valleys were observed at Φ = 75° and 255° for all micelle categories, suggesting that peptide self-rotation can largely mediate packing energy by improving the side-chain geometry match, whereas interlayer displacement can further reduce packing energy by minimizing side-chain steric collision. The lowest-energy micelle structure was selected from 504 candidates in each category. The four lowest-energy micelle structures were identified at Φ = 210° and λ = 1.6 Å for the P_N category, Φ = 45° and λ = 0 Å for the P_C category, Φ = 255°, λ = 0.8 Å for the AP_N category, and Φ = 60° and λ = 0.4 Å for the AP_C category (marked by a red circle in the packing-energy landscapes; Fig. S2). Note that it is not feasible to systematically search and optimize >1000 Aβ micelles using the explicit-solvent model, and it could be also problematic to search, compare, and determine stable Aβ micelles among four P_N, P_C, AP_N, and AP_C categories using the implicit-solvent model. Thus, we use a two-step strategy to predict the atomic structures of Aβ micelles. We only use the GBSW model to search for the best candidate within each micelle category, instead of choosing the best candidate among four categories. The four best candidates selected from each category were further examined for structural stability by means of all-atom, explicit-solvent MD simulations. Thus, the lowest-packing-energy structures determined in the GBSW model are not necessarily the most stable structures in the explicit solvent due to the lack of hydrogen bonds between peptides and waters.
Structural stability of Aβ micelles with a distinct peptide organization
Visual inspection of the explicit-solvent MD trajectories shows that both of the micelle structures with a parallel peptide orientation (i.e., the P_N and P_C models) quickly lost their initial spherical shape within 8 ns and tended to transform into elongated, ellipsoidal shapes (Fig. 2 b and Fig. S4). The deformation is quantified by largely increased root mean-square deviations (RMSD = 10.5 Å for the P_N model and 11.5 Å for the P_C model) and radius of gyration (Rg = 27.2 Å for the P_N model and 28.1 Å for the P_C model; Fig. S3). The structural instability and transition for both parallel models could be attributed to a charged N-/C-terminus. Since all charged C- or N-termini are deeply embedded in the core of the micelle due to the parallel organization, a strong intermolecular electrostatic repulsion would tend to push the peptides away from each other. Moreover, unfavorable hydrophobic-water interactions induced by the hydrophobic C-terminus at the outer surface of the P_C micelle, or unfavorable VDW interactions induced by the large C-terminus embedded inside the P_N micelle could also unfold the whole micelle structures. All of these effects can lead to structural instability of parallel micelle structures in solution. On the other hand, two micelles with antiparallel peptide organization were able to maintain the overall spherical integrity without peptide disassociation throughout the 44-ns simulations (see detailed snapshots in Fig. S4). Backbone RMSDs gradually increased and stabilized at 7.2 Å for the AP_N micelle and 8.3 Å for the AP_C micelle, and averaged Rg approached similar values of 24.5 Å for the AP_N micelle and 24.9 Å for the AP_C micelle (Fig. S3). Both antiparallel micelles were slightly expanded from their initial energy-minimized structures by optimizing side-chain contacts. A residue-based root mean-square fluctuation (RMSF) analysis revealed that both structures showed small fluctuations for the central residues (Gly29-Ala30-Ile31-Ile32-Gly33), and large fluctuations for the two terminal residues and the Lys28 residue (Fig. S3 c), indicating that the charged residues are more flexible than the neutral residues. In contrast to the parallel peptide organization, electrostatic repulsion between the N- and C-termini is partially neutralized via antiparallel peptide packing. Overall, a structural comparison among the four micelles with distinct parallel and antiparallel peptide orientations suggests that high structural stability is a result of a delicate balance among surface hydrophobicity, geometrical packing, and side-chain arrangements.
Figure 2.
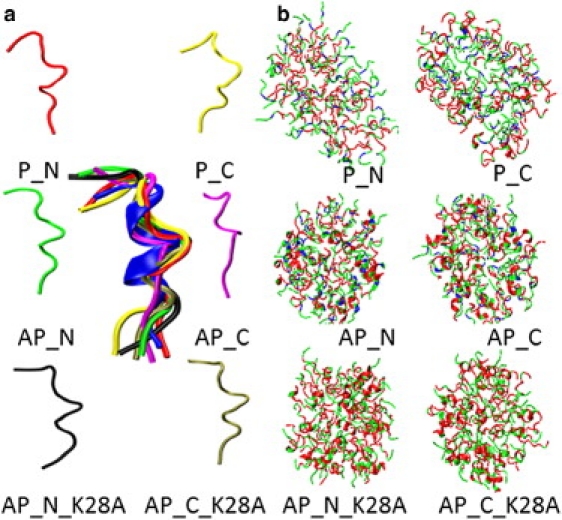
(a) Aβ25-35 monomers averaged from the last 5-ns simulations at 10 ps interval and their structural superimposition with initial NMR model (blue). (b) Structural comparison of final snapshots of each micelle from MD simulations. Color: nonpolar residues (red), polar residues (green), and positively charged residues (blue). Detailed structural evolution of each micelle is shown in Fig. S4.
Although both antiparallel micelles displayed relatively high stability, they also underwent local secondary structure adjustments to some extent. The population of secondary structures was averaged and compared between the first and last 4 ns of the simulation by means of the STRIDE algorithm (33). It can be seen from Table 1 that, although peptides in the AP_N micelle mainly adopted turn and coil conformations throughout the 44-ns simulation, all peptides experienced structural conversions for a reduced turn propensity (54.4% vs. 48.2%), an increased helix propensity (10.1% vs. 14.5%), a slightly increased coil propensity (34.6% vs. 35.8%), and a similar β-strand propensity (1.0% vs. 1.5%). A similar tendency for structural transition was also observed for the AP_C micelle, in which turns were reduced by 10.2% while the helix increased by 6.7%. It appears that the reduced turn conformation slowly converts into a helix conformation. Though it is well known that the cross-β structure is a characteristic conformation in Aβ fibrils formed by either full-length Aβ peptides or other Aβ fragments, our simulation shows that the micelles could exist as stable conformations without the appearance of β-strands (i.e., <2%) within the 44-ns simulations. This suggests that the micelles are most likely to appear at the very early stage of Aβ aggregation, serving as a peptide reservoir to interact with monomers or other low-order oligomers to form nuclei seeds or high-order oligomers with ordered β-structures.
Table 1.
Comparison of secondary structure distributions for four stable micelles between the first and last 4 ns of a simulation
| Model | Time(ns) | Secondary structure (%) |
|||
|---|---|---|---|---|---|
| Turn | β-strand | Helix | Coil | ||
| AP_N | 0–4 | 54.4 | 1.0 | 10.1 | 34.6 |
| 40–44 | 48.2 | 1.5 | 14.5 | 35.8 | |
| AP_N_K28A | 0–4 | 46.6 | 2.3 | 15.8 | 35.4 |
| 40–44 | 37.6 | 2.5 | 21.5 | 38.5 | |
| AP_C | 0–4 | 57.5 | 1.1 | 8.8 | 32.7 |
| 40–44 | 47.3 | 1.6 | 15.5 | 35.7 | |
| AP_C_K28A | 0–4 | 49.4 | 1.2 | 16.4 | 33.0 |
| 40–44 | 37.3 | 1.6 | 22.6 | 38.6 | |
Effect of Lys28 on the stability of Aβ micelles
Two WT simulations showed that Lys28 residues display relative large fluctuations in the stable AP_N and AP_C micelles, presumably due to large electrostatic repulsion. We performed two additional MD simulations by substituting the positively charged Lys28 with the Ala residue for both AP_N and AP_C micelles to examine the effect of charged residues on the overall structural stability and the local structural adjustment of the micelles. Visual inspection of the MD trajectories for both mutants reveals that, overall, the micelle structures were well maintained during the 44-ns simulations. The AP_N_K28A mutant (averaged RMSD = 7.1 Å) experienced almost the same structural deviation as the WT of AP_N (RMSD = 7.2 Å), and the AP_C_K28A mutant underwent a smaller structural change (RMSD = 6.7 Å) than the AP_C (RMSD = 8.3 Å; Fig. S3 a and Table S1). Both mutants experienced minor structural expansion (Fig. S3 b and Table S1), resulting in more compact spherical structures. The trajectories of these mutants were further analyzed in terms of individual residue fluctuations (Fig. S3 c). As expected, the most dramatic reduction in residue fluctuations occurred at the mutation site of position 28. A comparison of the RMSD, Rg, and RMSF data between the WTs and K28A mutants suggests that the enhanced stability of the mutants is attributed to the reduction of electrostatic repulsion and the low fluctuations near residue 28.
It is also interesting to monitor the local secondary structure transition for K28A mutants. As can be seen in Table 1, although the most populated secondary structure of both mutants was still the turn conformation, in similarity to the WTs the reduction in turns concurrently induced the increment in the α-helix by 5.7% for AP_N_K28A and 6.2% for AP_C_K28A, and in the coils by 3.1% for AP_N_K28A and 5.6% for AP_C_K28A. The K28A mutation did not enhance the formation of β-strand, further suggesting that unlike other amyloid oligomers, it probably does not need to contain a large portion of β-structure in the micelle at the very early stage of aggregation. Fig. 2 a shows six Aβ25-35 monomer structures averaged from each micelle at 10 ps intervals in the last 5 ns of the MD simulations and their structural superimposition relative to the initial NMR structure. It can be seen that the average Aβ25-35 monomeric building blocks extracted and averaged from micelles are very similar to each other; however, these similar monomers were assembled into micelles displaying different structural stability and dynamics (Fig. 2 b), emphasizing the importance of interpeptide interactions in micelle formation.
To quantitatively correlate the structural stability of micelles with the underlying driving forces upon the mutation of Lys28 to Ala, Fig. 3 shows the time evolution of side-chain contacts and hydrogen bonds between peptides for four micelles (AP_N, AP_C, AP_N_K28A, and AP_C_K28A). It can be seen that all four micelles have a similar number of intermolecular hydrogen bonds between peptides, whereas the WTs (AP_N and AP_C) have a fewer side-chain contacts than the mutants (AP_C_K28A and AP_N_K28A). Side-chain contacts for the WTs decreased in the first ∼20 ns and then remained constant for the rest of the simulations, whereas side-chain contacts for the mutants experienced subtle changes. The reduction of these side-chain contacts in WTs compared to the K28A mutants can be attributed to unfavorable electrostatic interactions between the Lys28 residues. Moreover, the average number of side-chain contacts from the last 10 ns was significantly larger than the average number of hydrogen bonds, suggesting that interpeptide side-chain contacts are likely to play a pronounced role in promoting the formation of micelles, whereas hydrogen bonds serve to assist the structural conversion to a more ordered β-structure conformation. In previous studies, Cheon et al. (34) and Auer et al. (35) also observed that Aβ25-35 monomers first coalesced into metastable disordered spherical assemblies driven by hydrophobic interactions and then converted into β-sheet-rich assemblies driven by hydrogen bonds. A dynamic interaction network thus plays an important role in controlling structural conversion and aggregation pathways among these states.
Figure 3.
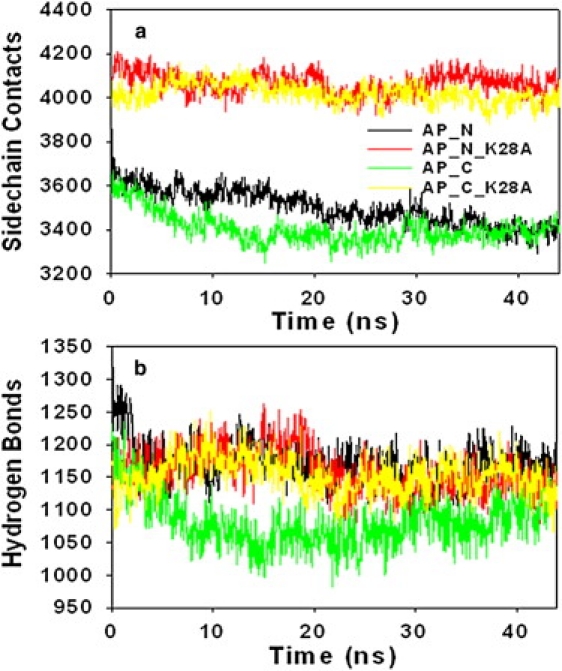
Time evolution of (a) side-chain contacts and (b) hydrogen bonds between peptides for micelles AP_N (black), AP_C (green), AP_N_K28A (red), and AP_C_K28A (yellow).
Peptide-peptide and peptide-water interactions
The structural stability and conformational dynamics of Aβ micelles are governed by two energetic contributions from peptide-peptide interactions and peptide-water interactions. Fig. 4 shows Aβ-Aβ peptide interactions with the decomposed VDW, and electrostatic contributions and Aβ solvation interactions. All interaction energies are averaged at every 5 ps from the last 5 ns of the simulations using the GBSW method. Overall, a comparison of the various energy distributions among the micelles reveals some interesting common features, as discussed below.
Figure 4.
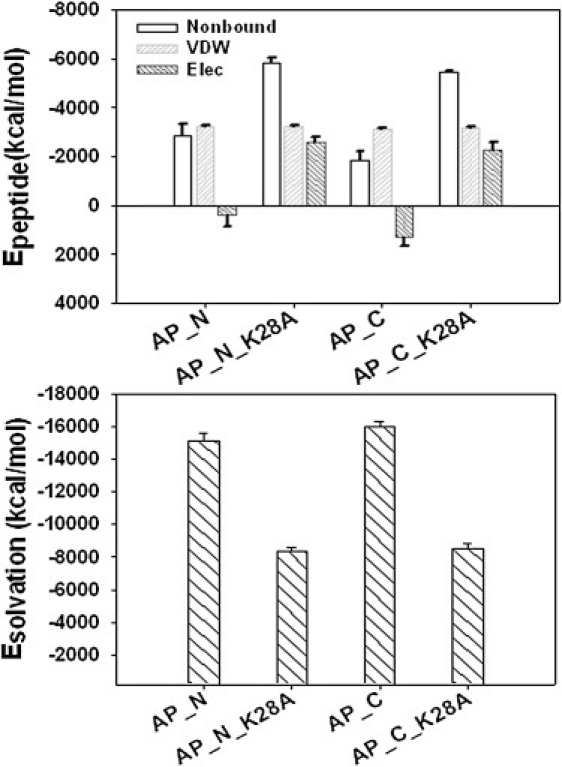
(top) Average peptide-peptide interaction energy and (bottom) peptide solvation energy for AP_N, AP_C, and the corresponding K28A mutants, calculated by the GBSW method.
First, the Aβ solvation energy makes a dominant contribution in stabilizing the micelle structures, much more so than the Aβ peptide-peptide interactions. All Aβ solvation energies have very small standard deviations, indicating that these interactions are very stable, independently of Aβ conformations. Strong solvation interactions are attributed to the formation of a tightly bound water layer at the micelle interface, as evidenced by the large number of hydrogen bonds formed at the surface (HBAP_N = 1019, HBAP_C = 918, HBAP_N_K28A = 667, and HBAP_C_K28A = 607) and the slow self-diffusivity of interfacial waters (∼0.385 ×10−5 cm2/s; DBulk = 3.37×10−5 cm2/s; Table S3). However, there is the lack of waters in the interior of the micelles due to dense side-chain contacts. Waters play different roles during the different stages of amyloid aggregation, especially before and after formation of a stable aggregate. Expulsion of waters from peptides is the obligatory first step to facilitate peptide association and subsequent fibrillization by reducing the free-energy barrier arising from dehydration entropic effects (36). Once the stable aggregates form, the highly hydrated water layer around the aggregate surface helps to prevent peptide disassociation. These results are in accord with recent experimental (36) and computational (37) results indicating that solvation energy is an important driving force for amyloid aggregation.
Second, for the WT micelles, the VDW interactions clearly played a dominant role in peptide-peptide interactions, contributing all favorable interactions to peptide association. Burial of the charged residues of Lys28 and N-/C-terminus was largely compensated for by VDW interactions; hydrophobic interactions among Ala30, Ile31, Ile32, and Leu33; and some attractive interactions between Lys28 and the C-terminus. However, upon substitution of Lys28 with Ala, electrostatic energy became much more favorable by decreasing 2957.5 kcal/mol for the AP_N_K28A micelle and 3584.5 kcal/mol for the AP_C_K28A micelle, providing additional force to associate Aβ peptides. The neutralization of Lys28 by Ala not only eliminated the unfavorable electrostatic interactions of Lys28-Lys28 buried deep in the micelle core, enhancing peptide-peptide interactions from repulsive (positive) forces to attractive (negative) forces, it also enhanced favorable hydrophobic interactions with Ala30, Ile31, and Ile32, leading to the increment of side-chain contacts from ∼3400 to ∼4000. Despite the electrostatic penalty for WT antiparallel micelles, substantial stabilizing energy from solvation and VDW interactions was still retained. Thus, the K28A mutation in Aβ25-35 micelles has a limited influence on fibril formation. Karsai et al. (38) found that WT and Lys28-acetylated Aβ25-35 peptides can form structurally similar amyloid fibrils at a similar aggregation rate. Unlike Aβ25-35, substitution of Lys28 in the full-length Aβ40/42 can completely eliminate intrastrand salt bridges with Asp23 and Glu22, retard the formation of the β-hairpin motif, and decrease the propensity for amyloid formation, because the formation of the basic β-strand-turn-β-strand motif with an intact Asp23-Lys28 salt bridge near the turn is an essential step in full-length Aβ fibril formation. This indicates that amyloid fibril formation by Aβ25-35 or Aβ40/42 proceeds through different pathways and is driven by distinct forces. Thus, aside from well-known hydrophobic interactions as the driving force of Aβ oligomerization/fibrillization, Aβ micelle formation is a result of the delicate balance between maximization of favorable peptide-peptide interactions and optimization of interfacial water interactions with micelles.
Biological implications
Aβ aggregates exist in many temporary structural forms, including a native state, unfolded state, and misfolded intermediate states (39), and spherical soluble oligomers have been observed for many different types of amyloids (16). Studies have reported Aβ40/42 micelle species consisting of 25–80 Aβ monomers with diameters of 30–70 Å depending on the experimental conditions (5,17,24). Unlike the full-length Aβ, Aβ25-35 can form micelles immediately after solution, as shown by AFM and EM images (14,40–43). However, a detailed structural characterization of these micelles, including size, number of peptides, and peptide arrangement, is lacking. Here, we modeled Aβ25-35 micelles of ∼55 Å consisting of 62 peptides with different peptide organizations in solution. The formation of stable parallel and antiparallel structures by Aβ fragments depends on specific complementary side-chain pairs, hydrogen-bonding patterns, and favorable hydrophobic and electrostatic interactions. Because of its inherent spherical morphology, a parallel structure usually suffers from larger free energy than an antiparallel structure due to an entropy penalty. It is very unlikely for all peptides to aggregate into a micelle with the same peptide orientation. Simulation results showed that for the stable Aβ micelles, the antiparallel orientation between peptides was more energetically favorable than the parallel orientation. From an energetic perspective, an antiparallel orientation can minimize unfavorable electrostatic interactions induced by charged residues and terminal residues, whereas from an entropic perspective, it seems unlikely that all peptides will pack in the same parallel way during the assembly process, as opposed to random orientation. Our antiparallel models present the most favorable situation in terms of free energy. Moreover, we examined two additional antiparallel micelles (AP_N with Φ = 75° and λ = 1.6 Å, and AP_C with Φ = 255° and λ = −0.6 Å) and two parallel micelles (P_N with Φ = 60° and λ = −1.2 Å, and P_C with Φ = 360° and λ = 0 Å) selected from the energy landscape (Fig. S2). Both of the antiparallel structures consistently displayed similar high structural stability within 40 ns, whereas the parallel structures transformed into nonspherical shapes within 8 ns (Fig. S5). Cheon et al. (34) studied the early stage of the Aβ16-22 and Aβ25-35 oligomerization process using Monte Carlo simulations. They found that both sequences could quickly coalesce into spherical aggregates with no significant β-structure present, followed by structural reorganization into a single-layer β-sheet. A similar two-step oligomerization from disordered micelles to ordered β-sheets via a coalescence-reorganization mechanism was further confirmed by Auer et al. (35). Consistent with our micelle models, at the initial coalescence step, all peptides in the micelles are not organized in a parallel manner. Moreover, a number of studies have shown an antiparallel β-sheet orientation for double-layer Aβ40/42 linear oligomers and annular oligomers (44–46).
The fact that micelles or spherical aggregates of various sizes are often observed by AFM and EM during the very early stage of amyloid fibrillization, and then disappear as the nucleus or fibrils form (5), indicates that these micelles are intermediates in the pathway to fibril formation. Due to the early appearance and transitory nature of micelles, micelle structures do not necessarily adopt characteristic β-structures; rather, they show a mixture of α-helix, random coil, and extended conformations. Our micelle models possessed a mixture of turn, coil, helix, and β-strand conformations. Although only a small portion of β-structure was present in the micelles, a slow trend from turn to α-helix was observed. Using transmission EM and solid-state NMR, Chimon and co-workers (15) found that the formation of spherical intermediates (5–35 nm in diameter) required an assembly procedure from Aβ monomers to spherical aggregates with random Aβ conformation to spherical intermediates with high β-sheet structure. Chebaro et al. (47) found that Aβ16-35 monomers and dimers were dominated by coil and turn conformations with little β-sheet structure.
Several studies on Aβ aggregation have revealed that Aβ micelles have a high binding affinity to the antibody. It is generally accepted that if the Aβ oligomer has a strong binding affinity to the antibody, it may be a more biologically relevant (toxic) species, because only toxic species can be specifically recognized by the antibody. To identify the biological relevance of our micelle models, we docked six micelles to an antiamyloid antibody (PDB: 3BAE) (48) using Patchdock (49) and Firedock (50). The docking results (Fig. 5) showed that the antibody displayed a higher binding affinity for stable micelles with antiparallel organization (AP_N and AP_C) than for two unstable micelles with parallel peptide organizations (P_N and P_C; Fig. 5). This is consistent with experimental data (16,24) indicating that all spherical oligomers from Aβ, α-synuclein, IAPP, polyQ, and prion106-126 can be specifically recognized by the antibody and induce a dramatic increase in membrane conductance. We note that these micelles can also be converted into other protofibrils/fibrils with varied morphologies over time when in equilibrium with Aβ monomers and/or oligomers.
Figure 5.
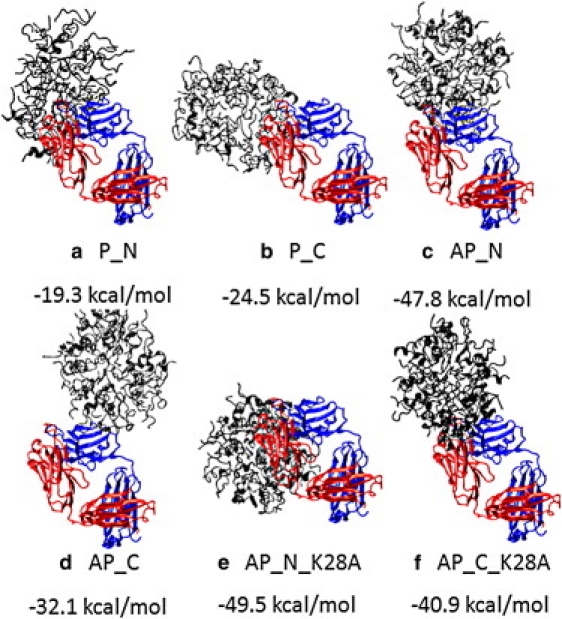
Six micelles (gray) obtained from the peptide-packing program and MD simulations were docked to a Fab region of the antiamyloid antibody (PDB: 3BAE). Unstable micelles with parallel peptide organization (a and b) generally have smaller binding energy than stable micelles with antiparallel peptide organization (c–f).
Given these experimental and computational findings, we propose a general model to describe the sequence of assembly along with different aggregates of monomers, micelles, nucleus, and fibrils (Fig. 6). Aβ monomers can quickly aggregate into compact micelles driven by hydrophobic interactions at the very early stage of aggregation (a→b), as observed in many AFM and EM images. These amorphous micelles can serve as a reservoir to interact with other species and slowly transform into ordered β-structure-rich oligomers with different structural morphologies (b→c), followed by a fast growth process for peptide elongation and protofibril lateral association to mature fibrils (c→d). Alternatively, Aβ monomers can also form small oligomers directly, not via the micelle state (a→c). This procedure corresponds to a slow lag phase for seed formation. Since both of these hypothetical pathways lead to final fibrils, they are not mutually exclusive.
Figure 6.
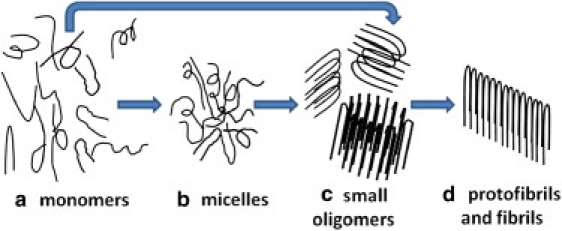
Schematic model for Aβ fibrillogenesis involving different Aβ aggregates and pathways during the aggregation process.
Conclusions
Aβ micelles are often observed during the early stage of amyloid formation; however, because of their small sizes and transitory nature, their atomic structures and the biological role they play in amyloid aggregation and toxicity are still unclear. In this work, we used an in-house-developed peptide-packing program coupled with explicit-solvent MD simulations to predict, model, and validate the atomic structures of Aβ micelles by optimizing side-chain packing (orientation and displacement) and interfacial hydrophobicity. The MD simulations demonstrate the existence of stable Aβ micelles with an antiparallel peptide organization in solution, which undergo an early and slow conformation change from turn to α-helix. It is unlikely for peptides to be organized in a parallel manner to form a micelle, due to the high entropy penalty and large, unfavorable charge repulsion. An energetic comparison among all micelle models also reveals that the high structural stability of Aβ micelles originates from competitive Aβ-Aβ and Aβ-water interactions involving the surface hydrophobicity of the Aβ micelle, interior interpeptide packing, and the structure and dynamics of the interfacial waters. Moreover, given their strong binding affinity to the antibody, these stable micelles may be biologically relevant species. Two stable WT micelles with different packing structures also suggest distinct intermediates during the aggregation process. On the other hand, it behooves us to emphasize that simulations cannot afford to explore all possibilities for micelle structures, due to the limited sampling spaces. Other structural possibilities for micelles are not excluded by our Aβ25-35 micelles. This work provides quantitative biophysical insights into the formation of Aβ micelles during the early stage of the aggregation process, and its implications for the transformation into mature fibrils.
Supporting Material
Five figures and three tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00603-X.
Supporting Material
Acknowledgments
This work was supported by a National Science Foundation CAREER Award (CBET 0952624), a 3M Non-Tenured Faculty Award, and the American Chemical Society Petroleum Research Fund (48188-G5). This study utilized the high-performance Biowulf PC cluster at the Ohio Supercomputer Center and the ATOM Biowulf cluster at the University of Akron.
References
- 1.Frozza R.L., Horn A.P., Salbego C.G. A comparative study of β-amyloid peptides Aβ1-42 and Aβ25-35 toxicity in organotypic hippocampal slice cultures. Neurochem. Res. 2009;34:295–303. doi: 10.1007/s11064-008-9776-8. [DOI] [PubMed] [Google Scholar]
- 2.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 3.Kirkitadze M.D., Bitan G., Teplow D.B. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glabe C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paravastu A.K., Leapman R.D., Tycko R. Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayed R., Pensalfini A., Glabe C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009;284:4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng J., Yu X., Wang Q. Molecular modeling of two distinct triangular oligomers in amyloid β-protein. J. Phys. Chem. B. 2010;114:463–470. doi: 10.1021/jp907608s. [DOI] [PubMed] [Google Scholar]
- 9.Takeda T., Klimov D.K. Probing energetics of Aβ fibril elongation by molecular dynamics simulations. Biophys. J. 2009;96:4428–4437. doi: 10.1016/j.bpj.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy G., Straub J.E., Thirumalai D. Dynamics of locking of peptides onto growing amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2009;106:11948–11953. doi: 10.1073/pnas.0902473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen H.D., Hall C.K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA. 2004;101:16180–16185. doi: 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawaya M.R., Sambashivan S., Eisenberg D. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 13.Zheng J., Ma B., Nussinov R. Structural stability and dynamics of an amyloid-forming peptide GNNQQNY from the yeast prion sup-35. Biophys. J. 2006;91:824–833. doi: 10.1529/biophysj.106.083246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karsai A., Murvai U., Kellermayer M.S. Oriented epitaxial growth of amyloid fibrils of the N27C mutant β 25-35 peptide. Eur. Biophys. J. 2008;37:1133–1137. doi: 10.1007/s00249-007-0253-0. [DOI] [PubMed] [Google Scholar]
- 15.Chimon S., Shaibat M.A., Ishii Y. Evidence of fibril-like [β]-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's [β]-amyloid. Nat. Struct. Mol. Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 16.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 17.Yong W., Lomakin A., Benedek G.B. Structure determination of micelle-like intermediates in amyloid β-protein fibril assembly by using small angle neutron scattering. Proc. Natl. Acad. Sci. USA. 2002;99:150–154. doi: 10.1073/pnas.012584899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabaté R., Estelrich J. Evidence of the existence of micelles in the fibrillogenesis of β-amyloid peptide. J. Phys. Chem. B. 2005;109:11027–11032. doi: 10.1021/jp050716m. [DOI] [PubMed] [Google Scholar]
- 19.Petkova A.T., Leapman R.D., Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 20.Yan L., Philippe D., Guanghong W. Thermodynamics and dynamics of amyloid peptide oligomerization are sequence dependent. Proteins. 2009;75:954–963. doi: 10.1002/prot.22305. [DOI] [PubMed] [Google Scholar]
- 21.Dumoulin M., Dobson C.M. Probing the origins, diagnosis and treatment of amyloid diseases using antibodies. Biochimie. 2004;86:589–600. doi: 10.1016/j.biochi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Habicht G., Haupt C., Fändrich M. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc. Natl. Acad. Sci. USA. 2007;104:19232–19237. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valincius G., Heinrich F., Lösche M. Soluble amyloid β-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: implications for cell toxicity. Biophys. J. 2008;95:4845–4861. doi: 10.1529/biophysj.108.130997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kayed R., Sokolov Y., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 25.Millucci L., Raggiaschi R., Santucci A. Rapid aggregation and assembly in aqueous solution of A β (25-35) peptide. J. Biosci. 2009;34:293–303. doi: 10.1007/s12038-009-0033-3. [DOI] [PubMed] [Google Scholar]
- 26.Esposito C., Tedeschi A., D'ursi A.M. Exploring interaction of β-amyloid segment (25-35) with membrane models through paramagnetic probes. J. Pept. Sci. 2006;12:766–774. doi: 10.1002/psc.811. [DOI] [PubMed] [Google Scholar]
- 27.Suh E.C., Jung Y.J., Lee K.E. A β 25-35 induces presynaptic changes in organotypic hippocampal slice cultures. Neurotoxicology. 2008;29:691–699. doi: 10.1016/j.neuro.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Wei G., Shea J.-E. Effects of solvent on the structure of the Alzheimer amyloid-β (25-35) peptide. Biophys. J. 2006;91:1638–1647. doi: 10.1529/biophysj.105.079186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma B., Nussinov R. The stability of monomeric intermediates controls amyloid formation: Aβ25-35 and its N27Q mutant. Biophys. J. 2006;90:3365–3374. doi: 10.1529/biophysj.105.075309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaminsky Y.G., Marlatt M.W., Kosenko E.A. Subcellular and metabolic examination of amyloid-β peptides in Alzheimer disease pathogenesis: evidence for Aβ (25-35) Exp. Neurol. 2010;221:26–37. doi: 10.1016/j.expneurol.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Im W., Lee M.S., Brooks C.L. Generalized born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 32.D'Ursi A.M., Armenante M.R., Picone D. Solution structure of amyloid β-peptide (25-35) in different media. J. Med. Chem. 2004;47:4231–4238. doi: 10.1021/jm040773o. [DOI] [PubMed] [Google Scholar]
- 33.Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 34.Cheon M., Chang I., Favrin G. Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLOS Comput. Biol. 2007;3:e173. doi: 10.1371/journal.pcbi.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auer S., Meersman F., Vendruscolo M. A generic mechanism of emergence of amyloid protofilaments from disordered oligomeric aggregates. PLOS Comput. Biol. 2008;4:e1000222. doi: 10.1371/journal.pcbi.1000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukherjee S., Chowdhury P., Gai F. Effect of dehydration on the aggregation kinetics of two amyloid peptides. J. Phys. Chem. B. 2009;113:531–535. doi: 10.1021/jp809817s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krone M.G., Hua L., Shea J.E. Role of water in mediating the assembly of Alzheimer amyloid-β Aβ16-22 protofilaments. J. Am. Chem. Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karsai A., Nagy A., Kellermayer M.S. Effect of lysine-28 side-chain acetylation on the nanomechanical behavior of Alzheimer amyloid β25-35 fibrils. J. Chem. Inf. Model. 2005;45:1641–1646. doi: 10.1021/ci0501701. [DOI] [PubMed] [Google Scholar]
- 39.Houry W.A., Sauder J.M., Scheraga H.A. Definition of amide protection factors for early kinetic intermediates in protein folding. Proc. Natl. Acad. Sci. USA. 1998;95:4299–4302. doi: 10.1073/pnas.95.8.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hensley K., Carney J.M., Butterfield D.A. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karsai Á., Grama L., Kellermayer M.S.Z. Potassium-dependent oriented growth of amyloid β25–35 fibrils on mica. Nanotechnology. 2007;18:345102. [Google Scholar]
- 42.Liu R., McAllister C., Sierks M.R. Residues 17-20 and 30-35 of β-amyloid play critical roles in aggregation. J. Neurosci. Res. 2004;75:162–171. doi: 10.1002/jnr.10859. [DOI] [PubMed] [Google Scholar]
- 43.Hughes E., Burke R.M., Doig A.J. Inhibition of toxicity in the β-amyloid peptide fragment β -(25-35) using N-methylated derivatives: a general strategy to prevent amyloid formation. J. Biol. Chem. 2000;275:25109–25115. doi: 10.1074/jbc.M003554200. [DOI] [PubMed] [Google Scholar]
- 44.Zheng J., Jang H., Nussinov R. Modeling the Alzheimer Aβ17-42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng J., Jang H., Nussinov R. Annular structures as intermediates in fibril formation of Alzheimer Aβ17-42. J. Phys. Chem. B. 2008;112:6856–6865. doi: 10.1021/jp711335b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchete N.-V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 47.Chebaro Y., Mousseau N., Derreumaux P. Structures and thermodynamics of Alzheimer's amyloid-β Aβ (16-35) monomer and dimer by replica exchange molecular dynamics simulations: implication for full-length Aβ fibrillation. J. Phys. Chem. B. 2009;113:7668–7675. doi: 10.1021/jp900425e. [DOI] [PubMed] [Google Scholar]
- 48.Miles L.A., Wun K.S., Parker M.W. Amyloid-β-anti-amyloid-β complex structure reveals an extended conformation in the immunodominant B-cell epitope. J. Mol. Biol. 2008;377:181–192. doi: 10.1016/j.jmb.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 49.Schneidman-Duhovny D., Inbar Y., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mashiach E., Schneidman-Duhovny D., Wolfson H.J. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36:W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.