
Figure 1.
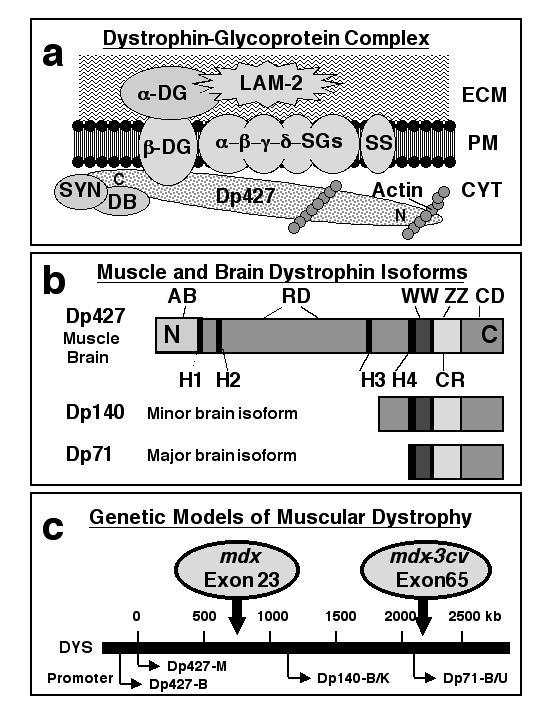
Diagrammatic representation of the dystrophin-glycoprotein complex, the structure of muscle and brain dystrophin isoforms and the genetic animal models of muscular dystrophy. In muscle, dystrophin forms a tightly associated complex with various surface components which provides a stabilizing linkage between the sub-sarcolemmal membrane cytoskeleton and the extracellular matrix. In panel (a) is shown the proposed spatial organization of this peripheral complex consisting of dystrophin (Dp427), α-, β-, γ-, and δ-sarcoglycan (SG), the sarcolemma-spanning backbone structure provided by α- and β-dystroglycan (DG), sarcospan (SS), various syntrophins (SYN) and dystrobrevins (DB), as well as laminin-2 (LAM-2) and cortical actin. Panel (b) outlines the various domains of dystrophin molecules with the N-terminal actin-binding domain (AB), hinge regions (H), the central spectrin-like rod domain (RD), as well as C-terminal binding domains such as the WW domain, the ZZ domain, the cysteine-rich region (CR) and the extreme carboxy-terminal domain (CD). While skeletal muscle fibres contain the Dp427 isoform of dystrophin, brain tissues express besides the full-length Dp427 molecule also two shorter isoforms termed Dp71 and Dp140 (b). Four of the seven promoters which drive the tissue-specific expression of dystrophins are shown in panel (c) illustrating that a point mutation in exon 23 or a mutation in exon 65 results in the absence of Dp427 in mdx mice and the absence of all brain isoforms of dystrophin in mdx-3cv mice.