
Figure 2.
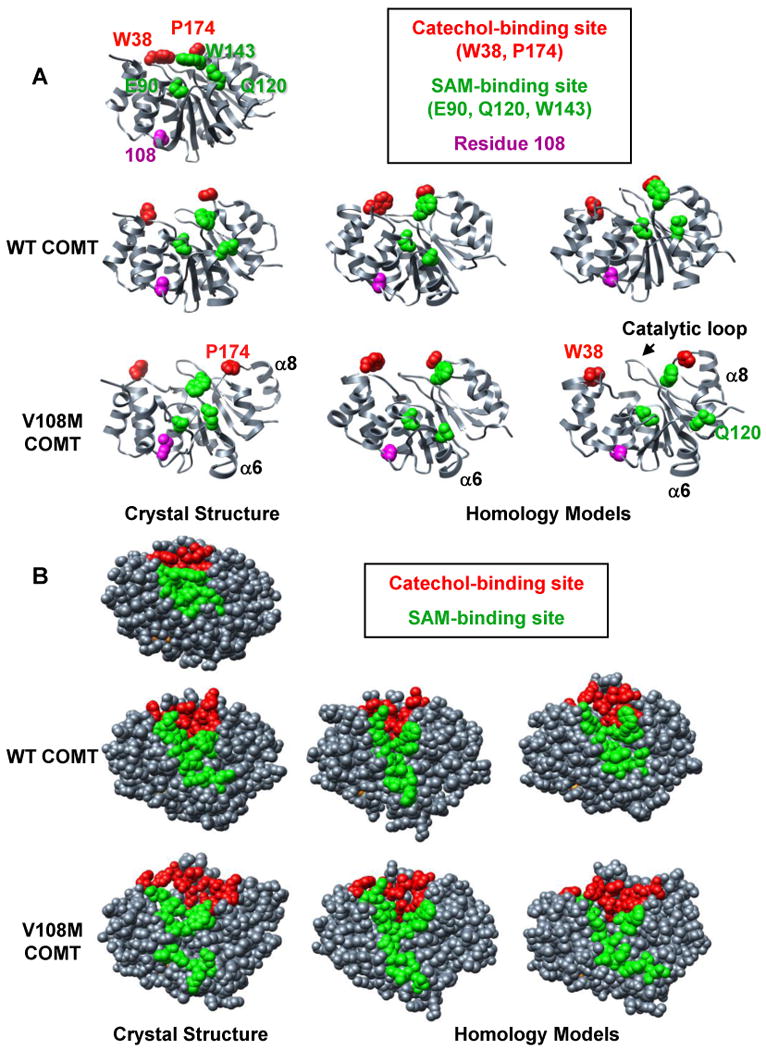
Comparison of MD simulations performed at 37°C using crystal structures and homology models of human wild-type and V108M COMT. Snapshots of structures from the final ns of MD simulations of (left) the crystal structures of human wild-type (3BWM.PDB, (54)) and V108M (3BWY.PDB, (54)) COMT, and (center, right) the homology models of human COMT based on the crystal structure of rat COMT (1VID.PDB, (53)) performed with C-scale values of either 0.0 (center) or 0.4 (right) (see Materials and Methods). (A) The 108M COMT structure is prone to disruption at 37°C. Altered packing around residue M108 reorients helix α6, pulling Q120 away from the SAM-binding site. Helix α8 (and P174) pulls away from the protein core resulting in an expanded conformation. These motions occur during simulations of both the crystal structure and homology models of the V108M COMT protein, but are not observed during the simulations of wild-type COMT. SAM-binding residues (E90, Q120, W143), catechol-binding residues (W38, P174) and residue 108 are shown in space-filling representation and colored in green, red, and magenta, respectively. (B) The 108M COMT active site is more exposed to solvent and distorted at 37°C than that of the wild-type protein. Residues in the SAM- and catechol-binding sites are colored green and red, respectively.