

Abstract
Background
Congenital fibrosis of the extraocular muscles type 1 (CFEOM1) is an autosomal dominant eye movement disorder linked to the pericentromere of chromosome 12 (12p11.2 - q12). Sarcospan is a member of the dystrophin associated protein complex in skeletal and extraocular muscle and maps to human chromosome 12p11.2. Mutations in the genes encoding each of the other components of the skeletal muscle sarcospan-sarcoglycan complex (α - δ sarcoglycan) have been shown to cause limb girdle muscular dystrophy (LGMD2C-F). To determine whether mutations in the sarcospan gene are responsible for CFEOM1 we: (1) attempted to map sarcospan to the CFEOM1 critical region; (2) developed a genomic primer set to directly sequence the sarcospan gene in CFEOM1 patients; and (3) generated an anti-sarcospan antibody to examine extraocular muscle biopsies from CFEOM1 patients.
Results
When tested by polymerase chain reaction, sarcospan sequence was not detected on yeast or bacterial artificial chromosomes from the CFEOM1 critical region. Sequencing of the sarcospan gene in CFEOM1 patients from 6 families revealed no mutations. Immunohistochemical studies of CFEOM1 extraocular muscles showed normal levels of sarcospan at the membrane. Finally, sarcospan was electronically mapped to bacterial artificial chromosomes that are considered to be outside of the CFEOM1 critical region.
Conclusions
In this report we evaluate sarcospan as a candidate gene for CFEOM1. We have found that it is highly unlikely that sarcospan is involved in the pathogenesis of this disease. As of yet no sarcospan gene mutations have been found to cause muscular abnormalities.
Background
CFEOM1 is an autosomal dominant disorder that has been linked to the pericentromere of chromosome 12, flanked by marker D12S1584 on the p arm and D12S1668 on the q arm [1, 2]. The clinical phenotype consists of congenital, bilateral ptosis and external ophthalmoplegia, with the eyes partially or completely fixed in a hypotrophic or downward position. On autopsy, CFEOM1 patients appear to be lacking the superior division of cranial nerve III, which innervates the levator and superior rectus muscles [3]. Whether this disease is caused by a primary defect in the nerve or the muscle remains unclear. The disease was initially linked to an 8 centiMorgan region spanning the centromere of chromosome 12, and then further refined to a critical region of 3 cM [1, 2]. Yeast and bacterial artificial chromosome (YAC and BAC) contigs have been generated and a positional cloning approach to identify the CFEOM1 causative gene is ongoing.
Sarcospan is a member of the dystrophin associated protein complex present in skeletal and extraocular muscle [4,5,6]. Sarcospan is most tightly associated with the transmembrane sarcoglycan subcomplex, mutation of which causes autosomal recessive limb girdle muscular dystrophy (LGMD2C-2F) [7,8,9,10,11]. Primary mutation of α - δ sarcoglycan leads to a variable degree of secondary instability of sarcospan and the non-mutant sarcoglycans. Sarcospan is homologous to the tetraspanin superfamily, members of which have been shown to facilitate both integral-membrane and membrane-proximal protein interactions involved in many different cellular processes [12]. Sarcospan had previously been identified as Krag, a gene that is co-amplified with Ki-ras in the Y1 murine adrenal carcinoma cell line [13, 14]. Portions of the gene's genomic structure were elucidated at that time, and the gene was localized to chromosome 12p11.2.
Given the genomic localization of sarcospan in the critical region defined for CFEOM1, and its association with other proteins known to be involved in muscular diseases, sarcospan has been proposed as a candidate disease gene for CFEOM1 [5]. We have refined the previously published genomic structure of sarcospan more fully and screened for mutations in six families with CFEOM1. We have also generated antibodies that recognize human sarcospan and examined extraocular muscle samples from CFEOM1 patients.
We find sarcospan to be unmutated in all six CFEOM1 families studied and sarcospan immunoreactivity to be identical in control and CFEOM1 extraocular muscle. Sarcospan is also shown to map electronically to BACs that are considered to be outside of the CFEOM1 critical region. These data make it unlikely that sarcospan, or other dystrophin associated proteins, are involved in the pathogenesis of CFEOM1.
Results
Genomic organization of the human sarcospan gene
Five independent clones were isolated from a human genomic phage library using hybridization probes that covered the entire coding region of sarcospan. Primers within the coding sequence were designed to cross the two known intron-exon junctions and other hypothesized junctions. The sarcospan open reading frame is encoded by three exons. The first exon contains 279 base pairs (bp) of coding sequence and is extremely G/C rich (71%). The second exon, as previously determined, is 87 bp in length. The third exon is very large and includes the last 363 bp of coding sequence and over 1500 bp 3'-untranslated region (UTR).
Analysis of the CFEOM1 critical region
Based on its cytogenetic localization and expression pattern, sarcospan is a candidate gene for autosomal dominant CFEOM1. Initially, to determine whether sarcospan was contained on YACs or BACs spanning the CFEOM1 critical region, YACs 766h7, 762e1, 951h6, 813h9, 782e8, 813g11, 832f4, 887g10, 916c8, 936f5, 946d5, 852c3, 723h3, 906f9, 798b12, 958b2, 943f6, 957b8, and 973h3 [2] and BACs B455J16, B50I19, B459A22, B392F18, B193D8, B855O3, B460N10, B351C12, B56H16, B937H2, B1035D8, B251K10, B152M7, B396F22, B45D10, B471G7, B520I18, and B367O10 (AECOM Genome Center Home Page chromosome 12 maps http://sequence.aecom.yu.edu/chr12/QARM1.pdf and http://sequence.aecom.yu.edu/chr12/Parm2.pdf) were tested. Positive control primers were able to amplify fragments of the YACs and BACs while sarcospan primers were not (data not shown).
Mutation Analysis
Although the coverage of the YAC and BAC physical maps was nearly complete, only direct sequencing of CFEOM1 patient DNA would yield definitive results about the status of the sarcospan gene in CFEOM1. Primer pairs to amplify all of exon 2 and the coding portions of exons 1 and 3 were designed. Because of the large 3'-UTR present in exon 3, a primer pair was designed to amplify the coding portions of the exon including the 5' splice junction and 40 bp of 3' non-coding sequence (Figure 1).
Figure 1.

Schematic representation of the genomic organization of human sarcospan. Exons are indicated by solid boxes, the 3'-UTR by a dashed box, and introns by a solid line. The exact size of the first intron is not known; the second intron is approximately 6 kb. Genomic primers are represented by arrows showing their position relative to intron/exon borders and coding sequences.
Two affected individuals from each of six families with CFEOM1 were analyzed by direct sequencing. No mutations or polymorphisms were found.
Immunofluorescence Analysis
Samples of CFEOM1 affected and unaffected extraocular muscle were analyzed for the pattern of sarcospan staining. Muscle was simultaneously stained with anti-spectrin antibody so that the samples' membrane integrity could be assessed, and a comparison could be made between the levels of sarcospan in normal and CFEOM1 samples. All samples showed normal sarcolemmal staining; no abnormalities were observed (Figure 2).
Figure 2.
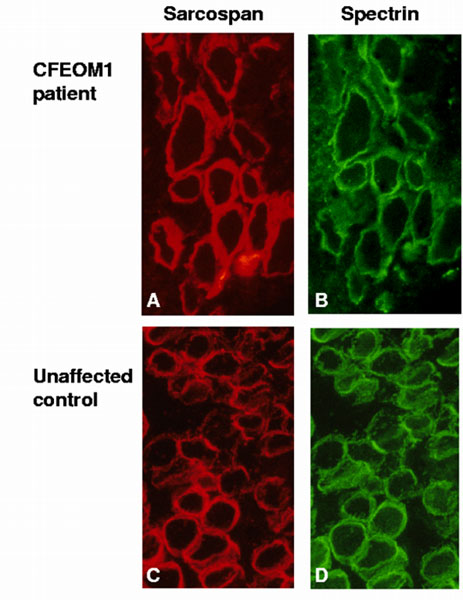
Sarcospan staining of CFEOM1 and unaffected extraocular muscle. Sarcospan is present at the membrane of extraocular muscle from CFEOM1 (A) and unaffected control (B) patients. Spectrin is also shown as an indicator of membrane integrity (C, D).
BAC Mapping of Sarcospan
Comparison of the sarcospan coding sequence (GenBank accession number AF016028) to the human genome sequence revealed that sarcospan is contained on the BACs B283G6 and B612B6 (Chromosome 12p map page 1 http://sequence.aecom.yu.edu/chr12/Parm2.pdf). These BACs are located 48.1 cM from the telomere of 12p, while the CFEOM1 critical region is between 53.3 and 56.5 cM (Chromosome 12p map page 3 (search for D12S1584) http://sequence.aecom.yu.edu/chr12/Parm2.pdf and Chromosome 12q map page 1 (search for D12S1668) http://sequence.aecom.yu.edu/chr12/QARM1.pdf), thus placing sarcospan outside of the CFEOM1 critical region.
Discussion
The critical region for the congenital eye movement disorder, CFEOM1, has been reduced to a 3 cM region at the centromere of human chromosome 12 [2]. In an effort to identify the gene which when mutated gives rise to CFEOM1, a combination of positional and candidate gene approaches has been undertaken. The sarcospan gene is composed of three exons that are located on human chromosome 12p11.2 and the sarcospan protein has been shown to be an integral component of the sarcoglycan complex [5, 15, 16]. Mutations in any one of the other four members of this complex have been shown to cause muscular dystrophy. As the extraocular muscle is one of the few muscles spared in Duchenne Muscular Dystrophy, the dystrophin associated protein complex present in extraocular muscle may respond differently to mutation of the complex than other striated muscle [17, 18]. By virtue of its genomic localization and expression in extraocular muscle, sarcospan is a candidate gene for autosomal dominant CFEOM1. We therefore chose to study sarcospan more closely within the context of this disease.
Immunofluorescence data showed comparable patterns of sarcospan staining in CFEOM1 and control patient samples implying that there was no haplo-insufficiency, altered accumulation or increased degradation of the sarcospan protein in these patients. Direct sequencing of the three coding exons of sarcospan in CFEOM1 patients confirmed that they were normal, rendering alteration of the sarcospan protein as the primary genetic defect in CFEOM1 unlikely. This conclusion is supported by the electronic localization of sarcospan to BACs outside of the CFEOM1 critical region.
The normal staining pattern of sarcospan in the autosomal dominant CFEOM1 patient muscle indicates that the primary genetic mutation is likely to be found in another, unrelated gene. Patients with autosomal dominant limb girdle muscular dystrophy type 1C, caused by mutations in caveolin-3 [19, 20], or autosomal recessive LGMD2B, caused by mutations in dysferlin, show normal dystrophin and sarcoglycan-sarcospan complex staining patterns [21]. Sarcospan immunoreactivity is altered in both Duchenne Muscular Dystrophy and the sarcoglycanopathies (LGMD2C-F) and appears to be very sensitive to disturbances in the dystrophin associated protein complex [16]. Thus the finding that sarcospan is normal in CFEOM1 patient muscle suggests that sarcospan itself and the rest of the dystrophin associated protein complex are not involved in the pathogenesis of the disease.
A sarcospan null mouse has recently been generated and appears to display a normal phenotype [22]. This does not, however, rule out the possibility of sarcospan playing a primary role in a muscle disorder. There may, for example, be a homologous tetraspanin protein that compensates for the absence of sarcospan in mice, but not in humans. There is, of course, also a difference between complete absence of a protein and a protein with an altered sequence. This may hold especially true, as sarcospan appears to be member of the tetraspanin family of proteins, which have been described as molecular facilitators; alterations of such a protein's sequence could affect the proteins with which it interacts. We are therefore expanding our patient analysis to include patients with other forms of muscular abnormalities including unlinked muscular dystrophies.
The effort to identify the CFEOM1 disease gene is continuing with analysis of other genes and expressed sequence tags from the critical region at the centromere of chromosome 12.
Conclusions
The DNA sequence analysis and protein immunofluorescence results that show sarcospan to be normal in CFEOM1 patients, combined with the localization of sarcospan to BACs that are outside of the CFEOM1 critical region, make it unlikely that sarcospan is involved in CFEOM1.
Materials and Methods
Screening of Human Genomic DNA Library
Two hybridization probes spanning the entire sarcospan coding sequence were generated by PCR amplification of sarcospan cDNA amplifying 366 bp (5'-ATGGGCAAGAACAAGCAGCCACG-3' and 5'-TTTCATAGAAAATTGAATACATGTCC-3') and 466 bp (5'-GGGCTGGGATCATTGTCTGCT-3' and 5'-GGAATTCTTAGATCTTTTGCTGGGG-3'). The bands were excised from 0.8% low-melt agarose and 20 ng of DNA were labeled with [α-32P]-dCTP using the Life Technologies Random Primers DNA Labeling System. A human genomic phage library (Clontech) was screened by hybridization with the radiolabeled products according to standard protocols. Briefly, filters were pre-hybridized at 65°C in hybridization buffer (5x SSC, 50 mM NaPO4, pH 7.4, 2.5x Denhardt's Solution) with 100 μg/ml denatured salmon sperm DNA for two hours. Probes were denatured at 95°C for 5 minutes and added to fresh hybridization buffer at 200,000 cpm/ml buffer for 16 hours. Following three one-hour washes in 2x SSC, 0.1% SDS, filters were exposed for 18 - 100 hours. Positively hybridizing plaques were purified by limiting dilution and phage DNA was isolated and purified with the Lambda Maxi Kit (Qiagen).
Determination of intron-exon boundaries
Five unique, partially overlapping genomic phage clones were identified. Phage DNA was sequenced with exon specific primers on an ABI automated sequencer. Acquired data was analyzed with the Sequencher software (Genecodes). The intron-exon borders were defined by divergence of the cDNA and genomic sequences, and by their adherence to splice donor and acceptor consensus sequences.
Patient material
Genomic DNA extracted from blood was used to screen two affected family members from each of six previously described pedigrees [1, 2]. The patients included in our analysis are AIII13, AIV1; BII4, BIV1; HIII17, HIII19; AAIII5, AAIII10; ACIV5, ACV1; and ADIII3, ADIII8. Extraocular muscle (inferior rectus) from patient AVI1 was obtained during a diagnostic biopsy. Age match control extraocular muscle (inferior rectus) from an unrelated, unaffected individual was obtained during an autopsy.
This study was approved by the Children's Hospital institutional review board, and informed consent was obtained from all adult participating subjects and from parents or legal guardians of participating minors.
YAC and BAC maps
Development of the YAC map was described previously by Engle, et al. [1]. BACs were isolated from RPCI-11 male and RPCI-13 female BAC libraries as described [23, 24].
PCR amplification from YAC contig
Primers from exons 1 and 3 were used to test the YACs from the critical region defined for CFEOM1. The exon 1 primers amplified a 116 bp fragment (5'-AAGGAGTGCGGGGAGGAG-3' and 5'-GCCATGAGGAAGCCCACC-3') and the exon 3 primers amplified a 150 bp fragment (5'-TTTGCCGCCCACCACTATTC-3' and 5'-TGAAAGTGCCAGTGACGC-3'). A total of 28 cycles (94°C /1 minute; 57°C/1 minute; 72°C/1 minute) following a 4 minute denaturation was used.
PCR amplification from BAC contig
Primers for exons 2 and 3 were used to test the BACs from the critical region defined for CFEOM1. Amplifications were performed as described below. Positive control reactions included template DNA isolated from human genomic phage that contained the appropriate sarcospan exons, and primers from the markers used to order the BAC map to ensure the quality of the DNA. Reactions were analyzed on agarose gels and scored for presence or absence of the appropriately sized product.
Primer sets and PCR conditions for direct sequencing
Three primer pairs amplifying the coding portions of exons 1 and 3 and all of exon 2 were designed (Table 1). Optimal polymerase chain reaction conditions were determined for each of the sets using 60-100 ng genomic DNA. Optimal buffer conditions for the exon 1 primer set were obtained with the FailSafe PCR PreMix Selection Kit (Epicentre Technologies) and Buffer F was used in conjunction with the FailSafe PCR Enzyme mix. PCR amplifications were performed in a 50 μl reaction volume with 60-100 ng genomic DNA, 200 μM of each dNTP and 1.25 units PCR enzyme mix. A total of 35 cycles (94°C/20 sec; 61°C/20 sec; 72°C/30 sec) following a 4 minute denaturation step was used. Exons 2 and 3 were amplified using PfuTurbo polymerase (Stratagene) at 2.5 Units per 50 μl reaction, containing 5 μl of the accompanying 10x PCR buffer, 200 μM of each dNTP, 100 ng of each primer and 60-100 ng genomic DNA. A total of 35 cycles (94°C/20 sec; 56°C/20 sec; 72°C/1 min) was used to amplify both exons 2 and 3. PCR amplification products were purified using the QIAquick PCR Purification Kit (Qiagen) and both strands were sequenced on an ABI automated sequencer. Data was analyzed with the Sequencher software (Genecodes).
Table 1.
Primer sequences used in the genomic amplification of exons
| Exon | PCR product | Forward Primer | Reverse Primer |
| size (bp) | (5'-3') | (5'-3') | |
| 1 | 465 | CCTCCATAATTCGAATACCAG | CTGGGTTAGTCTCAACTCGAC |
| 2 | 275 | CGGACAGCTTTATAACATGGATG | CTTAAGAAGGCACTCCTTTATTTTG |
| 3 | 493 | AATTCGCTTTGCAAATCATCATCC | GTTTGTTTAACCTCAGCTACTC |
Antibodies
Anti-human-sarcospan antibodies were generated by subcutaneous injection of New Zealand white rabbits with the synthetic N-terminal peptide MGKNKQPRGQQRQGGC (QCB). The final amino acid was added for purification purposes. The resulting serum was purified over a peptide affinity column made by covalent coupling of the unlinked peptide to SulfoLink beads (Pierce). Anti-human spectrin antibodies (NCL-SPEC1) were obtained from Vector Laboratories.
Immunofluorescence Analysis
CFEOM1 patient and normal muscle sections from extraocular muscle were stained with anti-human sarcospan and anti-human spectrin antibodies. Sections were fixed for 1 minute in ice cold methanol; blocked for 45 minutes in blocking buffer (15% horse serum, 0.025% Triton, 1x PBS); incubated overnight at 4°C with anti-sarcospan (1:750) and anti-spectrin (1:100) antibodies in block buffer; washed 3 × for 15 minutes in wash buffer (0.025% Triton, 1x PBS); incubated for 2 hours at 4°C with Cy3 conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch) and Alexa-488 conjugated anti-mouse secondary antibody (Molecular Probes Inc.) diluted 1:300 in blocking buffer; washed 3 × for 15 minutes in wash buffer and mounted with Immumount (Shandon). Samples were examined using a Zeiss Axiophot microscope.
Abbreviations
CFEOM1: Congenital fibrosis of the extraocular muscles type 1
LGMD: limb girdle muscular dystrophy
YAC: yeast artificial chromosome
BAC: bacterial artificial chromosome
UTR: untranslated region
Acknowledgments
Acknowledgements
We would like thank K. Montgomery and R. Kucherlapati of the Albert Einstein College of Medicine Human Genome Research Center, Bronx, NY for the use of the center's unpublished BAC maps. We would also like to thank the members of the Kunkel and Engle laboratories - especially S.E. Lacy, the MRRC Core Sequencing Facility (supported by NIH-P30-HD18655) and C.F. O'Brien. LMK is an investigator of the Howard Hughes Medical Institute.
Contributor Information
Kristine F O'Brien, Email: kfobrien@mac.com.
Elizabeth C Engle, Email: engle@rascal.med.harvard.edu.
Louis M Kunkel, Email: kunkel@genetics.med.harvard.edu.
References
- Engle EC, Kunkel LM, Specht LA, Beggs AH. Mapping a gene for congenital fibrosis of the extraocular muscles to the centromeric region of chromosome 12. Nat Genet. 1994;7:69–73. doi: 10.1038/ng0594-69. [DOI] [PubMed] [Google Scholar]
- Engle EC, Marondel I, Houtman WA, de Vries B, Loewenstein A, Lazar M, Ward DC, Kucherlapati R, Beggs AH. Congenital fibrosis of the extraocular muscles (autosomal dominant congenital external ophthalmoplegia): genetic homogeneity, linkage refinement, and physical mapping on chromosome 12 [published erratum appears in Am J Hum Genet 1996 Jan;58(1):252]. Am J Hum Genet. 1995;57:1086–1094. [PMC free article] [PubMed] [Google Scholar]
- Engle EC, Goumnerov BC, McKeown CA, Schatz M, Johns DR, Porter JD, Beggs AH. Oculomotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Ann Neurol. 1997;41:314–325. doi: 10.1002/ana.410410306. [DOI] [PubMed] [Google Scholar]
- Andrade FH, Porter JD, Kaminski HJ. Eye muscle sparing by the muscular dystrophies: lessons to be learned? Microsc Res Tech. 2000;48:192–203. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<192::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin- glycoprotein complex. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- Porter JD, Merriam AP, Hack AA, Andrade FH, McNally EM. Extraocular muscle is spared despite the absence of an intact sarcoglycan complex in gamma- and delta-sarcoglycan deficient mice. Neuromuscal Disorder. [DOI] [PubMed]
- Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tome FM, Romero NB, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Bönnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [published erratum appears in Nat Genet 1996 Jan;12(1):110] [DOI] [PubMed] [Google Scholar]
- Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, et al. Beta-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–265. doi: 10.1038/ng1195-257. [DOI] [PubMed] [Google Scholar]
- Nigro V, de Sa Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos-Bueno MR, Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet. 1996;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bonnemann CG, Gussoni E, Denton PH, et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy [see comments]. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- Scott AF, Elizaga A, Morrell J, Bergen A, Penno MB. Characterization of a gene coamplified with Ki-ras in Y1 murine adrenal carcinoma cells that codes for a putative membrane protein. Genomics. 1994;20:227–230. doi: 10.1006/geno.1994.1157. [DOI] [PubMed] [Google Scholar]
- Heighway J, Betticher DC, Hoban PR, Altermatt HJ, Cowen R. Coamplification in tumors of KRAS2, type 2 inositol 1,4,5 triphosphate receptor gene, and a novel human gene, KRAG. Genomics. 1996;35:207–214. doi: 10.1006/geno.1996.0340. [DOI] [PubMed] [Google Scholar]
- Crosbie RH, Lebakken CS, Holt KH, Venzke DP, Straub V, Lee JC, Grady RM, Chamberlain JS, Sanes JR, Campbell KP. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol. 1999;145:153–165. doi: 10.1083/jcb.145.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosbie RH, Lim LE, Moore SA, Hirano M, Hays AP, Maybaum SW, Collin H, Dovico SA, Stolle CA, Fardeau M, Tome FM, Campbell KP. Molecular and genetic characterization of sarcospan: insights into sarcoglycan-sarcospan interactions. Hum Mol Genet. 2000;9:2019–2027. doi: 10.1093/hmg/9.13.2019. [DOI] [PubMed] [Google Scholar]
- Emery AEH. Some unanswered questions in Duchenne muscular dystrophy. Neuromuscal Disorder. 1994;4:301–303. doi: 10.1016/0960-8966(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Porter JD, Baker RS. Muscles of a different "color": the unusual properties of the extraocular muscle may predispose or protect them in neurogenic and myogenic disease. Neurology. 1996;46:30–37. doi: 10.1212/wnl.46.1.30. [DOI] [PubMed] [Google Scholar]
- Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati MA, Volonte D, Galbiati F, Cordone G, Bricarelli FD, Lisanti MP, Zara F. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18:365–368. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- McNally EM, de Sa Moreira E, Duggan DJ, Bönnemann CG, Lisanti MP, Lidov HGW, Vainzof M, Passos-Bueno MR, Hoffman EP, Zatz M, Kunkel LM. Caveolin-3 in muscular dystrophy. Hum Mol Genet. 1998;7:871–877. doi: 10.1093/hmg/7.5.871. [DOI] [PubMed] [Google Scholar]
- Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RC, Marie SK, Anderson LV, Bönnemann CG, McNally EM, Nigro V, Kunkel LM, Zatz M. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet. 1996;5:1963–1969. doi: 10.1093/hmg/5.12.1963. [DOI] [PubMed] [Google Scholar]
- Lebakken CS, Venzke DP, Hrstka RF, Consolino CM, Faulkner JA, Williamson RA, Campbell KP. Sarcospan-deficient mice maintain normal muscle function. Mol Cell Biol. 2000;20:1669–1677. doi: 10.1128/MCB.20.5.1669-1677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert Einstein College of Medicine Chromosome 12 Mapping Overview http://sequence.aecom.yu.edu/chr12/descriptions.htm
- Renault B, Hovnanian A, Bryce S, Chang JJ, Lau S, Sakuntabhai A, Monk S, Carter S, Ross CJ, Pang J, Twells R, Chamberlain S, Monaco AP, Strachan T, Kucherlapati R. A sequence-ready physical map of a region of 12q24.1. Genomics. 1997;45:271–278. doi: 10.1006/geno.1997.4888. [DOI] [PubMed] [Google Scholar]