

Abstract
The catalytic oxidation of alkenes by most iron porphyrins using a variety of oxygen sources, but generally not dioxygen, yields the epoxide with minor quantities of other products. The turnover numbers for these catalysts are modest, ranging from a few hundred to a few thousand depending on the porphyrin structure, axial ligands, and other reaction conditions. Halogenation of substituents increases the activity of the metalloporphyrin catalyst and/or makes it more robust to oxidative degradation. Oxidation of cyclohexene by 5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorophenyl)porphyrinato iron(III), ([FeIII(tppf20)]) and H2O2 is typical of the latter: the epoxide is 99% of the product and turnover numbers are about 350.[1–4] Herein, we report that dynamic organic nanoparticles (ONPs) of [FeIII(tppf20)] with a diameter of 10 nm, formed by host–guest solvent methods, catalytically oxidize cyclohexene with O2 to yield only 2-cyclohexene-1-one and 2-cyclohexene-1-ol with approximately 10-fold greater turnover numbers compared to the non-aggregated metalloporphyrin in acetonitrile/methanol. These ONPs facilitate a greener reaction because the reaction solvent is 89% water and O2 is the oxidant in place of synthetic oxygen sources. This reactivity is unexpected because the metalloporphyrins are in close proximity and oxidative degradation of the catalyst should be enhanced, thus causing a significant decrease in catalytic turnovers. The allylic products suggest a different oxidative mechanism compared to that of the solvated metalloporphyrins. These results illustrate the unique properties of some ONPs relative to the component molecules or those attached to supports.
Keywords: alkenes, catalytic oxidation, nanoparticles, oxygen, porphyrinoids
Introduction
Organic nanoparticles
Inorganic nanoparticles with various capping groups or imbedded into polymers or other matrixes are widely used, or proposed, for a diverse array of catalytic applications.[5, 6] The inorganic cores of these conventional nanoparticles are robust and structurally static. In contrast, the structures, properties, and functions of aggregates of organic molecules organized into nanoparticles, organic nanoparticles (ONPs), by weak intermolecular interactions are much less understood because the organization of the molecules in the ONPs can be dynamic.[7]
Most methods to make nanoaggregates of small organic molecules have their historical roots in the formation of colloidal dispersions of organic systems.[8] The methods to make nanoscaled aggregates of dyes such as porphyrinoids[7, 9–17] include: a) the rapid exchange of solvent, b) host–guest solvents whereby aggregation occurs by mixing of solutions containing the chromophoric molecules with miscible solvents in which they are not soluble (e.g. THF/H2O) and by stabilizing with surfactants or amphipathic molecules, c) interfacial precipitation, and d) the rapid expansion of supercritical solvents. The former two methods result in dispersions in solution and the latter two methods result in many types of nanostructures that are kinetically trapped from further aggregation by deposition on surfaces. There is considerable interest in understanding the intermolecular processes governing formation of organic colloids and ONPs.[7, 9, 18] Hierarchical organization of molecules into nanostructured aggregates can have a profound effect on their photonic and catalytic properties. For example, formation of ONPs of porphyrinoids and other chromophoric systems offers the potential to enhance or modulate the photonic properties of the molecules through quantum mechanical effects.[19, 20] Additionally, understanding the spontaneous formation of suspensions of molecular aggregates is becoming an important topic for the formulation of hydrophobic drugs.[21] The formation of narrowly dispersed ONPs may arise from a thermodynamic limitation due to nanoparticle surface energies, and a kinetic limitation whereby the rates of formation of the individual aggregates influence the availability of material;[9, 13, 22] the recent work of van Keuren et al. indicats the importance of transient formation of unstable clusters.[15]
Porphyrin catalysts
The discovery by Groves and co-workers[23–29] that iron porphyrins in organic solvents with oxygen sources such as iodosylbenzene can mimic the oxidative catalysis observed for cytochrome P450[30–35] led to a huge amount of research on the reactivity and mechanism of this reaction. Different metalloporphyrins exhibit different catalytic reactivities, which include different products or product ratios.[26, 36–41] Other major findings include: a) appropriate modification of the porphyrin macrocycle alters the reactivity in terms of site selectivity,[28, 42–44] b) halogenation generally makes the metalloporphyrins more efficient catalysts,[1–4, 45–55] c) axial ligands can alter the reactivity,[56–59] d) the solvent can affect the reactivity,[1–4] and e) other oxygen sources such as H2O2, and O2, can be used with some systems.[59–62] Various metalloporphyrins are now used in laboratory scale reactions. Heterogeneous porphyrin systems include those in lipids, micelles, zeolites, or on supports such as silica, or Montmorillonite clay.[37, 42, 51, 63–65] Several reaction types are catalyzed by metalloporphyrins, but perhaps the best studied are oxidation reactions. The catalytic oxidation of cyclohexene by iron tetraarylporphyrins is a standard reaction that has been investigated thoroughly over the last few decades, and the epoxide is the major product under a range of experimental conditions. The elegant work by Hupp et al. on self-assembled metalloporphyrin catalysts show that rigid arrays can yield remarkably robust systems, but require macrocycles that are difficult to obtain in high yields.[18, 40, 66]
In general, the turnover number for iron porphyrin catalysts in solution is a few hundred because of the degradation of the catalyst, but this increases to about 350 when 5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorophenyl)porphyrinato iron(III) ([FeIII(tppf20)]) is used because this is a more active catalyst and may be somewhat more resistant to oxidative degradation. There are numerous studies on the catalytic activity of this porphyrin,[61, 67, 68] including the catalytic oxidation of cyclooctene by [FeIII(tppf20)] in acetonitrile/methanol, which yields over 98% of the epoxide and traces of the 2-cyclooctene-1-one and the 2-cyclooctene-1-ol.[1–4] Under the same conditions we find similar reactivity for cyclohexene (Scheme 1).
Scheme 1.
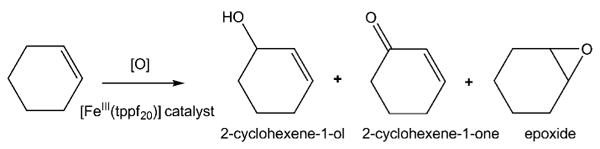
Oxidation of cyclohexene.
Given the enhanced catalytic activity of [FeIII(tppf20)], there are a considerable number of reports on other halogenated metalloporphyrins. Gray et al. reported significantly different oxidation chemistry for a derivative of [FeIII(tppf20)] wherein the eight β-pyrrole positions are also halogenated.[48–50] The differences in the catalytic oxidation of perhalogenated porphyrins arise from the distortions in the otherwise planar macrocycle and the electronic effects. When the β positions are chlorinated the activity of ethylbenzene oxidation by using oxygen at 100°C is increased but not the stability to oxidative degradation, but the stability increases when [FeIII(tppf20)] is linked to polystyrene.[61]
Organic nanoparticles of porphyrins
The formation of the all-organic nanoparticles of porphyrins and metalloporphyrins by host–guest solvent methods depends on the complex interplay between intermolecular forces and kinetics.[9, 10, 15, 22] The size and structure of the ONPs depends on: a) intermolecular forces between the porphyrins, host solvent, guest solvent, and the polyethylene glycol stabilizer, b) the ratio of host to guest solvents, and c) the vigorousness of mixing. Therefore, the structure of the porphyrin and the specific metal ion influence the size and the organization of the chromophores within the nanoaggregates. For [FeIII(tppf20)] in THF/water as host–guest solvents, large ONPs with a diameter of about 80 nm are formed by magnetically stirring and are likely composed of smaller subdomains of 5–20 nm. Sonication while adding the guest solvent results in the formation of ONPs with a diameter of approximately 10 nm, which are less prone to reorganization or disaggregation (Figure 1). Since the ONPs are self-organized solely by intermolecular forces, these are dynamic systems in that they can reorganize or disaggregate in response to environmental changes.[7, 9]
Figure 1.
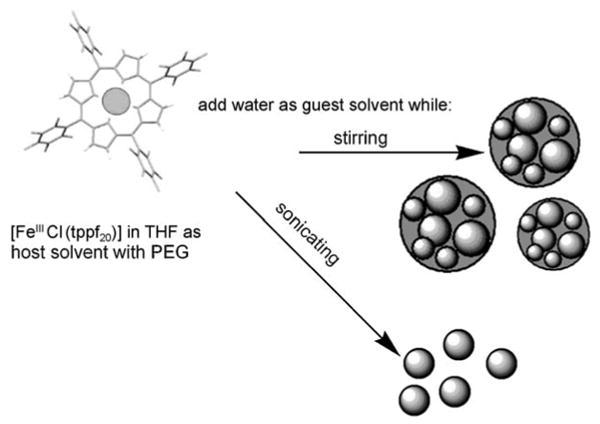
Preparation of ONPs. Water (5.0 mL) is added to a solution of [FeIIICl(tppf20)] in THF (0.4 mL, 1.0 mm) and PEG (0.2 mL) while stirring or sonicating to yield ONPs with a diameter of 80 nm and 10 nm, respectively.
We report herein that aqueous suspensions of organic nanoparticles of commercially available [FeIII(tppf20)], formed by the methods described above, have significantly enhanced catalytic properties compared to the component molecules and those on supports, and lead to different product distributions. This also allows the study of the fundamental differences in the chemistry of nanoscaled aggregates versus solvated molecules or solid-state materials.
Results and Discussion
A solution of [FeIII(tppf20)] in acetonitrile/methanol (3:1) catalytically oxidizes cyclohexene to the epoxide by using H2O2 with a turnover number (TON) of about 350. Previous reports describing the use of cyclooctene as a substrate parallel these results in the exclusive formation of the epoxide, and in the fact that only H2O2 can be used.[1–4] In contrast, 10 nm diameter ONPs of [FeIII(tppf20)] catalytically oxidize cyclohexene by using O2 to yield exclusively 2-cyclohexene-1-one and 2-cyclohexene1-ol with about 10-fold greater TON than the completely solvated metalloporphyrin, though at a much slower rate (Table 1).
Table 1.
[FeIII(tppf20)] ONP catalysis of cyclohexene oxidation.
| Solution[a] or ONPs[b] | Conditions | Yield (oxide) [%] |
Yield (ene-1-ol) [%] |
Yield (ene-1-one) [%] |
TON | Comments[d,e] |
|---|---|---|---|---|---|---|
| solution | CH3CN/CH3OH H2O2 | 98 | <1 | <1 | 350 | 15 min |
| solution | CH3CN/CH3OH H2O2 | 95±5 | 5±1 | <1 | not reported | cyclooctene, 15 min, ref. [1-4] |
| 10 nm ONPs | H2O2 | < 1 | 30 | 70 | 175 | ca. 5 min |
| 10 nm ONPs | 6.5 mL O2 | < 1 | 26 | 74 | 500 | O2 limiting reagent |
| 10 nm ONPs | 125 mL O2 | < 1 | 28 | 72 | 3500 | 16 h |
| 10 nm ONPs | 99.6% D2O 125 mL O2 | < 1 | 20 | 80 | no D in products other than the exchangeable alcohol proton | |
| 10 nm ONPs | 10% H218O 125 mL O2 | < 1 | 34 | 66 | 18O in 10% of ketone and < 1% in alcohol; via acetal | |
| 10 nm ONPs | H2O, 125 mL 98% 18O2 | < 1 | 23 | 76 | 18O in <8% of ketone and >90% of alcohol | |
| 10 nm ONPs | 125 mL O2 0.5 mL cyclohexene | < 1 | 28 | 72 | 3500 | large excess of substrate; 16 h |
| 10 nm ONP | 125 mL O2 no PEG | < 1 | 25 | 75 | 430 | 8 h |
| 30 nm ONPs | 125 mL O2 | 1 | 29 | 70 | 3100 | 6 h |
| 35 nm ONPs[c] | H2O/DMF C6H5IO | 70 | 11 | 19 | 16500 | 8 h |
| 120 nm ONPs[c] | H2O/DMF C6H5IO | 85 | 6 | 10 | 12000 | 8 h |
Solution reactions: 0.1 mm catalyst in acetonitrile/methanol (3:1, 2.75 mL); porphyrin/cyclohexene/H2O2 = 1:2000:3000. This is similar to the cyclooctene oxidations reported previously.[1–4]
ONP reactions: ONP suspension (2.5 mL, 70 μm, 1.75×10−7 mol of porphyrin); porphyrin/substrate/H2O2 = 1:2000:3000. Alternatively, the porphyrin-ONP suspension (2.5 mL) was mixed with cyclohexene (200 μL) and O2 (125 mL, 1 atm); porphyrin/substrate/O2 1:16 000:40 000.
ONPs made from DMF as host solvent under conditions used to obtain this size nanoparticle.[10]
t1/2 for total products formed.
All reactions were run exhaustively. TON = molproducts/molporphyrin has an error of ±5%. Products were extracted into CH2Cl2 and analyzed by using an Agilent 5975 series GC-MS. Control reactions: neither H2O2 nor O2 react directly with cyclohexene under these conditions. (See Experimental Section and the Supporting Information.).
The TON is defined as the total amount of products (ketone/alcohol 3:1) formed per porphyrin, and since the porphyrin slowly decomposes, these reactions are run until [metalloporphyrin]<0.2 mm. This represents a greener alternative to effect these organic transformations since dioxygen is efficiently used as oxidant in place of H2O2 or other synthetic oxygen sources, and the reaction solvent is 89% water.
The increased TON is contrary to expectations because the metalloporphyrins are in close proximity in the ONPs, which should enhance oxidative degradation of the catalyst and cause a significant decrease in catalytic turnovers. There have been significant efforts to isolate porphyrinoid catalysts (see above). Furthermore, the allylic oxidation products suggest a different mechanism compared to that of the corresponding solvated metalloporphyrin.[1–4] Control reactions in the absence of an oxygen source or metalloporphyrin result in no product formation. Adding 3% each of water and polyethylene glycol (PEG) to the homogeneous reaction mixture has no effect on the product ratios or TON, that is, only the epoxide is formed, H2O2 is required, and a TON of 350. The UV/Vis spectra of the exhausted reaction mixtures reveals that eventually the metalloporphyrin in solution or as an ONP decomposes[69] (see the Supporting Information). Unlike the solution phase reactions, the slow addition of H2O2 to the ONP suspension through a syringe pump results in modest yields of the allylic products, whereas addition of a 30% H2O2 solution in one aliquot degrades the porphyrin within a few minutes as observed by UV/Vis spectra. These observations indicate that the hierarchical organization of the metalloporphyrins in the ONPs is key to the observed activity.
Nanoparticle structure
The detailed structural arrangement of the [FeIII(tppf20)] within the ONPs is not known because the intermolecular forces used to self-organize the molecules into nanoparticles are weak, non-specific, and reversible.[9, 10] As discussed above, the nanoarchitecture of porphyrin molecules within the ONPs strongly depends on the component molecules, solvents, and mode of preparation.[9–12, 70, 71] Densely packed, 10 nm diameter ONPs of [FeIII(tppf20)] (ca. 1.75 nm × 1.75 nm × 1.0 nm ≈ 3 nm3) can contain up to about 200 porphyrins, but this represents an upper limit. Experiments that combine ONPs composed of different porphyrins indicate that in solution under ambient conditions, the porphyrins do not exchange between the ONPs. It is also likely that some PEGs and host–guest solvents may be present inside the ONPs. Neither AFM nor XRD indicate crystalline material.
Note that for all of the ONPs of iron porphyrins we have studied to date, the rates of the oxidation reactions are about 60 times slower than those for the corresponding metalloporphyrin in solution. The data in Table 1 show that catalytic activity depends on particle size and host solvent, which likely indicates differences in the nanoarchitectures of the porphyrins in the ONPs. For example, the oxidation of cyclohexene by large ONPs (ca. 120 nm diameter) of [FeIII(tppf20)] by using DMF as a host solvent requires H2O2 or iodosylbenzene and results in epoxidation similar to the homogeneous system, yet has an approximately 30-fold greater TON than the solution phase reaction.
Catalytic insights
There is still considerable discussion over the mechanisms of hydrocarbon oxidation by iron porphyrins and other metalloporphyrins,[1–4, 29, 34, 48, 72, 73] wherein the two dominant proposed mechanisms are: a) a radical hydrogen-abstraction–oxygen-rebound mechanism and b) an oxygen- (or hydroxyl-)insertion reaction that proceeds through a cationic ROH2+ species. It appears that different mechanisms are operative with different iron porphyrin systems depending on factors such as macrocycle ligand field, axial ligands, and solvent. Because the ONPs are dynamic in that the porphyrins can reorganize upon changing the environment, probing the mechanism of catalysis is particularly challenging, and detailed mechanistic studies that parallel several decades of metalloporphyrin research is beyond the scope of one manuscript.
For the ONP systems, the intermolecular interactions such as π stacking have significant effects on electronic structure of the macrocycle as shown by the substantially broadened UV/Vis spectra. Substrate accessibility and orientation to the reactive centers may be different compared to the solvated metalloporphyrin. Considering the close proximity of the [FeIII(tppf20)] in the ONPs, the formation of μ-oxo and/or dioxo dimers may also play an important role, as the former species is known to have increased catalytic activity in benzylic oxidations at elevated temperatures.[61] In the absence of steric effects on substrate binding, the C–H-bond energies should correlate with expected oxidation rates (i.e., allylic oxidations should be more facile than those at the 2° positions). Since the addition of about 3% each of water and PEG to the solution-phase reaction in acetonitrile/methanol has no effect on the product ratio or the TON, the organization of the metalloporphyrins inside the ONPs is the primary cause of the observed differences in the catalytic process. The rapid decomposition of [FeIII(tppf20)] in the ONPs in the H2O2 reactions is consistent with the close proximity of the metalloporphyrins, and the allylic products indicate that the oxidative mechanism with this oxygen source is similar to the O2 reactions. The PEG used to stabilize the suspension from precipitation plays a role in the reactivity of the ONPs, since about 10 nm diameter ONPs formed without PEG (which do not precipitate for about 3 days) result in the same 3:1 ketone/alcohol formation by using O2, but with a TON of only around 430. Thus, in addition to a different catalytic mechanism, the observed activity is the result of the slower rate of self-oxidation for the O2 reactions, the organization of the metalloporphyrins in the ONPs, and the partition of the hydrophobic substrate from the aqueous solvent into the ONPs.
Isotope experiments
Note that under a variety of conditions the ketone/alcohol product ratio is about 3:1. Experiments probing the source of the incorporated oxygen were used to garner insights into the mechanism of ONP catalysis. These preliminary investigations are consistent with previously reported radical mechanisms for reactions that result in allylic oxidations, for example, Mn, Sn, or Ru porphyrins, and are akin to the P450 reactions.[30, 32, 74] The oxygen in the products may originate from the water and/or the O2. GC-MS analysis of reactions run with nanoparticles that were preparated with water containing 10% H218O indicates that about 10% 18O in the ketone and no additional 18O in the alcohol is observed within the error of the experiment. When 98% 18O2 is used in the reaction <8% of the ketone and around 90% of the alcohol products contain 18O. These results indicate that the oxygen in the alcohol comes primarily from O2, but oxygen in the ketone also originates from water. The most likely explanation for the observed incorporation of oxygen from water into the ketone is the reversible formation of the acetal or hydrate. Reactions run in 99.6% D2O result in no incorporation of deuterium into the products, but some exchange with the alcohol proton. The role of water in the mechanism is supported by the narrow pH window in which the ONP catalyst is active, see below.
The presence of an alcohol to form an active axially bound [(tppf20)Fe(HOCH3)]+ adduct is reported to be important for cyclooctene-epoxidation reactions by this complex in solution,[1–4] and in the case of the ONPs the alcohol moiety on the PEG can initially serve in this capacity. During the course of the reaction, the 2-cyclohexene-1-ol may become an axial ligand and be activated towards further oxidation to yield the major product, the ketone, and water may be involved in this second step. Hydroxide or water may serve as axial ligands, but since no products are found outside the range of pH 6.5–7.0 hydroxide may quench the reaction by irreversibly binding to the iron centre or by changing the redox potential.[73] The lack of D or 18O incorporation from water into the alcohol argues against free radical reactions, wherein the intermediates escape from the ONP cage. The cyclohexene epoxide is cleanly converted by these ONPs to the gem diol, thus the epoxide is not an intermediate of the alcohol and ketone formation. Only traces of the ketone are observed when the 2-cyclohexene-1-ol is used as a substrate because its octanol/water partition coefficient is 40-fold less than the on for cyclohexene (k2-cyclohexene-1-ol = 18 vs. kcyclohexene = 730) and thus, it does not partition into the ONPs as well.
Iron centre
The nearly 20 ppm shift for the β-pyrrole resonances (δ = 80 ppm to δ = 62 ppm; 10 mm in CD3CN/MeOH 3:1) in the NMR spectra reported[4] indicates that the paramagnetism of [FeIII(tppf20)] in solution is reduced upon addition of methanol to form [(tppf20)Fe(HOCH3)]+. Our NMR studies on [FeIII(tppf20)] in acetonitrile/methanol indicate the likely presence of more than one paramagnetic species in solution under aerobic conditions since we cannot accurately calculate the effective magnetic moment, μeff, from paramagnetic shifts of the solvent resonances,[75] which also slowly change with time. This may be an indication of a shift from a 5/2 to a 3/2 system and/or for the formation of oxygenated species. Similarly, the μeff of the iron porphyrin in the ONPs under aerobic conditions and during catalysis has been difficult to ascertain by the Evans method NMR experiments.[76] A diminished paramagnetic shift for the β-pyrrole H is observed relative to the solution-phase complex under identical conditions (δ = 42 ppm, 7 mm in CD3CN/MeOH 3:1), and a smaller difference in the residual solvent resonances is observed. Though it is clear that at least some metalloporphyrins remain paramagnetic, the reduction in the paramagnetic shifts may be an indicator of antiferomagnetically coupled oxo-bridged dimers.[77] In principle these oxidation reactions can be influenced by a magnetic field if there are paramagnetic species, porphyrin or organic intermediate, in the rate-determining step in the reaction mechanism,[78, 79] but we observed no differences in the product ratios between reactions stirred magnetically (ca. 800 G) or mechanically by a shaker.
Our working hypothesis is that the close proximity of the iron porphyrins in the ONPs facilitates the formation of μ-dioxo-bridged dimers and/or μ-oxo-bridged dimers that have known enhanced catalytic activity in terms of alkane hydroxylation.[38, 61] Under the reaction conditions, the FeIV–oxo monomers may be in equilibrium.[29] Coordination of water, hydroxide, and the alcohol moiety of PEG may affect the stability or reactivity of the iron–oxo complexes. The importance of axial ligands is consistent with the observation that imidazole axial ligands block the incorporation of the oxygen from water in epoxidations by [FeIII(tppf20)].[80] The absence of cyclohexene oxide as a significant product in nanoparticle catalysis argues against the presence of significant amounts of solvated [FeIII(tppf20)] in the nanoparticle catalysis. However, since homogeneous reactions in acetonitrile/methanol (3:1) with a few percent water and PEG yield only the epoxide, axial coordination by these solvents does not explain the ONP results. The role of protic solvents on the reaction mechanism of the reaction in solution has been discussed in terms of proton transfer before the heterolytic cleavage of the O–O bond in the adduct hydrogen peroxide.[4] These results indicate that the hierarchical structure of the macrocycles in the ONPs is the dominant factor in the mechanistic differences. The products and greater TONs may be consistent with a radical-initiated reaction mechanism as suggested by Labinger et al.,[48–50] or shown by the use of [FeIII(tppf20)] in super critical CO2 and 20 atm O2.[67, 81] When O2 is the limiting reagent in the ONP system the TON is reduced significantly, indicating the importance of oxygen in the catalysis and that the extent of radical-chain reactions is limited.
Deformation of the otherwise planar macrocycle by steric crowding of the peripheral substituents has been proposed as a major source of reactivity differences in the perhalogenated metalloporphyrins relative to metal complexes of tppf20 and other arylporphyrins.[50] Nonplanar metalloporphyrins are known to have significantly different photonic properties including the dynamics of axial ligand binding.[82–84] The porphyrin in [FeIII(tppf20)] is not distorted[75] and these macrocycles likely adopt a nearly planar conformation in the ONPs because the intermolecular forces between the nanoparticle components are too weak to force the macrocycles into energetically unfavorable conformations. In addition, nonplanar porphyrins are characterized by broad red-shifted Soret bands, and the optical spectra of the ONPs are broad but contain several underlying peaks shifted to the red and the blue of those of the parent complex consistent with porphyrin J and H aggregation.
ONP mechanism
One plausible explanation for the increased TON is that the metalloporphyrins on the exterior of the ONPs are rigidly held in a structure that diminishes, but does not eliminate, the oxidation of one macrocycle by another. When the porphyrins on the exterior of the ONPs eventually decompose they fall off because of the greatly increased polarity of the oxidized product, thereby exposing the next layer of catalytically active molecules. This onion-type mechanism may account for the slow rates of catalysis by the present ONPs since fewer catalytic sites are available at a given time. Since cyclohexene is hydrophobic and the reaction solvent is mostly water, the substrate rapidly partitions into the ONPs as indicated by UV/Vis spectral shifts. This significantly increases the concentration of the substrate proximal to the catalytic sites.
There are two consequences of this concentrator effect:[85, 86] the presence of more substrate diminishes the probability of inter-porphyrin oxidation. Since the ketone is the dominant product, an initially formed alcohol may be further oxidized before it escapes the ONP cage (see above). This partitioning of the substrate is supported by the observation that ONPs of iron(III) tetra(4-phenylsulfonate)porphyrin, made by adding DMF or acetonitrile/methanol to an aqueous solution of the porphyrin, are inactive in this reaction, and suggests that the cyclohexene substrate does not partition into these water containing ONPs. The reduced TONs but similar product ratios of suspensions of ONPs without PEG, see above, may indicate that PEG also serves as a phase-transfer agent. The last observation further emphazises the importance of the organization and composition of the metalloporphyrins in the ONPs.
Conclusions
These results illustrate that ONP materials composed of porphyrins can display unique properties, or in this case, unexpected catalytic activities relative to the component molecules. These functionalities arise from the self-organized architecture inside the ONPs. Other nanoscaled materials of porphyrins, such as tubes, rods, and crystals, can be formed from simple commercially available compounds or from more complex molecular designs.[18, 87] Self-assembled porphyrinic materials generally require specifically designed recognition motifs in predefined geometries to affect specific architectures. Conversely, the construction of self-organized materials, such as ONPs, does not require complex exocyclic moieties.[12, 71, 88, 89] Thus, the self-organization strategy obviates the need for macrocycles that are synthetically challenging and the result of low-yield procedures. Porphyrins bearing the same substituent at the four meso positions are easy to prepare in large scales, and can be prepared in green, solventless reactions.[90] The [FeIII(tppf20)] ONP catalyst system represents an advance in green chemistry since despite numerous efforts in catalyst discovery and design there are still few molecular-based catalysts that can perform oxidation reactions under mild conditions by activation of O2 and in water. The metalloporphyrins in the ONP catalysts reported herein are organized by weak intermolecular interactions, so the structure is dynamic.
The dynamic organization of the molecules may enable the ONPs to adapt to a variety of substrates with different topologies. Preliminary work shows that the allylic ketones and alcohols of R(+)-limonene are formed under the same conditions. Although other inorganic and metallic systems can be superior alkene oxidation catalysts than the present metalloporphyrin ONPs in terms of TONs, and the epoxide is a versatile intermediate, there are numerous organic transformations requiring mild allylic oxidations. Because allylic oxidations are widely used in small scale reactions and in commercial organic synthesis, more efficient and greener methods to accomplish this transformation are of interest.[91–93] The allylic oxidation of alkenes by SeO2 (to yield the alcohol) and other reagents (to yield the ketone) have been used in organic synthesis for many decades, and the mechanisms of allylic reactions proceed through an array of complex mechanisms.[94] Our chemistry reported here is greener in that it is less toxic than SeO2 reactions.
As stated in the previous reports and here, the exact organization of the porphyrins in the aggregates are unknown, but differences in the electronic spectra indicate varying degrees of H versus J aggregates depending on preparative methods.[9] The spectroscopic signatures and particle sizes are quite different for the same metalloporphyrin, in this case [FeIII(tppf20)], prepared from different solvent systems, different solvent ratios, different mixing conditions, and different temperatures. Previously reported catalysis with the [FeIII(tppf20)] nanoparticles used DMF as the host solvent and iodosylbenzene as the oxygen source to form the epoxide as the major product.[10] We hypothesize that: a) there are differences in the axial coordination, b) that the iodosylbenzene partitions rapidly into the ONPs, and c) that the resulting Fe–oxo species is different than what is formed upon dioxygen binding.
Given the great variety of porphyrinoids and their metal complexes, the full potential of self-organized organic nanoparticles is yet uncharted.
Experimental Section
Materials and instrumentation
[FeIII(tppf20)], cyclohexene oxide, 2-cyclohexene-1-ol, 2-cyclohexene-1-one, and polyethylene glycol monomethyl ether (PEG164) were purchased from Aldrich Chemical Co. The solvents (tetrahydrofuran, toluene, 99.9% acetonitrile, 99.9%, methanol, and HPLC-grade dichloromethane), cyclohexene and 30% H2O2 were purchased from Fisher Scientific Co. Nanopure water was obtained by using Barnstead Nanopure water system. D2O (99.6%), was obtained from Cambridge Isotope laboratories Inc. H218O (10%) and 18O2 (98%) were obtained from Sigma–Aldrich.
Product analyses were performed by using GC-MS Agilent 5975 series system with HP-5 column (HP-5MS 30 m × 0.250 mm, 0.25 micron nominal, 5% phenylmethyl siloxane). Electronic spectra were recorded on Cary Bio-3 UV/Vis spectrophotometer. A Precision Detector PD2000DLS Cool-Batch dynamic light scattering instrument was used in batch mode at 25°C to determine the particle size. A Veeco Nanoscope III Multi-mode AFM was used to examine the ONPs on surfaces. A Fisher SF15 sonicator was used for nanoparticle preparations.
Reactions
Reactions were performed at ambient temperature. All reactions were run a minimum of five times except the isotope experiments, which were repeated three times, and the reported data represent the average of these reactions. All reactions were agitated by using a magnetic stirring bar unless otherwise noted. Although purchased as the chloride, since the counter ion on the metalloporphyrin is unknown in the ONP solution and in equilibrium in the solution-phase reactions, it is not specified. For the homogeneous, protic solvent reactions, a 1.0 mm stock solution of the iron porphyrin complex in acetonitrile/methanol (3:1) was used. The reaction was initiated in a 9 mL screw-capped vial by mixing of the 1.0 mm porphyrin stock solution (250 μL) with acetonitrile/methanol (2.5 mL, 3:1 for a final concentration of 0.1 mm) and cyclohexene (50 μL). Whereupon 30% H2O2 (80 μL) was slowly added to the reaction through a Teflon cannula securely fitted through a hole in the cap by using a syringe pump over the course of 80 min (1 μL min−1). The reaction was stirred for four hours (UV/Vis spectra analysis indicated that most of the porphyrin has decomposed by ca. 30 min). Ratio of porphyrin/substrate/H2O2 = 1:2000:3000 equivalents. An aliquot of the reaction was analyzed by GC-MS and product yields were determined relative to an added internal standard (toluene). Of the three possible products, cyclohexene oxide was obtained in greater than 99% (see the Supporting Information). The results are shown in Table 1.
For ONP reactions, 5.6 mL batches of the [FeIII(tppf20)] ONPs were prepared in 10 mL vials (or test tubes) by adding nanopure water (5.0 mL) to a mixture of PEG (0.2 mL) and a 1.0 mm solution of [FeIII(tppf20)] in THF (0.4 mL) while sonicating (the ONP suspension is 70 mm, 4.0 × 10−7 mol of porphyrin). The solution was further sonicated for 1 min. The prepared nanoparticles are stable for more than four weeks and were stored in a refrigerator at about 4°C. Each batch of nanoparticles was checked by dynamic light scattering (DLS) and UV/Vis absorption spectroscopy. The solutions appear slightly cloudy. The ONP solution had a pH 6.5–7.0. The porphyrin–ONP stock solution (2.5 mL, 70 mm, 1.75 × 10−7 mol of porphyrin) was mixed in a 9.5 mL screw-capped vial with cyclohexene (25 μL) and 30% H2O2 (40 μL), which was slowly added to the reaction through a teflon cannula securely fitted through a hole in the cap over the course of 40 min. Ratio of porphyrin/substrate/H2O2 = 1:2000:3000 equivalents. The reaction mixture was stirred for about 24 h. The reaction mixture (2.6 mL) was extracted thoroughly once with dichloromethane (2.8 mL) and the layers were allowed to separate. The water fraction and some of the organic fraction was removed to leave a total volume of 2.0 mL (this assures the same volume for every assay). Toluene (20 μL, 1.88 × 10−4 mol) was added to the 2.0 mL extract as an internal standard, whereupon 4.0 μL were diluted with dichloromethane (1.0 mL); 2.0 μL of this last solution was injected into the GC-MS.
Reactions by using O2 were run as follows: The porphyrin–nanoparticles stock solution (2.5 mL, 70 μm, 1.75 × 10−7 mol of porphyrin) was mixed with cyclohexene (200 μL) in a 25 mL pear-shaped flask fitted to a 125 mL separatory funnel filled with O2 (1 atm, filled by flushing the vessel three times with O2). The O2 was added by opening the stopcock. The reactions were run for 24 h (porphyrin/substrate/O2 = 1:16 000:40 000). For oxygen reactions, the pear-shaped flask was cooled in an ice bath for about 20 min with the stopcock of the separatory funnel open to condense all volatile organic species. The reaction volume (2.7 mL) was extracted once with dichloromethane (8 mL) and the layers were allowed to separate. The water fraction and some of the organic fraction was removed to leave a total volume of 6.0 mL (this assures the same volume for every reaction assay). To this volume toluene (20 μL, 1.88 × 10−4 mol) was added as an internal standard and 4.0 mL of the solution were diluted with dichloromethane (1.0 mL); 2.0 μL of this solution was injected into the GC-MS.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CHE-0554703), the National Institutes of Health (SCORE S06M60654). Hunter College Chemistry infrastructure is supported by the National Science Foundation, the National Institutes of Health, including the RCMI program (G12-RR-03037), and the City University of New York.
Footnotes
Supporting information for this article (results from control reactions, characterization of the ONPs, standardizations, and representative GC of the products from a catalytic reaction with O2) is available on the WWW under http://dx.doi.org/10.1002/chem.200901086.
References
- 1.Stephenson NA, Bell AT. J Am Chem Soc. 2005;127:8635–8643. doi: 10.1021/ja043380n. [DOI] [PubMed] [Google Scholar]
- 2.Stephenson NA, Bell AT. Inorg Chem. 2006;45:5591–5599. doi: 10.1021/ic0521067. [DOI] [PubMed] [Google Scholar]
- 3.Stephenson NA, Bell AT. Inorg Chem. 2006;45:2758–2766. doi: 10.1021/ic0521776. [DOI] [PubMed] [Google Scholar]
- 4.Stephenson NA, Bell AT. Inorg Chem. 2007;46:2278–2285. doi: 10.1021/ic060757c. [DOI] [PubMed] [Google Scholar]
- 5.Astruc D, Lu F, Aranzaes JR. Angew Chem. 2005;117:8062–8083. doi: 10.1002/anie.200500766. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:7852–7872. doi: 10.1002/anie.200500766. [DOI] [PubMed] [Google Scholar]
- 6.Scott RWJ, Wilson OM, Crooks RM. J Phys Chem B. 2005;109:692–704. doi: 10.1021/jp0469665. [DOI] [PubMed] [Google Scholar]
- 7.Horn D, Rieger J. Angew Chem. 2001;113:4460–4492. [Google Scholar]; Angew Chem Int Ed. 2001;40:4330–4361. doi: 10.1002/1521-3773(20011203)40:23<4330::aid-anie4330>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 8.LaMer VK, Dinegar RH. J Am Chem Soc. 1950;72:4847–4854. [Google Scholar]
- 9.Drain CM, Smeureanu G, Patel S, Gong X, Garno J, Arijeloye J. New J Chem. 2006;30:1834–1843. [Google Scholar]
- 10.Gong X, Milic T, Xu C, Batteas JD, Drain CM. J Am Chem Soc. 2002;124:14290–14291. doi: 10.1021/ja027405z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drain CM, Bazzan G, Milic T, Vinodu M, Goeltz JC. Isr J Chem. 2005;45:255–269. [Google Scholar]
- 12.Drain CM, Goldberg I, Sylvain I, Falber A. Top Curr Chem. 2005;245:55–88. [Google Scholar]
- 13.Brick MC, Palmer HJ, Whitesides TH. Langmuir. 2003;19:6367–6380. [Google Scholar]
- 14.Qian DJ, Nakamura C, Wakayama T, Miyake J. J Porphyrins Phthalocyanines. 2003;7:415–419. [Google Scholar]
- 15.Van Keuren E, Bone A, Ma C. Langmuir. 2008;24:6079–6084. doi: 10.1021/la800290s. [DOI] [PubMed] [Google Scholar]
- 16.Sane A, Taylor S, Sun Yp, Thies MC. Chem Commun. 2003:2720–2721. doi: 10.1039/b306967b. [DOI] [PubMed] [Google Scholar]
- 17.Sane A, Thies MC. J Phys Chem B. 2005;109:19688–19695. doi: 10.1021/jp0581072. [DOI] [PubMed] [Google Scholar]
- 18.Lee SJ, Hupp JT. Coord Chem Rev. 2006;250:1710–1723. [Google Scholar]
- 19.Rangel-Rojo R, Matsuda H, Kasai H, Nakanishi H. J Opt Soc Am B. 2000;17:1376–1382. [Google Scholar]
- 20.Nitschke C, O'Flaherty SM, Kroll M, Blau WJ. J Phys Chem B. 2004;108:1287–1295. [Google Scholar]
- 21.Mülller RH, Benita S, Böhm BHL. Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs. Scientific Publishers; Stuttgart: 1998. [Google Scholar]
- 22.Park J, Privman V, Matijevic E. J Phys Chem B. 2001;105:11630–11635. [Google Scholar]
- 23.Groves JT, Nemo TE. J Am Chem Soc. 1983;105:5786–5791. [Google Scholar]
- 24.Groves JT, Nemo TE, Myers RS. J Am Chem Soc. 1979;101:1032–1033. [Google Scholar]
- 25.Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. J Am Chem Soc. 1981;103:2884–2886. [Google Scholar]
- 26.Groves JT, Watanabe Y. Inorg Chem. 1986;25:4808–4810. [Google Scholar]
- 27.Groves JT, Viski P. J Org Chem. 1990;55:3628–3634. [Google Scholar]
- 28.Groves JT. Proc Natl Acad Sci USA. 2003;100:3569–3574. doi: 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groves JT. J Inorg Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. [Google Scholar]
- 31.Mayer JM. In: Biomimetic oxidations catalyzed by transition-metal complexes. Meunier B, editor. Imperial College Press; London: 2000. pp. 1–43. [Google Scholar]
- 32.Newcomb M, Hollenberg PF, Coon MJ. Arch Biochem Biophys. 2003;409:72–79. doi: 10.1016/s0003-9861(02)00445-9. [DOI] [PubMed] [Google Scholar]
- 33.Chandrasena REP, Vatsis KP, Coon MJ, Hollenberg PF, Newcomb M. J Am Chem Soc. 2004;126:115–126. doi: 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- 34.Meunier B, deVisser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 35.Silaghi-Dumitrescu R. J Biol Inorg Chem. 2004;9:471–476. doi: 10.1007/s00775-004-0543-2. [DOI] [PubMed] [Google Scholar]
- 36.Dolphin D, Traylor TG, Xie LY. Acc Chem Res. 1997;30:251–259. [Google Scholar]
- 37.Barloy L, Battioni P, Mansuy D. J Chem Soc Chem Commun. 1990:1365–1367. [Google Scholar]
- 38.Mansuy D. Coord Chem Rev. 1993;125:129–141. [Google Scholar]
- 39.Wang CQ, Shalyaev KV, Bonchio M, Carofiglio T, Groves JT. Inorg Chem. 2006;45:4769–4782. doi: 10.1021/ic0520566. [DOI] [PubMed] [Google Scholar]
- 40.Merlau ML, Cho SH, Sun SS, Nguyen ST, Hupp JT. Inorg Chem. 2005;44:5523–5529. doi: 10.1021/ic0505596. [DOI] [PubMed] [Google Scholar]
- 41.Groves JT. J Porphyrins Phthalocyanines. 2000;4:350–352. [Google Scholar]
- 42.Suslick KS. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 4. Academic Press; New York: 2000. pp. 41–60. [Google Scholar]
- 43.Simonneaux G, Le Maux P, Ferrand Y, Rault-Berthelot J. Coord Chem Rev. 2006;250:2212–2221. [Google Scholar]
- 44.Ungashe SB, Groves JT. Adv Inorg Biochem. 1994;9:317–351. [PubMed] [Google Scholar]
- 45.Ellis PE, Lyons JE. Catal Lett. 1989;3:389–397. [Google Scholar]
- 46.Lyons JE, Ellis PE. Catal Let. 1991;8:45–51. [Google Scholar]
- 47.Bartoli JF, Brigaud O, Battioni P, Mansuy D. J Chem Soc Chem Commun. 1991:440–442. [Google Scholar]
- 48.Grinstaff MW, Hill MG, Labinger JA, Gray HB. Science. 1994;264:1311–1313. doi: 10.1126/science.8191283. [DOI] [PubMed] [Google Scholar]
- 49.Labinger JA. Catal Lett. 1994;26:95–99. [Google Scholar]
- 50.Grinstaff MW, Hill MG, Birnbaum ER, Schaefer WP, Labinger JA, Gray HB. Inorg Chem. 1995;34:4896–4902. [Google Scholar]
- 51.Doro FG, Smith JRL, Ferreira AG, Assis MD. J Mol Catal A Chem. 2000;164:97–108. [Google Scholar]
- 52.Traylor TG, Kim C, Fann WP, Perrin CL. Tetrahedron. 1998;54:7977–7986. [Google Scholar]
- 53.Traylor TG, Xu F. J Am Chem Soc. 1990;112:178–186. [Google Scholar]
- 54.Moore KT, Horvath IT, Therien MJ. Inorg Chem. 2000;39:3125–3139. doi: 10.1021/ic000284o. [DOI] [PubMed] [Google Scholar]
- 55.Nam W, Han HJ, Oh SY, Lee YJ, Choi MH, Han SY, Kim C, Woo SK, Shin W. J Am Chem Soc. 2000;122:8677–8684. [Google Scholar]
- 56.Battioni P, Renaud JP, Bartoli JF, Reinaartiles M, Fort M, Mansuy D. J Am Chem Soc. 1988;110:8462–8470. [Google Scholar]
- 57.Higuchi T, Shimada K, Maruyama N, Hirobe M. J Am Chem Soc. 1993;115:7551–7552. [Google Scholar]
- 58.Traylor TG, Popovitz-Biro R. J Am Chem Soc. 1988;110:239–243. [Google Scholar]
- 59.Yamaguchi K, Watanabe Y, Morishima I. J Am Chem Soc. 1993;115:4058–4065. [Google Scholar]
- 60.Rosenthal J, Pistorio BJ, Chng LL, Nocera DG. J Org Chem. 2005;70:1885–1888. doi: 10.1021/jo048570v. [DOI] [PubMed] [Google Scholar]
- 61.Evans S, Smith JRL. J Chem Soc Perkin Trans 2. 2001:174–180. [Google Scholar]
- 62.Herron N, Tolman CA. J Am Chem Soc. 1987;109:2837–2839. [Google Scholar]
- 63.Bedioui F. Coord Chem Rev. 1995;144:39–68. [Google Scholar]
- 64.Battioni P, Lallier JP, Barloy L, Mansuy D. J Chem Soc Chem Commun. 1989:1149–1151. [Google Scholar]
- 65.Benaglia M, Danelli T, Fabris F, Sperandio D, Pozzi G. Org Lett. 2002;4:4229–4232. doi: 10.1021/ol0267230. [DOI] [PubMed] [Google Scholar]
- 66.Merlau ML, Mejia MdP, Nguyen ST, Hupp JT. Angew Chem. 2001;113:4369–4372. [Google Scholar]; Angew Chem Int Ed. 2001;40:4239–4242. doi: 10.1002/1521-3773(20011119)40:22<4239::AID-ANIE4239>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 67.Kokubo Y, Wu XW, Oshima Y, Koda S. J Supercrit Fluids. 2004;30:225–235. [Google Scholar]
- 68.Moore KT, Fletcher JT, Therien MJ. J Am Chem Soc. 1999;121:5196–5209. [Google Scholar]
- 69.Prakash P, Francisca Mary LJ. J Serb Chem Soc. 2005;70:1105–1111. [Google Scholar]
- 70.Drain CM, Batteas JD, Smeureanu G, Patel S. In: Dekker Encyclopedia of Nanoscience and Nanotechnology. Schwartz JA, Contescu CI, Putyera K, editors. Vol. 5. Marcel Dekker; New York: 2004. pp. 3481–3502. [Google Scholar]
- 71.Drain CM, Chen X. In: Encyclopedia of Nanoscience and Nanotechnology. Nalwa HS, editor. Vol. 9. American Scientific Press; New York: 2004. pp. 593–616. [Google Scholar]
- 72.Limberg C. Angew Chem. 2003;115:6112–6136. [Google Scholar]; Angew Chem Int Ed. 2003;42:5932–5954. doi: 10.1002/anie.200300578. [DOI] [PubMed] [Google Scholar]
- 73.Wolak M, van Eldik R. Chem Eur J. 2007;13:4873–4883. doi: 10.1002/chem.200601148. [DOI] [PubMed] [Google Scholar]
- 74.Lyons JE, Ellis PE., Jr . In: Metalloporphyrins in catalytic oxidations. Sheldon RA, editor. Marcel Dekker; New York: 1994. pp. 297–324. [Google Scholar]
- 75.Oh Y, Shin BC, Swenson D, Goff HM, Kang SK. Acta Crystallogr Sect C. 2004;60:m57–m59. doi: 10.1107/S0108270103028051. [DOI] [PubMed] [Google Scholar]
- 76.a) Evans DF. J Chem Soc. 1959:2003–2005. [Google Scholar]; b) Grant DH. J Chem Educ. 1995;72:39–40. [Google Scholar]
- 77.Kumar Patra A, Mukherjee R. Polyhedron. 1999;18:1317–1322. [Google Scholar]
- 78.Hughes ME, Corden BB. J Am Chem Soc. 1989;111:4110–4111. [Google Scholar]
- 79.Perito RP, Corden BB. J Am Chem Soc. 1987;109:4418–4419. [Google Scholar]
- 80.Lee KA, Nam W. J Am Chem Soc. 1997;119:1916–1922. [Google Scholar]
- 81.Lim MH, Jin SW, Lee YJ, Jhon GJ, Nam W, Kim C. Bull Korean Chem Soc. 2001;22:93–96. [Google Scholar]
- 82.Drain CM, Kirmaier C, Medforth CJ, Nurco DJ, Smith KM, Holten D. J Phys Chem. 1996;100:11984–11993. [Google Scholar]
- 83.Drain CM, Gentemann S, Roberts JA, Nelson NY, Medforth CJ, Jia S, Simpson MC, Smith KM, Fajer J, Shelnutt JA, Holten D. J Am Chem Soc. 1998;120:3781–3791. [Google Scholar]
- 84.Retsek JL, Drain CM, Kirmaier C, Nurco DJ, Medforth CJ, Smith KM, Sazanovich IV, Chirvony VS, Fajer J, Holten D. J Am Chem Soc. 2003;125:9787–9800. doi: 10.1021/ja020611m. [DOI] [PubMed] [Google Scholar]
- 85.Hecht S, Ihre H, Frechet JMJ. J Am Chem Soc. 1999;121:9239–9240. [Google Scholar]
- 86.Frechet JMJ. Proc Natl Acad Sci USA. 2002;99:4782–4787. doi: 10.1073/pnas.082013899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qian DJ, Nakamura C, Ishida T, Wenk SO, Wakayama T, Takeda S, Miyake J. Langmuir. 2002;18:10237–10242. [Google Scholar]
- 88.Garno JC, Xu C, Bazzan G, Batteas JD, Drain CM. In: Metal-Containing and Metallosupramolecular Polymers and Materials. Schubert US, Newcome GR, Manners I, editors. Vol. 928. ACS; Washington: 2006. pp. 168–183. [Google Scholar]
- 89.Milic T, Garno JC, Batteas JD, Smeureanu G, Drain CM. Langmuir. 2004;20:3974–3983. doi: 10.1021/la0359023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drain CM, Gong X. Chem Commun. 1997:2117–2118. [Google Scholar]
- 91.Andrus MB, Lashley JC. Tetrahedron. 2002;58:845–866. [Google Scholar]
- 92.Crich D, Zou Y. J Org Chem. 2005;70:3309–3311. doi: 10.1021/jo0500333. [DOI] [PubMed] [Google Scholar]
- 93.Yu JQ, Corey EJ. J Am Chem Soc. 2003;125:3232–3233. doi: 10.1021/ja0340735. [DOI] [PubMed] [Google Scholar]
- 94.Singleton DA, Hang C. Tetrahedron Lett. 1999;40:8939–8943. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.