

Abstract
Oxidation of proteins and peptides is a common phenomenon, and can be employed as a labeling technique for mass-spectrometry-based proteomics. Nonspecific oxidative labeling methods can modify almost any amino acid residue in a protein or only surface-exposed regions. Specific agents may label reactive functional groups in amino acids, primarily cysteine, methionine, tyrosine, and tryptophan. Nonspecific radical intermediates (reactive oxygen, nitrogen, or halogen species) can be produced by chemical, photochemical, electrochemical, or enzymatic methods. More targeted oxidation can be achieved by chemical reagents but also by direct electrochemical oxidation, which opens the way to instrumental labeling methods. Oxidative labeling of amino acids in the context of liquid chromatography(LC)–mass spectrometry (MS) based proteomics allows for differential LC separation, improved MS ionization, and label-specific fragmentation and detection. Oxidation of proteins can create new reactive groups which are useful for secondary, more conventional derivatization reactions with, e.g., fluorescent labels. This review summarizes reactions of oxidizing agents with peptides and proteins, the corresponding methodologies and instrumentation, and the major, innovative applications of oxidative protein labeling described in selected literature from the last decade.
Keywords: Protein oxidation, Radical, Protein footprinting, Peptide cleavage
Introduction
The development of analytical methods for oxidatively modified amino acid residues has benefited from the study of oxidative damage to proteins related to ageing and disease [1, 2]. This review focuses on applications of oxidative protein labeling in mass-spectrometry (MS)-based proteomics.
Proteomics experiments typically aim at the identification and subsequent quantitation of as many proteins as possible in a sample. Often, however, a subset of proteins of interest needs to be quantified, for example, as potential biomarkers. Labeling, whether oxidative or nonoxidative, changes properties of peptides and proteins, which can be used to improve sample cleanup, high performance liquid chromatography (HPLC) separation, MS detection, and quantitation. A labeling method can be tailored to enhance detection of target proteins or peptides and/or suppress signals from undesired or uninteresting ones. For example, through the labeling of N-terminal peptides, sample complexity is greatly reduced [3], whereas important details about protein processing are revealed.
The main targets for derivatization in proteomics are amines [protein or peptide N-terminus and lysine (Lys)] and thiols [cysteine (Cys)], for which numerous specific labeling methods are known. Oxidative derivatization primarily targets redox-sensitive residues, comprising the aromatic tryptophan (Trp), tyrosine (Tyr), phenylalanine (Phe), and histidine (His) residues and the sulfur-containing Cys and methionine (Met) residues (Fig. 1). Although thiols are usually labeled through nonoxidative electrophilic substitution reactions, disulfide bond formation between two thiol groups can be promoted by oxidizing agents and is a common and reversible labeling method.
Fig. 1.

Structures of the most common amino acid oxidation products. X denotes either a halogen or a hydroxyl group
For oxidative labeling reactions a distinction is made between primary (direct) oxidation of amino acid residues followed by reaction with nucleophiles (e.g., electrochemical oxidation of Tyr followed by reaction with water) and reaction with oxidizing reagents (e.g., hydroxyl radicals). For aromatic residues such as Tyr and Trp the distinction between oxidation and electrophilic aromatic substitution is not always clear. Different reactions can result in the same products via different intermediates (e.g., via hydroxylation or halogenation of the aromatic rings).
Oxidative labeling is presented here as a distinct category of labeling techniques, based on the specific methods employed and the fact that less commonly targeted redox-active residues are modified. In contrast to regular chemical labeling methods, reactive intermediates of either the reactant or the target protein are formed by oxidation. Oxidative labeling is the most straightforward labeling method for aromatic residues, in particular Tyr and Trp, and provides access to purely instrumental labeling methods, such as electrochemical oxidation, which has no equivalent in conventional, chemical labeling methods. Electrochemical oxidation can be achieved with a stand-alone electrochemical cell but the electrospray emitter itself can also act as an electrode in the case of online liquid chromatography (LC)-electrospray–MS analysis.
The following sections present and discuss the main reactive agents, their production methods, and their most common reactions with peptides and proteins, illustrating various applications of oxidative labeling in MS-based proteomics.
Methods for oxidative modification
A distinction can be made between nonspecific labeling reactions and reactions directed at specific amino acids or functional groups. The directed reactions are used for site-specific labeling, whereas less specific methods, notably those mediated by free radicals (e.g., hydroxyl radicals), are useful for accessibility-based labeling to probe the three-dimensional structure of proteins (see “Applications”). However, studies have shown that amino acids with redox-active side chains are more susceptible than others [4]. Nonspecific labeling reactions are also widely used for mimicking in vivo oxidation (e.g., susceptibility to oxidants generated during a host defense reaction).
Oxidation agents and reaction with peptides and proteins
In this section, a review of the most widely used oxidizing agents is presented and the primary end products are listed with selected examples taken from the literature. Oxidizing agents are grouped in three main categories, namely, reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive halogen species.
Reactive oxygen species
ROS-mediated oxidation of amino acids in proteins is an important process occurring in vivo as part of the first-line defense of a host organism against infections [5–7]. ROS-mediated protein modification has therefore been extensively studied by biologists and biochemists owing to its implication in disease development [1, 2]. ROS include hydroxyl radicals 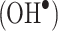 , superoxide anions
, superoxide anions 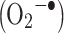 , peroxides (ROOR, including radical species derived from them), and ozone (O3). The effect of ROS on free amino acids and amino acid residues in proteins has been extensively studied and reviewed by Garrison [8] and more recently by Stadtman and Levine [9]. Typically, the study of ROS-mediated oxidation of proteins can be performed by generating the reactive species by chemical, photochemical, enzymatic, or electrochemical reactions of molecular oxygen (O2), hydrogen peroxide (H2O2), or water (H2O).
, peroxides (ROOR, including radical species derived from them), and ozone (O3). The effect of ROS on free amino acids and amino acid residues in proteins has been extensively studied and reviewed by Garrison [8] and more recently by Stadtman and Levine [9]. Typically, the study of ROS-mediated oxidation of proteins can be performed by generating the reactive species by chemical, photochemical, enzymatic, or electrochemical reactions of molecular oxygen (O2), hydrogen peroxide (H2O2), or water (H2O).
Hydroxyl radicals are by far the most commonly studied ROS. They can easily be generated (see “Production and use of oxidation agents”), and they have a broad range of reactivity, which includes not only the oxidation-sensitive Cys, Met, Trp, Tyr, Phe, and His side chains, but also aliphatic groups and the peptide backbone [4, 9]. Hydroxyl radicals can abstract electrons from the alpha carbon of any amino acid to form carbon radicals [4], which after reaction with O2 leads to peptide backbone cleavage. Alternatively beta-carbon radical formation leads to beta-scission, resulting in side chain cleavage [10]. Covalent modification of the side chains of aromatic amino acids by hydroxyl radicals results most commonly in hydroxylation (Fig. 1). Phe is converted to 2-hydroxy-Phe, 3-hydroxy-Phe, and 4-hydroxy-Phe, whereas Tyr yields mainly the ortho-hydroxylation product [11]. Tyr can also undergo cross-linking reactions to form dityrosine derivatives [12]. Trp residues are converted to a mixture of hydroxy-Trp isomers followed by further decomposition to kynurenine by a pyrrole ring opening reaction [13].
Ozone, as a powerful oxidizing agent, was shown to react with free amino acids and proteins similarly to other ROS by affecting sulfur-containing (Met and Cys) and aromatic (Trp, Tyr, Phe, and His) residues although Phe and Cys are much less reactive. Studies with ambient air ozone [14, 15] and aqueous ozone [16–19] have agreed on the following relative reactivity: Met > Trp > Tyr > His > Phe > Cys.
Performic acid oxidizes Met, Tyr, and Trp, leading to sulfoxide formation and aromatic hydroxylation, respectively. It is used specifically for oxidative cleavage of disulfide bonds, producing two cysteic acid residues [20, 21]. Sulfenic acid is an unstable intermediate oxidation product of Cys (Fig. 1). It may be further oxidized to unreactive sulfinic acid and sulfonic acid (cysteic acid). Sulfenic acid reacts readily with nucleophiles, including other Cys residues, with which it can form a disulfide bond [22].
ROS are highly reactive and can modify most amino acid residues. As shown later in this review, such nonspecific modifications are useful for protein surface mapping experiments. They are, however, of limited utility for selective, oxidative modifications.
Reactive nitrogen species
RNS mainly derived from nitric oxide 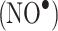 lead to nitration and nitrosation of proteins in vivo [2]. RNS, in comparison with ROS, preferentially oxidize sulfur-containing (Cys and Met) and aromatic (Tyr, Trp, Phe, and His) amino acids. In vivo nitration of Tyr is a well known widely studied phenomenon, whereas nitration of Trp has received less attention [23]. Tyr nitration occurs at the ortho positions and dinitration is possible [24].
lead to nitration and nitrosation of proteins in vivo [2]. RNS, in comparison with ROS, preferentially oxidize sulfur-containing (Cys and Met) and aromatic (Tyr, Trp, Phe, and His) amino acids. In vivo nitration of Tyr is a well known widely studied phenomenon, whereas nitration of Trp has received less attention [23]. Tyr nitration occurs at the ortho positions and dinitration is possible [24].
Peroxynitrite (ONOO-), which may be formed from 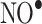 and superoxide anion
and superoxide anion 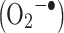 , is thought to be the primary agent for reaction with Cys, Met, and Trp, whereas for reaction with Tyr, Phe, and His secondarily formed radicals are thought to be involved [24]. Reaction with NO2 is believed to be the main in vivo Tyr nitration pathway [25–27].
, is thought to be the primary agent for reaction with Cys, Met, and Trp, whereas for reaction with Tyr, Phe, and His secondarily formed radicals are thought to be involved [24]. Reaction with NO2 is believed to be the main in vivo Tyr nitration pathway [25–27].
Reaction of Cys with NO in the presence of oxygen may lead to nitrosation of the thiol group [28]. In addition, formation of sulfenic acid is possible with RNS in a similar way as with ROS [24].
RNS are more selective than ROS and thus potentially more suitable for site-directed labeling of peptides and proteins. Subsequent reduction of nitrated Tyr or Trp to the corresponding aromatic amines is a useful approach for site-directed labeling [29, 30].
Reactive halogen species
Reactive halogen species are oxidized forms of chlorine, bromine, and iodine, including hypohalous acids, which readily react with aromatic amino acids. The oxidized halogen intermediate may be formed through reaction between halide anions and oxidizers, such as H2O2 (see “Chemical methods”). Reaction with Cys and Met to form sulfoxides is possible (Fig. 1) [22, 31] and cystine can yield N-dichlorocystine, but halogenation of free Cys is not observed. The extent of oxidation of Cys, Met, and His can be limited by using substoichiometric amounts of oxidizing agents.
Reaction with Tyr and Trp leads to single or multiple halogenations (see below). Halogenated intermediates (Fig. 2) react with other nucleophiles, including water, leading to hydroxylation or cleavage of adjacent peptide bonds. Oxidative halogenation and cleavage of peptide bonds C-terminal to Tyr and Trp often occur simultaneously and can be achieved by chemical or electrochemical methods (see “Direct electrochemical oxidation of peptides and proteins”). Some selectivity for the cleavage reaction is observed, depending on the size of the reactant and the redox activity of Tyr or Trp (at low pH the redox potential of Trp is slightly lower than that of Tyr [32]). N-Bromosuccinimide, N-iodosuccinimide, and N-chlorosuccinimide have been used to cleave peptide bonds C-terminal to Tyr [33–35] and Trp [36, 37] residues. In all cases cleavage is accompanied by halogenation. Reagents have been proposed that are selective for Trp (e.g., ortho-iodosobenzoic acid) but have never achieved widespread use [38].
Fig. 2.

a Proposed reaction pathway for oxidation reaction of Trp with bromine [39]. A first oxidation step with Br+ leads to the oxoindole (1), followed by a second oxidation, which induces internal cleavage of the peptide bond (2). In the presence of an excess of bromine, halogenation of the six-membered ring occurs (3). b The analogous reaction of Tyr with 3 equiv of N-bromosuccinimide (NBS) leads to a dibrominated, cleaved Tyr residue [138]
Halogenation of Tyr is most favorable at the ortho position (Figs. 1, 2b), yielding a mixture of singly and doubly halogenated Tyr. For Trp the 2 position of the pyrrole ring and any position on the six-membered aromatic ring are targeted [39], analogous to naturally occurring Trp bromination patterns [40] (Figs. 1, 2a). Oxidation by reactive halogen species usually leads first to 2-oxoindole formation (Fig. 2a, structure 1) through reaction with water. Subsequent oxidation reactions result in halogenation of the six-membered aromatic ring, presumably at the 5-position, and/or peptide bond cleavage, as illustrated in Fig. 2a [39]. The secondary oxidation of the 2-oxoindole leads to an intramolecular reaction to form an iminolactone, which is readily hydrolyzed, cleaving the tryptophanyl or tyrosyl peptide bond (Fig. 2a, structure 2). The reaction of Trp with 3 equiv of N-bromosuccinimide leads to monobrominated, cleaved 2-oxo-Trp [41], whereas the equivalent reaction with Tyr produces a dibrominated, cleaved Tyr residue [39] (Fig. 2b).
Hypohalous acids, such as HOCl and HOBr, produce mainly halogenated Tyr and Trp, as well as 2-oxo-Trp [42–44]. In addition, many other minor modifications of these and other residues have been described [45]. Mono- and dichlorination of Tyr with performic acid in the presence of Cl- have been observed, which may be ascribed to intermediate HOCl formation [46, 47].
Production and use of oxidizing agents
Chemical methods
Chemical oxidants can be added directly to a protein sample or generated after chemical, photochemical, electrochemical, or enzymatic activation (see below). Hydrogen peroxide is the most easily obtained oxidizing agent. Hydroxyl radicals are formed from hydrogen peroxide or from water by chemical, electrochemical, or photochemical activation. Online reactions coupled with electrospray–MS or reactions on the target plate for matrix-assisted laser desorption/ionization (MALDI)-MS [48] have the advantage that sample handling is avoided.
Various peroxide species are produced by combining hydrogen peroxide and precursors, such as nitrite to form peroxynitrite [49], or formic acid to form performic acid [47]. Reactive halogen species include hypohalous acids such as HOCl and N-halogensuccinimides. Others are produced by oxidizers such as hydrogen peroxide to form hypohalous acids with molecular halogens (I2 and Br2) or alkali halides. Iodination of Tyr is used in radiology, where radioactive iodine (125I) is incorporated in proteins, typically with 125I- which is activated by chloramine T [50].
Metal-catalyzed oxidation of proteins with transition metal complexes (e.g., ruthenium bipyridine or porphyrin-like compounds) is used for photochemically or chemically induced oxidation of Trp, Tyr, and Cys to their radical intermediates [51, 52]. Such methods are most commonly employed for protein cross-linking or to determine the metal-binding site of proteins [53, 54].
The Fenton reaction has been used to produce hydroxyl radicals from hydrogen peroxide by addition of Fe(II) for Trp and Tyr oxidation and cross-linking studies [55, 56]. An iron complex has been constructed which produces oxoferryl [Fe(IV)=O] in combination with hydrogen peroxide [57], which is claimed to be potentially more specific than other ROS such as hydroxyl radical owing to lower diffusion.
Reactive intermediates produced by irradiation
Hydroxyl radicals are photochemically generated either by photolysis of hydrogen peroxide or by radiolysis of water. Photolysis induced by a UV lamp [58] or a laser [59–65] and radiolysis induced by X-ray [66–70] and γ-ray [71] irradiation as well as pulsed electron beam methods [72] have proven to be useful to produce hydroxyl radicals for protein footprinting experiments (see “Protein surface mapping”).
Photochemical oxidation of hydroquinone with a UV laser on a MALDI target coated with TiO2 particles has been shown to produce benzoquinone, which reacts with thiols in peptides [73]. This parallels the electrochemical generation of benzoquinone in the electrospray emitter reported by the same group (see “Electrospray emitter”). Furthermore, metal complexes employed in protein oxidation can be photochemically activated, e.g., the ruthenium–bipyridine complex [52].
Electrochemically produced reactive intermediates
Electrochemistry is a useful method of producing ROS in a controlled manner. An electrochemical cell can be used to oxidize water to hydroxyl radicals at sufficiently high potential. Boron-doped diamond (BDD) electrodes are particularly suitable for hydroxyl radical production owing to their intrinsic high overpotential for the formation of molecular oxygen by oxidation of water (oxygen evolution) [74, 75]. The potential at which the radical formation is observed is dependent on the doping and impurity level of the BDD material employed [75]. Another major advantage of BDD when it comes to protein analysis is its low adsorption. Adsorption of proteins and peptides to the working electrode affects the reproducibility, as has been observed when working with commonly used carbon electrodes [76]. Hydroxyl radical formation at high voltage combined with the low adsorption properties makes BDD a suitable and efficient material for footprinting experiments [77] (see “Protein surface mapping”).
The Fenton reaction (see “Chemical methods”) can be initiated in an electrochemical cell [78]. Alternatively, the electrospray emitter of a mass spectrometer’s source can be employed to oxidize atmospheric oxygen. High voltage on the electrospray emitter tip (typically above 5 kV) can generate a corona discharge, which leads to hydroxyl and perhydroxyl radical formation and subsequent peptide oxidation in the gas phase [79], an approach that has been used for protein footprinting experiments [80, 81] (see “Protein surface mapping”). However, Boys et al. [82] showed recently that corona discharge can occur under regular electrospray ionization (ESI) conditions (3.5 kV, N2 nebulizer gas) and induce hemoglobin oxidation. Electrochemical production of RNS in an electrochemical cell has been shown to lead to Tyr nitration [83, 84] by oxidation of nitrite to nitrogen dioxide or peroxynitrite.
Enzymatically produced reactive intermediates
Peroxidases are often used for production of ROS or RNS in in vivo assays. Both the enzyme and an oxidizing reagent may be added to a protein sample, but more commonly peroxidases are supplied with hydrogen peroxide to induce in situ oxidation. For example, myeloperoxidases can produce HOCl from Cl- and H2O2, and NO2 from nitrite and H2O2 [85], leading to Tyr chlorination and nitration, respectively.
The tyramine labeling system [86] uses peroxidase activity to cross-link tyramine to Tyr residues in proteins. Tyramine-fluorophore labeling has been used to probe Tyr radical formation in vivo [87].
Direct electrochemical oxidation of peptides and proteins
Electrochemical cell
Electrochemistry has long been a domain reserved to specialists but the coupling of electrochemistry with MS has generated increased interest owing to its ability to monitor electron transfer processes online. Many groups have contributed to the development of methods and instrumentation for coupling of electrochemical cells to MS and LC-MS systems (see [88–90] for reviews).
Electrodes can donate or accept electrons and thus reduce or oxidize macromolecules. Metalloproteins have, for instance, been widely studied because of their crucial role in in vivo redox processes [91]. The electrochemistry of biomacromolecules is mainly used for detection purposes and in that aspect metalloproteins are easily oxidizable targets owing to their metal ion cofactor.
Electrochemistry is also used to mimic oxidative modifications occurring in vivo through enzymatic and ROS activity. Indeed, proteins contain electroactive amino acid residues whose side chains can interact with an electrode and undergo electron transfer reactions. Sulfur-containing Cys and Met residues as well as the aromatic Tyr, Trp, and His are susceptible to direct oxidation reactions in an electrochemical cell when a positive potential is applied to the working electrode. Reduction of disulfide bridges can, on the other hand, be achieved by applying negative potentials [92].
Electrochemical oxidation of aromatic amino acids produces reactive intermediates which mainly yield hydroxylated residues by reaction with water and, for Tyr, quinones after secondary oxidation reactions. Sulfur-containing residues can be oxidized through up to three oxygen atom insertions in the case of Cys [90]. Another common oxidation reaction for Cys is to form a disulfide bond with another Cys residue. Recent work in our group showed that cleavage of the protein backbone can occur after electrochemical oxidation of peptides and proteins [76, 93]. Similarly to the chemical cleavage with halogen species described in “Reactive halogen species,” electrochemical oxidation of peptides and proteins leads to the preferred cleavage of peptide bonds C-terminal to Tyr and Trp residues. The cleavage reaction yields a spirolactone derivative of Tyr and Trp (Fig. 2). This amino acid specificity makes it a promising method for the development of an alternative protein digestion technique. Interestingly, it has been shown that the method can be used to distinguish phosphorylated Tyr from Tyr, since phospho-Tyr cannot be oxidized and thus no cleavage occurs [93].
Proteins have not been extensively studied in terms of oxidative side-chain modifications using electrochemistry although it is a potentially useful method to mimic in vivo processes. Brabec and Mornstein [94, 95] first presented electrochemical oxidation of Tyr and Trp side chains in the protein backbone. Nitration of Tyr side chains in lysozyme has also been investigated by electrochemical oxidation of nitrite [84].
Carbon-based working electrodes are the most commonly used working electrodes in organic electrochemistry but have the main drawback of surface oxidation and fouling due to irreversible adsorption when working with large peptides and proteins. This strongly hampers accurate analysis and reproducibility. BDD electrodes have recently gained considerable attention owing to their chemical inertness, large potential window, and more importantly their lower adsorption properties as compared with glassy carbon [74, 75]. Shin et al. [96] showed strongly reduced adsorption of bovine serum albumin to BDD electrodes as compared with glassy carbon and Chiku et al. investigated the direct electrochemical oxidation [97] and conformational changes of bovine serum albumin [98] using BDD electrodes, indicating that this electrode material is suitable for protein analysis.
Electrospray emitter
The simplest configuration to perform electrochemical oxidation in combination with MS is to make use of the inherent electrochemical processes occurring within the electrospray emitter of the mass spectrometer. Blades et al. [99] were the first to compare the ESI source to an electrolytic cell by showing that Zn(II) and Fe(II) ions were generated in solution by electrochemical oxidation of the emitter tip. ESI by its nature involves electrochemical processes. In positive ionization mode, positively charged droplets formed at the emitter tip flow towards the aperture of the mass spectrometer owing to the applied electrical field. The way to balance charges and to allow current to flow through the entire circuit is by providing electrons at the liquid–metal interface by electrochemical oxidation of the metallic emitter tip and/or electrolytes present in solution to release electrons, although there is ongoing discussion about this process [100, 101]. The emitter can thus act as a working electrode where oxidation (positive ion mode) or reduction (negative ion mode) reactions take place, whereas the curtain plate or inlet capillary of the mass spectrometer plays the role of the counter electrode. Van Berkel and coworkers studied the fundamental aspects of the ESI process extensively [101] and “in source” redox reactions were mainly used to ionize compounds such as porphyrins [102], polyaromatic hydrocarbons [103], metallocenes [104], and fullerenes [105] in the form of radical cations in positive ion mode.
Girault and coworkers pioneered the use of in source redox reactions for peptide and protein analysis. In their work, hydroquinone was oxidized to benzoquinone, which reacts with the thiol groups of Cys in peptides and proteins within the emitter as shown in Fig. 3a [106–110]. In this way they developed a system for “online counting” of Cys residues with the goal to facilitate peptide identification through database searches, which is enhanced when at least one amino acid residue in the sequence is known [109, 111, 112]. The technique was shown to be successful on a set of peptides containing up to three Cys residues and on proteins, although the tagging efficiency decreased with increasing molecular size owing to steric hindrance and slower kinetics.
Fig. 3.

Oxidation reactions in an electrospray emitter. a Oxidation of hydroquinone (hQ) to benzoquinone (bQ) in the presence of Cys-containing peptides leads to quinone-labeled peptides [106]. b Oxidation of metals (e.g., from the emitter material itself) leads to metal ions that may coordinate with peptides [113]. c Oxidation of water reduces the solvent pH, leading to higher protein charge states [121]
The same group explored the use of sacrificial electrodes on microchip devices to generate metal ions in solution [Cu(II), Zn(II), Ni(II), Fe(II), and Ag(I)] that can complex to peptides (Fig. 3b) [113]. The use of copper and zinc electrodes appeared to be most efficient to probe metal ion-protein interactions that mimic those observed in biological systems. Copper sacrificial electrodes, for instance, were used to study copper–Cys interactions [114–116]. Copper is known to catalyze Cys oxidation in vivo [117] and it was shown that generating Cu(I) and Cu(II) ions in solution promoted the formation of inter- or intramolecular disulfide bridging. Alternatively, a sacrificial zinc electrode was used to tag phosphopeptides. Zn(II) ions generated at the emitter tip were shown to interact with phosphopeptides and complex phosphorylated Tyr and Ser [116, 118].
Redox reactions taking place at the electrospray emitter electrode can affect the composition of the solution in the capillary [119, 120]. Oxidation or reduction of water at the liquid–metal interface (Fig. 3c) can induce a variation of the pH in solution. Pan et al. [121] showed that this change of solution composition can be used to enhance ubiquitin, myoglobin, and cytochrome c detection in negative ion mode. Indeed reduction of water within the electrospray capillary progressively increased the basicity of the solution, through formation of OH− ions, thus altering the charge state of the different proteins and improving the detection in negative ESI-MS. The electrochemical modification of a solute inside the electrospray capillary is strongly dependent on the liquid flow rate. When the flow rate is increased, there is a lower yield of electrochemical reaction products [101].
Applications
Applications of chemical modifications and tagging strategies for protein research and proteomics have been covered in recent reviews [122–124]. Here we highlight applications where oxidation facilitates sample preparation and purification, and those where oxidative changes of the physicochemical properties with respect to chromatographic separation, ionization efficiency, or mass are utilized for improved or differential detection of labeled peptides.
Sample preparation
Oxidative modification for sample preparation is most commonly performed at the protein level. The oxidized functional group may be the end point of labeling, but oxidation methods can also be employed for the creation of functional groups for secondary labeling, e.g., biotinylation for affinity purification or fluorescent labeling for detection.
Figure 4 illustrates a variety of secondary labeling methods after oxidation of amino acids. Nitro-Tyr can be reduced with sodium disulfite [125], dithiothreitol in combination with heme [126], or by electrochemical reduction [127–130] and yield amino-Tyr. This amine group is useful for coupling labels (Fig. 4a) such as biotin [27, 30, 131], sulfhydryl groups [29], or fluorescent tags [132]. This strategy allows isolation of residues containing the modified amino acids. Zhang et al. [29] and Abello et al. [30] showed selective tagging of nitro-Tyr after prior acetylation of primary amines.
Fig. 4.

Secondary labeling reactions of oxidized amino acid residues. a Oxidative nitration, followed by reduction of the nitro group to an amine, and finally amide formation [27, 131]. b Reaction of the radical intermediate of Tyr with the spin-label 5,5-dimethylpyrroline-N-oxide (DMPO) [134] or a second Tyr residue [51]. c Secondary oxidation of hydroxylated Tyr to an orthoquinone, followed by reaction with an amine (e.g., a protein Lys), leading to a Schiff base intermediate, which is very reactive towards thiols [51]. d Reaction of sulfenic acid with a linker molecule containing secondary amine or azide functional groups for labeling [137]. e Tyr peptide bond cleavage leads to a lactone intermediate that can react with alcohols or amines [138]
Oxidative cross-linking of radical intermediates of Tyr with other Tyr residues (Fig. 4b) or with tyramine coupled to a fluorophore [51, 52, 87] produces dityrosine-containing compounds with distinct spectroscopic properties. Cross-linking reactions are of great interest owing to the fact that they involve residues (Tyr, Trp, Cys) that often play an essential role in protein function and biological recognition processes. Studies of such reactions have, for instance, revealed intermolecular interactions between G-protein-coupled receptors within intact cells [51] or that neutrophil-derived proteins with anti-inflammatory and bactericidal properties are more susceptible to attack by Tyr radicals [87]. The biological role of protein Tyr radicals can be assessed by trapping them with the spin-label 5,5-dimethylpyrroline-N-oxide before they react with other radicals (Fig. 4b). The trapped radicals are stable enough for subsequent tryptic digestion and MS analysis and can thus be localized in the protein [133, 134]. Such studies allow one to map the radicals in proteins under oxidative stress, leading to a better understanding of the mechanisms that are related to human diseases. Trapped Tyr residues can also be detected with antibodies and by molecular magnetic resonance imaging [135].
Tyr orthoquinone is produced by a second oxidation after oxidative hydroxylation (see “Electrochemical cell”). This group is susceptible to nucleophilic attack by thiols, as shown in Fig. 4c, or by other oxidized Tyr residues [51, 136]. Sulfenic acid is capable of nucleophilic attack, which has been used to couple a linker molecule that subsequently reacts with fluorescent labels [137] (Fig. 4d).
The spirolactone group of Tyr, which is produced upon peptide cleavage, is reactive towards alcohols and amines under basic conditions to form esters and amides (Fig. 4e) [138]. An analogous reaction with amines such as tris(hydroxymethyl)aminomethane has been described for the homoserine lactone produced by CNBr cleavage C-terminal to Met [139].
Protein digestion for proteomics, which is typically performed enzymatically, may be achieved by electrochemical or chemical oxidative cleavage of proteins as described in “Reactive halogen species” and “Electrochemical cell.” The distinct specificity and the reactive spirolactones of Tyr and Trp at the peptide C-terminus may provide novel peptide enrichment and identification strategies.
LC properties
Oxidative labeling may change peptide retention on the chromatographic stationary phases. Hydroxylation of Tyr and Trp and oxidation of Met and Cys to sulfoxide or sulfoacids decrease their hydrophobicity, thus reducing peptide retention on reversed phase (RP) columns. Hydrophobicity and elution order may be further affected by solvent composition and pH (e.g., nitro-Tyr has a longer retention time than Tyr in RP-HPLC under acidic conditions, but is eluted earlier under basic conditions [27]). Increased retention times are observed upon halogenation, so Tyr, bromo-Tyr, and dibromo-Tyr are eluted in this order in RP-HPLC [140].
Direct comparison of labeled versus unlabeled samples is possible, but as for peptide mass shifts (see “Mass change of peptides and fragments”), sample complexity and low abundance of peptides of interest can make retention time shifts hard to detect. To overcome this issue the COFRADIC method has been developed [3], which is a two-stage LC method with a primary separation, labeling of certain molecules or functional groups in fractions, and a secondary separation. Differential labeling will cause a shift in retention time for labeled peptides, whereas unlabeled peptides remain unaffected, making labeled peptides easily detectable. COFRADIC has been applied to various targets based on different labeling methods, including oxidation of Met with performic acid [141]. Met oxidation has also been used to reduce the hydrophobicity of peptides derived from membrane proteins, which are difficult to elute from RP stationary phases [142]. This is a case where oxidative modification of Met residues, which are often located in transmembrane segments, leads to increased peptide coverage, which is often an issue when analyzing membrane proteins by LC-MS. Reaction of Cys with benzoquinone, similar to the electrospray tagging reaction (see “Electrospray emitter”), has been employed to induce a hydrophilic shift in COFRADIC with RP-HPLC [143].
Mass-spectrometric properties
Ionization efficiency and charge state
Protein solution-phase conformation influences the charge state of a protein during the ESI process [144, 145]. The charge state itself influences the ionization efficiency of proteins since highly charged conformers are more easily amenable to ESI. A protein in its native folded conformation gives rise to lower-charge states (fewer available protonation sites) and a wider charge distribution compared with its unfolded equivalent. On the basis of this empirical relationship, ESI-MS has been used as a tool for monitoring protein conformational changes in solution [146], including for protein footprinting experiments [147] (see “Protein surface mapping”), where oxidative modifications affect the solution-phase structure. The nature of the oxidized residue can also affect the charge state as testified, for instance, by the lower proton affinity of 2-oxo-His compared with unmodified His [148]. Oxidation of Cys to cysteic acid can, on the other hand, contribute to an increase of the charge state in negative ion mode owing to its acidity. It is also noteworthy that the peptide dissociation patterns in MS/MS may be affected by oxidatively modified residues, which must be taken into account during the interpretation of MS/MS spectra [148] (see “Mass change of peptides and fragments”). Ionization efficiency can also be increased by taking advantage of inherent electrochemical processes taking place in the electrospray emitter as discussed in “Electrospray emitter.”
Mass change of peptides and fragments
Almost any labeling method, including oxidation, will increase the mass of an analyte. Differential MS analysis of oxidized versus unoxidized samples will therefore reveal modified peptides, provided that they are sufficiently abundant and can be ionized well. Alternatively, much more sensitive MS/MS methods can selectively detect modified peptides on the basis of characteristic product ions or neutral losses [149]. Table 1 lists precursor and neutral loss scanning applications that have been described for various (in vivo) modifications that can be mimicked by oxidative labeling.
Table 1.
Specific fragments of oxidatively modified amino acid residues used for tandem mass spectrometry based detection
| Residue | Modification | Precursor immonium ion (Da) | Neutral loss (Da) | References |
|---|---|---|---|---|
| Trp | Bromo | 237.002 | [155] | |
| 239.001a | ||||
| Hydroxy | 175.086 | [45] | ||
| Tyr | Hydroxy | 152.071 | [45] | |
| Bromo | 213.986 | [174] | ||
| Chloro | 170.037 | [45, 174] | ||
| Nitro | 181.061 | [175] | ||
| Dinitro | 226.046 | |||
| Iodo | 261.973 | [176] | ||
| Diiodo | 387.869 | |||
| Met | Sulfoxide | 63.998 | [177, 178] | |
| Cys | Sulfinic acid | 65.977b | [150] | |
| Cam sulfoxide | 107.004 | [179, 180] | ||
| Cam sulfone | 122.999 |
cam carbamidomethylcysteine produced by alkylation with iodoacetamide
a 81Br isotope
bNegative ionization
Collision-induced dissociation of peptides is influenced by the side-chain properties of the constituent amino acids. Notably, creation of acidic or basic groups affects the proton localization, so formation of sulfinic acid and sulfonic acid leads to greatly increased fragmentation at their C-terminal side [150], analogous to the preferential fragmentation induced by aspartic acid [151]. This observation may help in interpreting MS/MS spectra of unknown peptides [152]. In contrast, the preferential fragmentation normally observed for His is inhibited upon its oxidation to 2-oxo-His [148].
Although fragmentation of protonated peptides by collision-induced dissociation is the most common technique in proteomics, electron-transfer-induced dissociation has gained considerable interest since its introduction in 2004 [153], and applications targeting oxidized residues are easily envisaged. In a related method, iodinated Tyr is irradiated in the gas phase to produce radical intermediates which readily fragment upon collisional activation [154].
Mass defect and isotopic pattern signature
In addition to the overall mass increase upon labeling, the elemental composition of the labeling group may introduce a shift in the mass defect of the peptide or protein, which is on average +0.00055 amu per amu for protein-derived peptides. Labels with many hydrogen atoms cause an additional positive mass defect, whereas incorporation of heavier elements, including chlorine, bromine, and iodine, leads to an increasingly large negative mass defect (Table 2). The term “mass-deficient mass tag” (MaDMaT) was proposed by Steen and Mann [155]. The large mass defect of halogens may be used for the preparation of internal standards for quantitative proteomics [156]. MaDMaT-labeled peptides are easily distinguished from nonlabeled peptides in a mass spectrometer with sufficient mass resolution and accuracy [156]. Performic acid oxidation of Met [20] or halogen labeling [157, 158] expands the mass distribution of tryptic peptides, and the compositional information may be used to improve peptide identification.
Table 2.
Effect of oxidative modifications of Tyr on the mass defect and isotope pattern of angiotensin I (DRVYIHPFHL). The average and lowest monoisotopic masses of tryptic peptides (without missed cleavages) were calculated in 1-Da mass bins from a yeast protein database (http://www.yeastgenome.org; orf_trans.fasta database of 8 May 2009)
| Modification | Chemical composition | Monoisotopic mass (M+H+, Da) and mass defect | Average tryptic peptide monoisotopic mass (M+H+, Da) | Lowest tryptic peptide monoisotopic mass (M+H+, Da) | Isotope masses and relative intensities |
|---|---|---|---|---|---|
| None | C62H90N17O14 | 1,296.6848 (0.52 mmu) | 1,296.6475 (−0.0373 Da) | 1,296.5526 (−0.1322 Da) | 1,296.68 (100.0) |
| 1,297.69 (74.9) | |||||
| 1,298.69 (30.6) | |||||
| 1,299.69 (8.9) | |||||
| 1,300.70 (2.0) | |||||
| Hydroxy | C62H90N17O15 | 1,312.6797 (0.52 mmu) | 1,312.6706 (−0.0091 Da) | 1,312.5286 (−0.1511 Da) | 1,312.68 (100.0) |
| 1,313.68 (74.9) | |||||
| 1,314.69 (30.8) | |||||
| 1,315.69 (9.1) | |||||
| 1,316.69 (2.1) | |||||
| Chloro | C62H89N17O14Cl | 1,330.6458 (0.49 mmu) | 1,330.6723 (+ 0.0265 Da) | 1,330.5344 (−0.1114 Da) | 1,330.65 (100.0) |
| 1,331.65 (74.9) | |||||
| 1,332.65 (62.5) | |||||
| 1,333.65 (32.8) | |||||
| 1,334.65 (11.8) | |||||
| 1,335.65 (3.2) | |||||
| Bromo | C62H89N17O14Br | 1,374.5953 (0.43 mmu) | 1,374.6884 (+0.0931 Da) | 1,374.5483 (−0.0470 Da) | 1,374.60 (78.2) |
| 1,375.60 (58.6) | |||||
| 1,376.60 (100.0) | |||||
| 1,377.60 (63.9) | |||||
| 1,378.60 (24.9) | |||||
| 1,379.60 (7.1) | |||||
| 1,380.60 (1.6) | |||||
| Iodo | C62H89N17O14I | 1,422.5814 (0.41 mmu) | 1,422.7089 (+0.1275 Da) | 1,422.5259 (−0.0555 Da) | 1,422.58 (100.0) |
| 1,423.58 (74.9) | |||||
| 1,424.59 (30.6) | |||||
| 1,425.59 (8.9) | |||||
| 1,426.59 (2.0) | |||||
| Dibromo | C62H88N17O14Br2 | 1,452.5058 (0.35 mmu) | 1,452.7232 (+0.2174 Da) | 1,452.5795 (+0.0701 Da) | 1,452.51 (44.4) |
| 1,453.51 (33.3) | |||||
| 1,454.50 (100.0) | |||||
| 1,455.51 (68.7) | |||||
| 1,456.50 (69.3) | |||||
| 1,457.51 (39.3) | |||||
| 1,458.51 (14.6) | |||||
| 1,459.51 (4.1) |
The well-known specific isotope patterns of chlorine and bromine are used to confirm their presence and number from the relative isotope intensities (Table 2). In combination with its large mass defect, the characteristic isotope pattern makes detection of labeled peptides in mass spectra rather straightforward.
Differential isotopic labeling for quantitation
Isotopic labeling is a standard approach in mass-spectrometric quantitation, and becoming increasingly important in proteomics. Synthesis of labeled peptides as well as metabolic and enzymatic labeling are widely used, but the most common method is chemical labeling either at the peptide or at the protein level. The primary labeling targets are the same as for nonisotopic labeling, such as primary amines of Lys residues or protein/peptide N-termini [159, 160] or the thiol of Cys [161]. Labeling is performed by reaction with alkyl halide (nucleophilic substitution) or maleimide (thiols), or N-hydroxysuccinimidyl esters (amines). Trp is also a target for direct electrophilic aromatic substitution using the sulfenylation reagent 2-nitrobenzenesulfenyl chloride [162].
Oxidative labeling for quantitation is limited by the lack of specificity and completeness of the reactions, but it is certainly useful if incomplete labeling is acceptable. Other applications described to date comprise oxidative tagging with H182O during ESI [163] and metabolic isotopic labeling of amino acid targets followed by H2O2 oxidation, as shown for Met in the COFRADIC workflow [164].
Protein surface mapping
Protein footprinting is a technique that gained attention during the last decade owing to its ability to probe solvent-accessible residues. The technique of choice makes use of in situ generated hydroxyl radicals that induce oxidative chemical modification of surface-accessible reactive amino acid side chains. The term “footprinting” was introduced some 30 years ago by Galas and Schmitz [165], who performed DNA surface mapping. H/D exchange experiments are also used to probe surface-accessible residues by MS, but the need to quench the reaction at low pH to avoid back-exchange reactions during protein analysis limits their usefulness, since only a few proteases (e.g., pepsin) work under low pH conditions (see [147, 166] for reviews). We will focus here on the use of hydroxyl radicals that allowed considerable improvement in terms of spatial resolution compared with the bulky proteases used in other methods [167–169]. In addition, the fact that the small hydroxyl radicals nonspecifically attack a wide range of residues makes them better suited to probe solvent accessibility and thus a protein’s tertiary and quaternary structure. Sulfur-containing (Cys, Met, and cystine) and aromatic (Trp, Tyr, and Phe) residues represent the most reactive targets for oxidative covalent modification, but other side chains (His, Leu, Ile, Arg, Lys, Val, Pro, Gln, and Glu) have also been shown to be modified [66–68, 70, 77, 170, 171]. Details about the chemical reactions underlying the covalent modification of amino acid side chains and backbone cleavage can be found in a detailed review by Xu and Chance [4].
The general footprinting workflow (Fig. 5) consists in producing hydroxyl radicals, which can be achieved by several means (see below and “Production and use of oxidizing agents”) and different exposure times. After exposure, proteins are digested with proteases and the extent of oxidation is measured by MS. The location of oxidized amino acids is finally assigned by MS/MS analysis. The results obtained provide information about the solvent-exposed residues on the surface of the protein and can also lead to the determination of protein-ligand interaction sites by comparison of protein and protein-ligand complexes. When protein oxidation is measured as a function of the time of exposure to the hydroxyl radicals, conformational changes can be followed owing to increased amino acid accessibility by protein unfolding induced by primary oxidation events.
Fig. 5.

Workflow for hydroxyl radical footprinting of proteins and protein/ligand complexes. MS mass spectrometry. (Adapted from Xu and Chance [4])
A wide set of techniques to produce hydroxyl radicals have expanded the toolbox available for footprinting experiments (see “Production and use of oxidizing agents”). The Fenton reaction to produce hydroxyl radicals chemically [55, 56] has the main drawback that iron and EDTA may distort the native protein conformation. Photochemical formation of hydroxyl radicals by photolysis of hydrogen peroxide or by radiolysis of water is very efficient and provides the advantage (compared with the Fenton reaction) of fast (nano- to microseconds) radical generation, and the possibility of short exposure times (fewer oxidation-induced structural changes) allowing time-resolved studies. Radiolysis has the advantage of not requiring the addition of any reagent to the sample solution. Electrochemistry may also be used to generate reactive oxidants for the mapping of solvent accessibility. Whereas Brabec et al. [95, 172] have pioneered the use of electrochemical oxidation to probe protein conformational changes, new approaches for electrochemical-oxidation-based probing of the higher-order structure of proteins have been introduced by McClintock et al. [77] where hydroxyl radicals have been produced by oxidation of water on a BDD electrode. Oxidation in the electrospray source by corona discharge has also been performed [81, 173].
Protein footprinting experiments have found widespread application to probe protein structure and solvent-accessible residues in the native conformation. Moreover protein–protein, protein–ligand, and protein–drug interaction sites as well as conformational changes (e.g., protein folding and unfolding) have been studied extensively, as shown by the many applications that have been reviewed in recent years [4, 68, 80, 147, 171]. Although footprinting methods have matured, some drawbacks such as secondary oxidation reactions are still an issue. It was observed that buried sulfur-containing residues can be oxidized either by internal electron transfer reactions [58] or by secondary oxidations due to residual hydrogen peroxide or peptidyl hydroperoxides [65]. Oxidation of non-solvent-accessible residues can hamper data interpretation and must be considered when performing footprinting experiments. Adding catalase and/or Met as scavengers limits secondary oxidations that may occur particularly on (buried) sulfur-containing residues [65] but internal electron transfer reactions are still difficult to avoid.
Conclusion
Oxidative labeling of peptides and proteins has expanded the toolbox of biochemists and notable progress has been made in the past decade. A variety of strategies for targeting amino acid side chains have been developed, in particular for sulfur-containing and aromatic residues. Current labeling techniques targeting primary amines (N-termini, Lys) and Cys residues have found widespread applications in proteomics, whereas other amino acids remain inaccessible to labeling to a large extend. The increased interest in oxidative labeling stems from the possibility to target other amino acid residues, such as Tyr, Trp, and Met, which have essential roles in protein function and biological recognition processes. Although the technique still suffers in many cases from incomplete conversion and selectivity, several applications in biological systems have shown its usefulness. It is, for instance, to be expected that oxidative labeling will help to improve our understanding of protein–protein interactions with respect to protein function and signaling processes.
An interesting feature of oxidative labeling is its potential to introduce reactive groups in a site-specific manner, which can be targeted by secondary chemical reactions as shown for nitro-Tyr residues. Electrochemistry, which is far from being fully exploited, can efficiently induce such oxidative modifications. Many advances and developments have been observed in this field during the last decade and interesting applications can be envisaged especially owing to new developments in electrode materials and surface-modified electrodes. BDD electrodes, for example, are of interest owing to their reduced adsorption of biomolecules, opening the way for new applications of direct and ROS-mediated oxidation of peptides and proteins.
The increasing demand for MS-based proteomics methods and the relationship with in vivo oxidative protein damage through radical-induced modifications suggest that these techniques will find increasing use.
Acknowledgement
This work was financially supported by the Dutch Technology Foundation (STW, grant 07047).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Imlay JA. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 2.Dalle-Donne I, Scaloni A, Giustarini D, Cavarra E, Tell G, Lungarella G, Colombo R, Rossi R, Milzani A. Mass Spectrom Rev. 2005;24:55–99. doi: 10.1002/mas.20006. [DOI] [PubMed] [Google Scholar]
- 3.Gevaert K, Van Damme P, Martens L, Vandekerckhove J. Anal Biochem. 2005;345:18–29. doi: 10.1016/j.ab.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 4.Xu G, Chance MR. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 5.Bokoch GM, Knaus UG. Trends Biochem Sci. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 6.Leto TL, Geiszt M. Antioxid Redox Signal. 2006;8:1549–1561. doi: 10.1089/ars.2006.8.1549. [DOI] [PubMed] [Google Scholar]
- 7.Van der Vliet A. Free Radic Biol Med. 2008;44:938–955. doi: 10.1016/j.freeradbiomed.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrison WM. Chem Rev. 1987;87:381–398. [Google Scholar]
- 9.Stadtman ER, Levine RL. Amino Acids. 2003;25:207–218. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 10.Headlam HA, Davies MJ. Free Radic Biol Med. 2002;32:1171–1184. doi: 10.1016/s0891-5849(02)00814-6. [DOI] [PubMed] [Google Scholar]
- 11.Maskos Z, Rush JD, Koppenol WH. Arch Biochem Biophys. 1992;296:521–529. doi: 10.1016/0003-9861(92)90606-w. [DOI] [PubMed] [Google Scholar]
- 12.Giulivi C, Traaseth NJ, Davies KJ. Amino Acids. 2003;25:227–232. doi: 10.1007/s00726-003-0013-0. [DOI] [PubMed] [Google Scholar]
- 13.Maskos Z, Rush JD, Koppenol WH. Arch Biochem Biophys. 1992;296:514–520. doi: 10.1016/0003-9861(92)90605-v. [DOI] [PubMed] [Google Scholar]
- 14.Cohen SL. Anal Chem. 2006;78:4352–4362. doi: 10.1021/ac052263n. [DOI] [PubMed] [Google Scholar]
- 15.Lloyd JA, Spraggins JM, Johnston MV, Laskin J. J Am Soc Mass Spectrom. 2006;17:1289–1298. doi: 10.1016/j.jasms.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Verweij H, Christianse K, van Steveninck J. Chemosphere. 1982;11:721–725. doi: 10.1016/0167-4838(82)90111-x. [DOI] [PubMed] [Google Scholar]
- 17.Berlett BS, Levine RL, Stadtman ER. J Biol Chem. 1996;271:4177–4182. doi: 10.1074/jbc.271.8.4177. [DOI] [PubMed] [Google Scholar]
- 18.Kotiaho T, Eberlin MN, Vainiotalo P, Kostiainen R. J Am Soc Mass Spectrom. 2000;11:526–535. doi: 10.1016/S1044-0305(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 19.Cataldo F. Polym Degrad Stab. 2003;82:105–114. [Google Scholar]
- 20.Matthiesen R, Bauw G, Welinder KG. Anal Chem. 2004;76:6848–6852. doi: 10.1021/ac049032l. [DOI] [PubMed] [Google Scholar]
- 21.Samgina TY, Artemenko KA, Gorshkov VA, Poljakov NB, Lebedev AT. J Am Soc Mass Spectrom. 2008;19:479–487. doi: 10.1016/j.jasms.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 22.Nagy P, Ashby MT. J Am Chem Soc. 2007;129:14082–14091. doi: 10.1021/ja0737218. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez B, Rubbo H, Kirk M, Barnes S, Freeman BA, Radi R. Chem Res Toxicol. 1996;9:390–396. doi: 10.1021/tx950133b. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez B, Radi R. Amino Acids. 2003;25:295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 25.Prutz WA, Monig H, Butler J, Land EJ. Arch Biochem Biophys. 1985;243:125–134. doi: 10.1016/0003-9861(85)90780-5. [DOI] [PubMed] [Google Scholar]
- 26.Radi R. Proc Natl Acad Sci USA. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abello N, Kerstjens HAM, Postma DS, Bischoff R. J Proteome Res. 2009;8:3222–3238. doi: 10.1021/pr900039c. [DOI] [PubMed] [Google Scholar]
- 28.Di Simplicio P, Franconi F, Frosal¡ S, Di Giuseppe D. Amino Acids. 2003;25:323–339. doi: 10.1007/s00726-003-0020-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q, Qian WJ, Knyushko TV, Clauss TRW, Purvine SO, Moore RJ, Sacksteder CA, Chin MH, Smith DJ, Camp DG, Bigelow DJ, Smith RD. J Proteome Res. 2007;6:2257–2268. doi: 10.1021/pr0606934. [DOI] [PubMed] [Google Scholar]
- 30.Abello N, Barroso B, Kerstjens HAM, Postma DS, Bischoff R. Talanta. 2010;80:1503–1512. doi: 10.1016/j.talanta.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Shechter Y, Burstein Y, Patchornik A. Biochemistry. 1975;14:4497–4503. doi: 10.1021/bi00691a025. [DOI] [PubMed] [Google Scholar]
- 32.Harriman A. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 33.Schmir GL, Cohen LA, Witkop B. J Am Chem Soc. 1959;81:2228–2233. [Google Scholar]
- 34.Junek H, Kirk KL, Cohen LA. Biochemistry. 1969;8:1844–1848. doi: 10.1021/bi00833a010. [DOI] [PubMed] [Google Scholar]
- 35.Alexander NM. J Biol Chem. 1974;249:1946–1952. [PubMed] [Google Scholar]
- 36.Patchornik A, Lawson WB, Witkop B. J Am Chem Soc. 1958;80:4747–4748. [Google Scholar]
- 37.Patchornik A, Lawson WB, Witkop B. J Am Chem Soc. 1958;80:4748–4749. [Google Scholar]
- 38.Mahoney WC, Hermodson MA. Biochemistry. 1979;18:3810–3814. doi: 10.1021/bi00584a026. [DOI] [PubMed] [Google Scholar]
- 39.Burstein Y, Patchornik A. Biochemistry. 1972;11:4641–4650. doi: 10.1021/bi00775a001. [DOI] [PubMed] [Google Scholar]
- 40.Bittner S, Scherzer R, Harlev E. Amino Acids. 2007;33:19–42. doi: 10.1007/s00726-006-0441-8. [DOI] [PubMed] [Google Scholar]
- 41.Lawson WB, Patchornik A, Witkop B. J Am Chem Soc. 1960;82:5918–5923. [Google Scholar]
- 42.Pattison DI, Davies MJ. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 43.Pattison DI, Davies MJ. Biochemistry. 2004;43:4799–4809. doi: 10.1021/bi035946a. [DOI] [PubMed] [Google Scholar]
- 44.Fu X, Wang Y, Kao J, Irwin A, d’Avignon A, Mecham RP, Parks WC, Heinecke JW. Biochemistry. 2006;45:3961–3971. doi: 10.1021/bi052339+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mouls L, Silajdzic E, Haroune N, Spickett CM, Pitt AR. Proteomics. 2009;9:1617–1631. doi: 10.1002/pmic.200800391. [DOI] [PubMed] [Google Scholar]
- 46.Chowdhury SK, Eshraghi J, Wolfe H, Forde D, Hlavac AG, Johnston D. Anal Chem. 1995;67:390–398. doi: 10.1021/ac00098a026. [DOI] [PubMed] [Google Scholar]
- 47.Dai J, Zhang Y, Wang J, Li X, Lu Z, Cai Y, Qian X. Rapid Commun Mass Spectrom. 2005;19:1130–1138. doi: 10.1002/rcm.1901. [DOI] [PubMed] [Google Scholar]
- 48.Corless S, Cramer R. Rapid Commun Mass Spectrom. 2003;17:1212–1215. doi: 10.1002/rcm.1028. [DOI] [PubMed] [Google Scholar]
- 49.Robinson KM, Beckman JS. Methods Enzymol. 2005;396:207–214. doi: 10.1016/S0076-6879(05)96019-9. [DOI] [PubMed] [Google Scholar]
- 50.Stein R, Goldenberg DM, Thorpe SR, Basu A, Mattes MJ. Cancer Res. 1995;55:3132–3139. [PubMed] [Google Scholar]
- 51.Kodadek T, Duroux-Richard I, Bonnafous J-C. Trends Pharmacol Sci. 2005;26:210–217. doi: 10.1016/j.tips.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Antos JM, Francis MB. Curr Opin Chem Biol. 2006;10:253–262. doi: 10.1016/j.cbpa.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Lim J, Vachet RW. Anal Chem. 2003;75:1164–1172. doi: 10.1021/ac026206v. [DOI] [PubMed] [Google Scholar]
- 54.Bridgewater JD, Vachet RW. Anal Biochem. 2005;341:122–130. doi: 10.1016/j.ab.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 55.Dubinina EE, Gavrovskaya SV, Kuzmich EV, Leonova NV, Morozova MG, Kovrugina SV, Smirnova TA. Biochemistry (Mosc) 2002;67:343–350. doi: 10.1023/a:1014840617890. [DOI] [PubMed] [Google Scholar]
- 56.Shcherbakova I, Mitra S, Beer RH, Brenowitz M. Nucleic Acids Res. 2006;34:e48. doi: 10.1093/nar/gkl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ekkati AR, Kodanko JJ. J Am Chem Soc. 2007;129:12390–12391. doi: 10.1021/ja075075i. [DOI] [PubMed] [Google Scholar]
- 58.Sharp JS, Becker JM, Hettich RL. Anal Chem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- 59.Aye TT, Low TY, Sze SK. Anal Chem. 2005;77:5814–5822. doi: 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- 60.Charvátová O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. J Am Soc Mass Spectrom. 2008;19:1692–1705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hambly DM, Gross ML. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 62.Pan Y, Brown L, Konermann L. J Mol Biol. 2009;394:968–981. doi: 10.1016/j.jmb.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 63.Pan Y, Stocks BB, Brown L, Konermann L. Anal Chem. 2009;81:28–35. doi: 10.1021/ac8020449. [DOI] [PubMed] [Google Scholar]
- 64.Stocks BB, Konermann L. Anal Chem. 2008;81:20–27. doi: 10.1021/ac801888h. [DOI] [PubMed] [Google Scholar]
- 65.Saladino J, Liu M, Live D, Sharp JS. J Am Soc Mass Spectrom. 2009;20:1123–1126. doi: 10.1016/j.jasms.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maleknia SD, Brenowitz M, Chance MR. Anal Chem. 1999;71:3965–3973. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 67.Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Int J Radiat Biol. 2002;78:101–114. doi: 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- 68.Guan J-Q, Chance MR. Trends Biochem Sci. 2005;30:583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 69.Xu G, Chance MR. Anal Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 70.Xu G, Kiselar J, He Q, Chance MR. Anal Chem. 2005;77:3029–3037. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- 71.Tong X, Wren JC, Konermann L. Anal Chem. 2008;80:2222–2231. doi: 10.1021/ac702321r. [DOI] [PubMed] [Google Scholar]
- 72.Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS. Anal Chem. 2009;81:2496–2505. doi: 10.1021/ac802252y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiao L, Roussel C, Wan JJ, Kong J, Yang PY, Girault HH, Liu BH. Angew Chem Int Ed Engl. 2008;47:2646–2648. doi: 10.1002/anie.200703876. [DOI] [PubMed] [Google Scholar]
- 74.Kraft A. Int J Electrochem Sci. 2007;2:355–385. [Google Scholar]
- 75.Luong JHT, Male KB, Glennon JD. Analyst. 2009;134:1965–1979. doi: 10.1039/b910206j. [DOI] [PubMed] [Google Scholar]
- 76.Permentier HP, Bruins AP. J Am Soc Mass Spectrom. 2004;15:1707–1716. doi: 10.1016/j.jasms.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 77.McClintock C, Kertesz V, Hettich RL. Anal Chem. 2008;80:3304–3317. doi: 10.1021/ac702493a. [DOI] [PubMed] [Google Scholar]
- 78.Jurva U, Wikström HV, Bruins AP. Rapid Commun Mass Spectrom. 2002;16:1934–1940. doi: 10.1002/rcm.808. [DOI] [PubMed] [Google Scholar]
- 79.Morand K, Talbo G, Mann M. Rapid Commun Mass Spectrom. 1993;7:738–743. doi: 10.1002/rcm.1290070811. [DOI] [PubMed] [Google Scholar]
- 80.Maleknia SD, Downard K. Mass Spectrom Rev. 2001;20:388–401. doi: 10.1002/mas.10013. [DOI] [PubMed] [Google Scholar]
- 81.Wong JWH, Maleknia SD, Downard KM. J Am Soc Mass Spectrom. 2005;16:225–233. doi: 10.1016/j.jasms.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 82.Boys BL, Kuprowski MC, Noël JJ, Konermann L. Anal Chem. 2009;81:4027–4034. doi: 10.1021/ac900243p. [DOI] [PubMed] [Google Scholar]
- 83.Kendall G, Cooper HJ, Heptinstall J, Derrick PJ, Walton DJ, Peterson IR. Arch Biochem Biophys. 2001;392:169–179. doi: 10.1006/abbi.2001.2451. [DOI] [PubMed] [Google Scholar]
- 84.Matters D, Cooper HJ, McDonnell L, Iniesta J, Heptinstall J, Derrick P, Walton D, Peterson I. Anal Biochem. 2006;356:171–181. doi: 10.1016/j.ab.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 85.Mohiuddin I, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. J Surg Res. 2006;133:143–149. doi: 10.1016/j.jss.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 86.Larios JM, Budhiraja R, Fanburg BL, Thannickal VJ. J Biol Chem. 2001;276:17437–17441. doi: 10.1074/jbc.M100426200. [DOI] [PubMed] [Google Scholar]
- 87.Avram D, Romijn EP, Pap EHW, Heck AJR, Wirtz KWA (2004) Proteomics 4:2397–2407 [DOI] [PubMed]
- 88.Volk KJ, Yost RA, Brajter-Toth A. Anal Chem. 1992;64:21A–33A. doi: 10.1021/ac00190a024. [DOI] [PubMed] [Google Scholar]
- 89.Diehl G, Karst U. Anal Bioanal Chem. 2002;373:390–398. doi: 10.1007/s00216-002-1281-3. [DOI] [PubMed] [Google Scholar]
- 90.Permentier HP, Bruins AP, Bischoff R. Mini Rev Med Chem. 2008;8:46–56. doi: 10.2174/138955708783331586. [DOI] [PubMed] [Google Scholar]
- 91.Kaltashov IA, Zhang M, Eyles SJ, Abzalimov RR. Anal Bioanal Chem. 2006;386:472–481. doi: 10.1007/s00216-006-0636-6. [DOI] [PubMed] [Google Scholar]
- 92.Rivat C, Fontaine M, Ropartz C, Caullet C. Eur J Immunol. 1973;3:537–542. doi: 10.1002/eji.1830030903. [DOI] [PubMed] [Google Scholar]
- 93.Permentier HP, Jurva U, Barroso B, Bruins AP. Rapid Commun Mass Spectrom. 2003;17:1585–1592. doi: 10.1002/rcm.1090. [DOI] [PubMed] [Google Scholar]
- 94.Brabec V. J Electroanal Chem. 1980;116:69–82. [Google Scholar]
- 95.Brabec V, Mornstein V. Biochim Biophys Acta. 1980;625:43–50. doi: 10.1016/0005-2795(80)90106-3. [DOI] [PubMed] [Google Scholar]
- 96.Shin D, Tryk DA, Fujishima A, Merkoçi A, Wang J. Electroanalysis. 2005;17:305–311. [Google Scholar]
- 97.Chiku M, Ivandini TA, Kamiya A, Fujishima A, Einaga Y. J Electroanal Chem. 2008;612:201–207. [Google Scholar]
- 98.Chiku M, Nakamura J, Fujishima A, Einaga Y. Anal Chem. 2008;80:5783–5787. doi: 10.1021/ac800394n. [DOI] [PubMed] [Google Scholar]
- 99.Blades AT, Ikonomou MG, Kebarle P. Anal Chem. 1991;63:2109–2114. [Google Scholar]
- 100.De La Mora JF, Van Berkel GJ, Enke CG, Cole RB, Martinez-Sanchez M, Fenn JB. J Mass Spectrom. 2000;35:939–952. doi: 10.1002/1096-9888(200008)35:8<939::AID-JMS36>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 101.Van Berkel GJ, Kertesz V. Anal Chem. 2007;79:5510–5520. doi: 10.1021/ac071944a. [DOI] [PubMed] [Google Scholar]
- 102.Van Berkel GJ, McLuckey SA, Glish GL. Anal Chem. 1991;63:1098–1109. doi: 10.1021/ac00018a014. [DOI] [PubMed] [Google Scholar]
- 103.Van Berkel GJ, Zhou F. Anal Chem. 1995;67:3958–3964. [Google Scholar]
- 104.Xu X, Nolan SP, Cole RB. Anal Chem. 1994;66:119–125. [Google Scholar]
- 105.Dupont A, Gisselbrecht J-P, Leize E, Wagner L, Van Dorsselaer A. Tetrahedron Lett. 1994;35:6083–6086. [Google Scholar]
- 106.Rohner TC, Rossier JS, Girault HH. Electrochem Commun. 2002;4:695–700. [Google Scholar]
- 107.Roussel C, Dayon L, Jensen H, Girault HH. J Electroanal Chem. 2004;570:187–199. [Google Scholar]
- 108.Roussel C, Dayon L, Lion N, Rohner TC, Josserand J, Rossier JS, Jensen H, Girault HH. J Am Soc Mass Spectrom. 2004;15:1767–1779. doi: 10.1016/j.jasms.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 109.Dayon L, Roussel C, Prudent M, Lion N, Girault HH. Electrophoresis. 2005;26:238–247. doi: 10.1002/elps.200406144. [DOI] [PubMed] [Google Scholar]
- 110.Dayon L, Roussel C, Girault HH. J Proteome Res. 2006;5:793–800. doi: 10.1021/pr050365o. [DOI] [PubMed] [Google Scholar]
- 111.Hamdan M, Righetti PG. Mass Spectrom Rev. 2002;21:287–302. doi: 10.1002/mas.10032. [DOI] [PubMed] [Google Scholar]
- 112.Sechi S, Chait BT. Anal Chem. 1998;70:5150–5158. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 113.Rohner TC, Girault HH. Rapid Commun Mass Spectrom. 2005;19:1183–1190. doi: 10.1002/rcm.1899. [DOI] [PubMed] [Google Scholar]
- 114.Prudent M, Girault HH. J Am Soc Mass Spectrom. 2008;19:560–568. doi: 10.1016/j.jasms.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 115.Prudent M, Girault HH. Metallomics. 2009;1:157–165. doi: 10.1039/b817061d. [DOI] [PubMed] [Google Scholar]
- 116.Prudent M, Méndez M, Roussel C, Su B, Lion N, Rossier J, l S, Girault HH (2009) Chimia 63:283–287
- 117.Pecci L, Montefoschi G, Musci G, Cavallini D. Amino Acids. 1997;13:355–367. [Google Scholar]
- 118.Prudent M, Rossier JS, Lion N, Girault HH. Anal Chem. 2008;80:2531–2538. doi: 10.1021/ac7025139. [DOI] [PubMed] [Google Scholar]
- 119.Van Berkel GJ, Zhou F, Aronson JT. Int J Mass Spectrom Ion Process. 1997;162:55–67. [Google Scholar]
- 120.Zhou S, Edwards AG, Cook KD, Van Berkel GJ. Anal Chem. 1999;71:769–776. [Google Scholar]
- 121.Pan P, Gunawardena HP, Xia Y, McLuckey SA. Anal Chem. 2004;76:1165–1174. doi: 10.1021/ac035209k. [DOI] [PubMed] [Google Scholar]
- 122.Leitner A, Lindner W. J Chromatogr B. 2004;813:1–26. doi: 10.1016/j.jchromb.2004.09.057. [DOI] [PubMed] [Google Scholar]
- 123.Nakazawa T. Curr Proteomics. 2006;3:33–54. [Google Scholar]
- 124.Leitner A, Lindner W. Proteomics. 2006;6:5418–5434. doi: 10.1002/pmic.200600255. [DOI] [PubMed] [Google Scholar]
- 125.Sokolovsky M, Riordan JF, Vallee BL. Biochem Biophys Res Commun. 1967;27:20–25. doi: 10.1016/s0006-291x(67)80033-0. [DOI] [PubMed] [Google Scholar]
- 126.Balabanli B, Kamisaki Y, Martin E, Murad F. Proc Natl Acad Sci USA. 1999;96:13136–13141. doi: 10.1073/pnas.96.23.13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ohshima H, Celan I, Chazotte L, Pignatelli B, Mower HF. Nitric Oxide. 1999;3:132–141. doi: 10.1006/niox.1999.0216. [DOI] [PubMed] [Google Scholar]
- 128.Sodum RS, Akerkar SA, Fiala ES. Anal Biochem. 2000;280:278–285. doi: 10.1006/abio.2000.4528. [DOI] [PubMed] [Google Scholar]
- 129.Ishida N, Hasegawa T, Mukai K, Watanabe M, Nishino H. J Vet Med Sci. 2002;64:401–404. doi: 10.1292/jvms.64.401. [DOI] [PubMed] [Google Scholar]
- 130.Richards DA, Silva MA, Devall AJ. Anal Biochem. 2006;351:77–83. doi: 10.1016/j.ab.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 131.Nikov G, Bhat V, Wishnok JS, Tannenbaum SR. Anal Biochem. 2003;320:214–222. doi: 10.1016/s0003-2697(03)00359-2. [DOI] [PubMed] [Google Scholar]
- 132.Sharov VS, Dremina ES, Pennington J, Killmer J, Asmus C, Thorson M, Hong SJ, Li X, Stobaugh JF, Schoneich C. Methods Enzymol. 2008;441:19–32. doi: 10.1016/S0076-6879(08)01202-0. [DOI] [PubMed] [Google Scholar]
- 133.Detweiler CD, Lardinois OM, Deterding LJ, de Montellano PRO, Tomer KB, Mason RP. Free Radic Biol Med. 2005;38:969–976. doi: 10.1016/j.freeradbiomed.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 134.Deterding LJ, Lhattacharjee S, Ramirez DC, Mason RP, Tomer KB. Anal Chem. 2007;79:6236–6248. doi: 10.1021/ac070935z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gomez-Mejiba SE, Zhai Z, Akram H, Deterding LJ, Hensley K, Smith N, Tomer RA, Tomer KB, Mason RP, Ramirez DC. Free Radic Biol Med. 2009;46:853–865. doi: 10.1016/j.freeradbiomed.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burzio LA, Waite JH. Protein Sci. 2001;10:735–740. doi: 10.1110/ps.44201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reddie KG, Carroll KS. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 138.Hutinec A, Ziogas A, Rieker A. Amino Acids. 1996;11:345–366. doi: 10.1007/BF00807941. [DOI] [PubMed] [Google Scholar]
- 139.Compagnini A, Cunsolo V, Foti S, Saletti R. Eur J Mass Spectrom. 2001;7:123–130. [Google Scholar]
- 140.Wu W, Chen Y, d’Avignon A, Hazen SL. Biochemistry. 1999;38:3538–3548. doi: 10.1021/bi982401l. [DOI] [PubMed] [Google Scholar]
- 141.Gevaert K, Pinxteren J, Demol H, Hugelier K, Staes A, Van Damme J, Martens L, Vandekerckhove J. J Proteome Res. 2006;5:1415–1428. doi: 10.1021/pr060026a. [DOI] [PubMed] [Google Scholar]
- 142.Eichacker LA, Granvogl B, Mirus O, Müller BC, Miess C, Schleiff E. J Biol Chem. 2004;279:50915–50922. doi: 10.1074/jbc.M405875200. [DOI] [PubMed] [Google Scholar]
- 143.Dayon L, Girault HH. Anal Bioanal Chem. 2007;389:841–849. doi: 10.1007/s00216-007-1492-8. [DOI] [PubMed] [Google Scholar]
- 144.Chowdhury SK, Katta V, Chait BT. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 145.Loo JA, Edmonds CG, Udseth HR, Smith RD. Anal Chem. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- 146.Kaltashov IA, Eyles SJ. Mass Spectrom Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 147.Konermann L, Bradley BS, Yan P, Xin T. Mass Spectrom Rev. 2009 [Google Scholar]
- 148.Bridgewater JD, Srikanth R, Lim J, Vachet RW. J Am Soc Mass Spectrom. 2007;18:553–562. doi: 10.1016/j.jasms.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hung CW, Schlosser A, Wei JH, Lehmann WD. Anal Bioanal Chem. 2007;389:1003–1016. doi: 10.1007/s00216-007-1449-y. [DOI] [PubMed] [Google Scholar]
- 150.Men L, Wang Y. Rapid Commun Mass Spectrom. 2005;19:23–30. doi: 10.1002/rcm.1748. [DOI] [PubMed] [Google Scholar]
- 151.Paizs B, Suhai S. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 152.Sullivan AG, Brancia FL, Tyldesley R, Bateman R, Sidhu K, Hubbard SJ, Oliver SG, Gaskell SJ. Int J Mass Spectrom. 2001;210:665–676. [Google Scholar]
- 153.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu ZJ, Julian RR. J Am Soc Mass Spectrom. 2009;20:965–971. doi: 10.1016/j.jasms.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 155.Steen H, Mann M. Anal Chem. 2002;74:6230–6236. doi: 10.1021/ac025994l. [DOI] [PubMed] [Google Scholar]
- 156.Mirzaei H, Brusniak M-Y, Mueller LN, Letarte S, Watts JD, Aebersold R. Mol Cell Proteomics. 2009;8:1934–1946. doi: 10.1074/mcp.M800569-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hernandez H, Niehauser S, Boltz SA, Gawandi V, Phillips RS, Amster IJ. Anal Chem. 2006;78:3417–3423. doi: 10.1021/ac0600407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yao XD, Diego P, Ramos AA, Shi Y. Anal Chem. 2008;80:7383–7391. doi: 10.1021/ac801096e. [DOI] [PubMed] [Google Scholar]
- 159.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 160.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 161.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 162.Kuyama H, Makoto W, Chikako T, Eiji A, Koichi T, Osamu N. Rapid Commun Mass Spectrom. 2003;17:1642–1650. doi: 10.1002/rcm.1100. [DOI] [PubMed] [Google Scholar]
- 163.Chen M, Cook KD. Anal Chem. 2007;79:2031–2036. doi: 10.1021/ac061743r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gevaert K, Impens F, Ghesquiere B, Van Damme P, Lambrechts A, Vandekerckhove J. Proteomics. 2008;8:4873–4885. doi: 10.1002/pmic.200800421. [DOI] [PubMed] [Google Scholar]
- 165.Galas DJ, Schmitz A. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Engen JR. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sheshberadaran H, Payne LG. Proc Natl Acad Sci USA. 1988;85:1–5. doi: 10.1073/pnas.85.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Heyduk E, Heyduk T. Biochemistry. 1994;33:9643–9650. doi: 10.1021/bi00198a033. [DOI] [PubMed] [Google Scholar]
- 169.Zhong M, Lin L, Kallenbach NR. Proc Natl Acad Sci USA. 1995;92:2111–2115. doi: 10.1073/pnas.92.6.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sharp JS, Becker JM, Hettich RL. Anal Biochem. 2003;313:216–225. doi: 10.1016/s0003-2697(02)00612-7. [DOI] [PubMed] [Google Scholar]
- 171.Takamoto K, Chance MR. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 172.Brabec V, Bianco P, Haladjian J. Biophys Chem. 1982;16:51–59. doi: 10.1016/0301-4622(82)85007-2. [DOI] [PubMed] [Google Scholar]
- 173.Maleknia SD, Mark RC, Kevin MD. Rapid Commun Mass Spectrom. 1999;13:2352–2358. doi: 10.1002/(SICI)1097-0231(19991215)13:23<2352::AID-RCM798>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 174.Gaut JP, Byun J, Tran HD, Heinecke JW. Anal Biochem. 2002;300:252–259. doi: 10.1006/abio.2001.5469. [DOI] [PubMed] [Google Scholar]
- 175.Petersson AS, Steen H, Kalume DE, Caidahl K, Roepstorff P. J Mass Spectrom. 2001;36:616–625. doi: 10.1002/jms.161. [DOI] [PubMed] [Google Scholar]
- 176.Salek M, Lehmann WD. Proteomics. 2005;5:351–353. doi: 10.1002/pmic.200400949. [DOI] [PubMed] [Google Scholar]
- 177.Guan ZQ, Yates NA, Bakhtiar R. J Am Soc Mass Spectrom. 2003;14:605–613. doi: 10.1016/S1044-0305(03)00201-0. [DOI] [PubMed] [Google Scholar]
- 178.Schmidt F, Krah A, Schmid M, Jungblut PR, Thiede B. Rapid Commun Mass Spectrom. 2006;20:933–936. doi: 10.1002/rcm.2382. [DOI] [PubMed] [Google Scholar]
- 179.Steen H, Mann M. J Am Soc Mass Spectrom. 2001;12:228–232. doi: 10.1016/S1044-0305(00)00219-1. [DOI] [PubMed] [Google Scholar]
- 180.Yagüe J, Antonio N, Manuel B, Manel E, Patricia A, Casal JI. Proteomics. 2005;5:2761–2768. doi: 10.1002/pmic.200401189. [DOI] [PubMed] [Google Scholar]