

Abstract
The understanding of cellular processes and their pathophysiological alterations requires comprehensive data on the abundance, distribution, modification, and interaction of all cellular components. On the one hand, artificially introduced fluorescent fusion proteins provide information about their distribution and dynamics in living cells but not about endogenous factors. On the other hand, antibodies can detect endogenous proteins, posttranslational modifications, and other cellular components but mostly in fixed and permeabilized cells. Here we highlight a new technology based on the antigen-binding domain of heavy-chain antibodies (VHH) from Camelidae. These extremely stable VHH domains can be produced in bacteria, coupled to matrices, and used for affinity purification and proteome studies. Alternatively, these VHH domains can be fused with fluorescent proteins and expressed in living cells. These fluorescent antigen-binding proteins called “chromobodies” can be used to detect and trace proteins and other cellular components in vivo. Chromobodies can, in principle, detect any antigenic structure, including posttranslational modifications, and thereby dramatically expand the quality and quantity of information that can be gathered in high-content analysis. Depending on the epitope chosen, chromobodies can also be used to modulate protein function in living cells.
Figure.
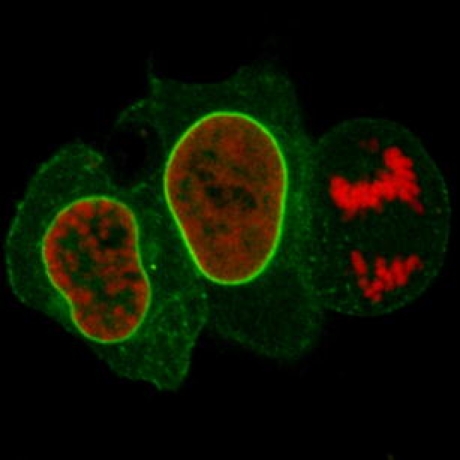
Detection of the nuclear lamina with lamin chromobody in living cells.
Keywords: Antibodies, Nanobodies, High-content analysis, Proteomics, Green fluorescent protein, Fluorescent proteins
Introduction/background
A multitude of biochemical and cell-based assays have been developed to study proteins and cellular processes. The discovery of the green fluorescent protein (GFP) [1, 2] as well as the subsequent isolation and engineering of fluorescent proteins with distinct and optimized spectral properties have revolutionized cell biology [3, 4]. Fluorescent proteins can be used as reporters of gene expression or genetically fused with proteins of interest to study their localization and dynamics in living cells. Fluorescent proteins have been used to study cellular processes ranging from DNA replication and cell cycle progression to DNA methylation and repair [5–9].
This approach is, however, limited to the visualization of artificially introduced fluorescent fusion proteins, whereas the endogenous proteins of interest remain invisible. Importantly, artificial fusion proteins may considerably differ from their endogenous counterparts in terms of expression level, activity, and localization [10, 11]. Finally, fluorescent fusions are limited to proteins and do not provide information about nonprotein components and posttranslational modifications.
In contrast, antibodies can detect practically any molecular component and cellular structure and are therefore powerful tools in biomedical research. Conventional antibodies consist of two identical heavy (H) and two light (L) chains, each comprising a variable domain at the N-terminus (abbreviated as VH and VL, respectively). Antibodies specifically recognize and bind their target molecules (antigens) via complementarity-determining regions located in the variable domains of the heavy and light chains. Generated by a highly sophisticated immune response system, antibodies are naturally secreted or are located at the cell surface as an essential part of the defense strategy against pathogens. However, their application within cells is limited by impaired disulfide bond formation in the reducing environment of the cytoplasm, inefficient assembly of the epitope-recognizing parts of the variable light and heavy chains, and difficult subcellular targeting [12, 13]. To make cellular target structures accessible for antibodies, cells have to be fixed and permeabilized. Therefore, antibodies only provide snapshots of the distribution of the antigen, and no information about the dynamics or mobility can be obtained.
Camelid antibodies
Based on conventional antibodies (IgG, 150 kDa), a number of recombinant antibody formats such as monovalent antibody fragments (Fab, 50 kDa) and single-chain variable fragments (scFv, 25 kDa) have been developed during the last decade, increasing the stability and the accessibility of antigen-binding molecules. However, as these antibody derivatives are composed of heavy- and light-chain domains, their application in living cells is still hampered by inefficient folding and disulfide bond formation. So far, only in a few cases have intracellular antibody derivates been successfully used for protein analysis in vivo [14–17].
One alternative to conventional antibodies is provided by nature itself. The humoral immune response of camels, dromedaries, and llamas includes a unique type of antibodies devoid of light chains, the so-called heavy-chain antibodies [18]. They recognize and bind their antigen via a single variable domain, the VHH (variable heavy chain of a heavy-chain antibody). With a molecular mass of approximately 13 kDa and a size in the range of approximately 2 nm × 4 nm, VHHs represent the smallest intact naturally occurring antigen-binding fragments and are therefore often called “nanobodies” [19, 20]. Nanobodies provide a series of advantages in comparison with other antibody fragments such as Fab and scFv. First, only a single domain has to be cloned and expressed to generate an intact antigen-binding fragment. Nanobodies are in vivo affinity matured entities that do not need in vitro affinity maturation steps as are often required with binders from naive or synthetic libraries. Second, specific nanobodies can be easily selected via phage display. Third, owing to their structure, nanobodies are extremely stable and soluble and they can be efficiently produced in heterologous expression systems including genetic fusions with fluorescent proteins [21, 22]. Last but not least, the antigen binding affinities of nanobodies in the nanomolar range are similar to those of scFvs [23, 24].
Chromobodies as molecular probes in living cells: closing the gap between fluorescent probes and end-point assays
The robustness and structural simplicity of nanobodies offer a wide field of new applications in molecular and cellular diagnostics. Most intriguingly and in contrast to conventional antibodies, nanobodies that are functional in living cells can be identified and selected from large VHH libraries.
On the basis of this unique characteristic, we established a new class of cellular biomarkers by genetically fusing nanobodies with fluorescent proteins for visualization of antigens in living mammalian cells. Owing to their chimeric structure, these recombinant derivatives were termed “chromobodies” (Fig. 1a). In a proof-of-concept study, chromobodies were introduced as promising, novel tools in cell biology research [25]. They were shown to detect antigens in chromatin and replication complexes as well as the cytoskeleton and allowed the visualization of dynamic changes during the cell cycle in real time, proving their applicability in different subcellular compartments. In addition a chromobody against the nuclear lamina (lamin chromobody) was generated and tested [25]. Despite transient binding, this lamin chromobody was sufficient to detect the nuclear lamina. Moreover, the transient binding mode seemed to minimize functional interference and allowed cells to pass through mitosis (Fig. 1b). Since the binding strength of chromobodies can be genetically engineered, they can be fine-tuned for optimal detection and minimal biological interference.
Fig. 1.

The chromobody technology. a Chromobodies. b Detection of the nuclear lamina with lamin chromobody in living cells. Confocal images of HeLa cells coexpressing lamin chromobody (green) and red fluorescent histone H2B as a mitosis marker. Cells in prophase, G2 phase, and anaphase are shown (from left to right), the latter displaying breakdown of the nuclear lamina, concomitant with chromosome separation (right). The scale bar represents 10 µm
Since chromobodies allow for targeting and tracing of a wide range of endogenous structures, including posttranslational modifications and nonprotein components, in their dynamic, cellular environment they should be capable of closing the analytical gap between live-cell-applicable, fluorescent fusion proteins, and antibody-based detection assays on fixed cells (end-point assays).
Chromobodies in high-content analysis
In the early stage of pharmaceutical drug development, high-content analysis (HCA) is an important technology, aiming for maximal readout of cellular parameters in terms of both quality and quantity. This way, compound libraries are screened and analyzed for desired effects and/or possible side effects at the cellular level. Based on high-throughput microscopy with automated image acquisition and computational pattern recognition in multiwell formats, HCA facilitates the simultaneous investigation of multiple cellular biomarkers in a statistically secured manner. However, HCA is limited by the availability of reliable biomarkers. To exemplify that chromobodies can complement and extend existing diagnostic approaches, we set up a first cellular assay for real-time monitoring of apoptosis in HeLa cells. We generated a cell line stably expressing the lamin chromobody to visualize the nuclear lamina. In this case, the cellular parameter of interest is the integrity of the nuclear lamina. Upon addition of apoptosis-inducing agents (such as staurosporine), fragmentation of the nuclear lamina can be monitored in real time. Using automated image acquisition and computational pattern recognition, we detected an increasing percentage of apoptotic cells, with higher staurosporine concentrations (Fig. 2).
Fig. 2.

Apoptosis assay with a lamin chromobody expressing cell line. a Time lapse imaging of untreated cells (upper row) and after addition of 5 μM staurosporine (STS). The nuclear lamina is visualized with a lamin chromobody (green). Selected images before and after (645 min) addition of STS are shown. b Quantitative evaluation with automated image acquisition and computational pattern recognition. Morphological changes of the lamina (fragmentation and vesicle formation) were monitored in 15-min intervals. The percentage of apoptotic cells after treatment with increasing concentrations of STS (0–5 µM) over a time period of 20 h is shown
Chromobodies may be used not only to track and visualize cellular components as described for the nuclear lamina, but also for pharmaceutical target validation. Chromobodies can, in principle, be generated against all surface epitopes to specifically block regulatory or catalytic domains, which resembles more closely the block of pharmaceutical, small molecules than presently used knockdown/knockout studies. Therefore, chromobodies enable new functional studies and may provide valuable information concerning target suitability at a very early stage of drug development.
Modulation of protein shape and function with nanobodies in living cells
Complex cellular processes are mostly regulated through synthesis, modification, and degradation of proteins. Besides these intensively investigated mechanisms, correct protein folding is essential for cellular processes. This is emphasized by the fact that severe diseases such as Creutzfeldt–Jakob disease and mad cow disease (BSE) are directly linked to protein misfolding. In both cases, changes in protein folding turn naturally harmless proteins into lethal, infectious particles (prions) [26]. Interestingly, several nanobodies were described that directly influence enzymatic function, protein folding, and protein aggregation or detect specific protein confirmations in vitro [27–30].
In a first pilot study we recently demonstrated that nanobodies can be utilized to manipulate and modulate the conformational state of proteins in living cells: Upon binding to GFP, two oppositely acting nanobodies cause functional three-dimensional changes resulting in decreased or increased fluorescence intensities of GFP in living cells (Fig. 3) [31]. We then exploited this effect to monitor shuttling of a transcription factor from the cytoplasm to the nucleus, depending on fluorescence changes that were induced upon nanobody binding. Again, these findings underline that nanobodies and chromobodies offer huge versatility and that many new applications can be expected.
Fig. 3.

Modulation of protein properties with nanobodies. a Structural surface representation of green fluorescent protein (GFP) (green; Protein Data Bank file 1EMB [40]), GFP bound by Minimizer nanobody (red; Protein Data Bank file 3G9A [31]), and GFP bound by Enhancer nanobody (red; Protein Data Bank file 3K1K [31]). Both nanobodies induce subtle conformational changes in the chemical environment of the GFP chromophore, thereby altering its fluorescent properties towards decreased fluorescence intensity (Minimizer) or enhanced fluorescence intensity (Enhancer). b Fluorescence modulation in living cells. Exemplary HeLa cells are shown, expressing nuclear localized GFP, GFP and Minimizer (red), and GFP and Enhancer (red). The scale bars represent 10 µm
Nanobodies for biochemical studies
Besides their unique applicability in living cells, nanobodies also proved to be valuable tools for the biochemical purification and analysis of cellular components and complexes. In contrast to conventional antibodies, nanobodies provide a number of advantages. First, they can be produced bacterially in unlimited amounts and reproducible quality, offering experimental reliability. Second, nanobodies are characterized by high robustness regarding heat as well as extreme salt and pH conditions [24, 32, 33]. Third, although comprising a small binding interface, the binding affinity of nanobodies is sufficiently high, and owing to the small surface fewer unspecific interactions seem to occur [33]. Furthermore, nanobodies can easily be coupled to beads, matrices, or even microarray chip surfaces, to generate antigen-specific nanotraps. Since nanotraps are covalently coupled single-chain peptides, elution of heavy and light chains as with conventional antibodies is prevented, an advantage for subsequent biochemical analysis.
On the basis of these favorable characteristics, we recently established a highly efficient system for the purification of GFP fusion proteins and their associated interaction partners from crude biological samples (Fig. 4, GFP-Trap) [33]. This new tool is now being widely used for protein–protein interaction studies, enzymatic assays, chromatin immunoprecipitations, and high-throughput proteomics [34–37].
Fig. 4.

Nanotraps for biochemical applications. a VHH-based nanotraps. b Example of the use of a GFP-specific nanotrap immunoprecipitation of GFP–profilerating cell nuclear antigen (PCNA) (approximately 60 kDa) from a crude cell lysate of GFP-PCNA expressing HEK 293T cells. Input (I) and bound (B) fractions were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by Coomassie staining. The GFP-Trap facilitates specific purification of GFP fusion proteins and their interaction partners
Outlook
Recombinant antibody technologies as established during the last decade enable a wide variety of novel applications for biomedical research especially for molecular and cellular biology. In particular, single domain antibody derivates such as the nanobodies, chromobodies, and nanotraps described here have several decisive advantages as they can be selected from recombinant libraries [38, 39]. Nanobodies can be produced in prokaryotes and eukaryotes and used for new functional studies in vitro and in vivo. Recent proof-of-principle studies demonstrated that the chromobody technology is applicable for real-time HCA, that nanobodies can be used to modulate protein properties in living cells, and that nanotraps represent a new class of highly efficient tools for biochemical and proteomic research. After these first demonstrations of the versatility of these new antibody formats, a plethora of new in vivo and in vitro applications are to be expected in the next years.
These developments will likely include chromobodies as biomarkers for HCA and live cell analysis providing an optical readout of essential cellular processes such as signal transduction, cell cycle progression, epigenetic regulation, and posttranslational modifications. At present, the rate-limiting factor is the still-limited availability of specific nanobodies as the best binders are still derived in a time-consuming procedure from in vivo matured heavy-chain antibodies. Thus the development of better selection and in vitro maturation procedures for the isolation and engineering of highly specific binders is of utmost importance.
Acknowledgements
This work was supported by the GO-Bio Program of the BMBF and the Deutsche Forschungsgemeinschaft (DFG).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Katrin Schmidthals and Jonas Helma contributed equally to this work.
Contributor Information
Ulrich Rothbauer, Phone: +49-89-218074232, FAX: +49-892-18074236, Email: u.rothbauer@lmu.de.
Heinrich Leonhardt, Phone: +49-892-18074232, FAX: +49-892-18074236, Email: h.leonhardt@lmu.de.
References
- 1.Tsien RY. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 2.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 3.Heim R, Tsien RY. Curr Biol. 1996;6:178–182. doi: 10.1016/S0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 4.Shaner NC, Steinbach PA, Tsien RY. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 5.Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, et al. J Cell Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schermelleh L, Spada F, Easwaran HP, Zolghadr K, Margot JB, et al. Nat Methods. 2005;2:751–756. doi: 10.1038/nmeth794. [DOI] [PubMed] [Google Scholar]
- 7.Easwaran HP, Leonhardt H, Cardoso MC. Cell Cycle. 2005;4:453–455. doi: 10.4161/cc.4.3.1525. [DOI] [PubMed] [Google Scholar]
- 8.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Proc Natl Acad Sci USA. 2005;102:8905–8909. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spada F, Haemmer A, Kuch D, Rothbauer U, Schermelleh L, et al. J Cell Biol. 2007;176:565–571. doi: 10.1083/jcb.200610062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalfie M, Kain S. Green fluorescent protein: properties, applications and protocols. Totowa: Wiley; 2005. [Google Scholar]
- 11.Leonhardt H, Rahn HP, Cardoso MC. J Cell Biochem Suppl. 1998;30–31:243–249. doi: 10.1002/(SICI)1097-4644(1998)72:30/31+<243::AID-JCB30>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Biocca S, Neuberger MS, Cattaneo A. EMBO J. 1990;9:101–108. doi: 10.1002/j.1460-2075.1990.tb08085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cattaneo A, Biocca S. Trends Biotechnol. 1999;17:115–121. doi: 10.1016/S0167-7799(98)01268-2. [DOI] [PubMed] [Google Scholar]
- 14.Biocca S, Pierandrei-Amaldi P, Cattaneo A. Biochem Biophys Res Commun. 1993;197:422–427. doi: 10.1006/bbrc.1993.2496. [DOI] [PubMed] [Google Scholar]
- 15.Cardinale A, Lener M, Messina S, Cattaneo A, Biocca S. FEBS Lett. 1998;439:197–202. doi: 10.1016/S0014-5793(98)01369-6. [DOI] [PubMed] [Google Scholar]
- 16.Kontermann RE. Methods. 2004;34:163–170. doi: 10.1016/j.ymeth.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Marasco WA, Chen S, Richardson JH, Ramstedt U, Jones SD. Hum Gene Ther. 1998;9:1627–1642. doi: 10.1089/hum.1998.9.11-1627. [DOI] [PubMed] [Google Scholar]
- 18.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, et al. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 19.Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Protein Eng. 1994;7:1129–1135. doi: 10.1093/protein/7.9.1129. [DOI] [PubMed] [Google Scholar]
- 20.Muyldermans S. J Biotechnol. 2001;74:277–302. doi: 10.1016/s1389-0352(01)00021-6. [DOI] [PubMed] [Google Scholar]
- 21.Muyldermans S, Baral TN, Retamozzo VC, De Baetselier P, De Genst E, et al. Vet Immunol Immunopathol. 2009;128:178–183. doi: 10.1016/j.vetimm.2008.10.299. [DOI] [PubMed] [Google Scholar]
- 22.Olichon A, Surrey T. J Biol Chem. 2007;282:36314–36320. doi: 10.1074/jbc.M704908200. [DOI] [PubMed] [Google Scholar]
- 23.Sheriff S, Constantine KL. Nat Struct Biol. 1996;3:733–736. doi: 10.1038/nsb0996-733. [DOI] [PubMed] [Google Scholar]
- 24.van der Linden RH, Frenken LG, de Geus B, Harmsen MM, Ruuls RC, et al. Biochim Biophys Acta. 1999;1431:37–46. doi: 10.1016/s0167-4838(99)00030-8. [DOI] [PubMed] [Google Scholar]
- 25.Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, et al. Nat Methods. 2006;3:887–889. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- 26.Prusiner SB. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 27.Lauwereys M, Arbabi GM, Desmyter A, Kinne J, Holzer W, et al. EMBO J. 1998;17:3512–3520. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumoulin M, Last AM, Desmyter A, Decanniere K, Canet D, et al. Nature. 2003;424:783–788. doi: 10.1038/nature01870. [DOI] [PubMed] [Google Scholar]
- 29.Lafaye P, Achour I, England P, Duyckaerts C, Rougeon F. Mol Immunol. 2009;46:695–704. doi: 10.1016/j.molimm.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Habicht G, Haupt C, Friedrich RP, Hortschansky P, Sachse C, et al. Proc Natl Acad Sci USA. 2007;104:19232–19237. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, et al. Nat Struct Mol Biol. 2010;17:133–138. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- 32.Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. FEBS Lett. 1997;414:521–526. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- 33.Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, et al. Mol Cell Proteomics. 2008;7:282–289. doi: 10.1074/mcp.M700342-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, et al. J Cell Biol. 2008;183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogowski K, Juge F, van Dijk J, Wloga D, Strub JM, et al. Cell. 2009;137:1076–1087. doi: 10.1016/j.cell.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 36.Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, et al. Science. 2009;325:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 37.Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, et al. Nature. 2009;462:935–939. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanha J, Xu P, Chen Z, Ni F, Kaplan H, et al. J Biol Chem. 2001;276:24774–24780. doi: 10.1074/jbc.M100770200. [DOI] [PubMed] [Google Scholar]
- 39.Monegal A, Ami D, Martinelli C, Huang H, Aliprandi M, et al. Protein Eng Des Sel. 2009;22:273–280. doi: 10.1093/protein/gzp002. [DOI] [PubMed] [Google Scholar]
- 40.Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, et al. Proc Natl Acad Sci USA. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]