

Abstract
Success in evolution depends critically upon the ability of organisms to adapt, a property that is also true for the proteins that contribute to the fitness of an organism. Successful protein evolution is enhanced by mutational pathways that generate a wide range of physicochemical mechanisms to adaptation. In an earlier study, we used a weak-link method to favor changes to an essential but maladapted protein, adenylate kinase (AK), within a microbial population. Six AK mutants (a single mutant followed by five double mutants) had success within the population, revealing a diverse range of adaptive strategies that included changes in nonpolar packing, protein folding dynamics, and formation of new hydrogen bonds and electrostatic networks. The first mutation, AKBSUB Q199R, was essential in defining the structural context that facilitated subsequent mutations as revealed by a considerable mutational epistasis and, in one case, a very strong dependence upon the order of mutations. Namely, whereas the single mutation AKBSUB G213E decreases protein stability by >25°C, the same mutation in the background of AKBSUB Q199R increases stability by 3.4°C, demonstrating that the order of mutations can play a critical role in favoring particular molecular pathways to adaptation. In turn, protein folding kinetics shows that four of the five AKBSUB double mutants utilize a strategy in which an increase in the folding rate accompanied by a decrease in the unfolding rate results in additional stability. However, one mutant exhibited a dramatic increase in the folding relative to a modest increase in the unfolding rate, suggesting a different adaptive strategy for thermostability. In all cases, an increase in the folding rates for the double mutants appears to be the preferred mechanism in conferring additional stability and may be an important aspect of protein evolution. The range of overlapping as well as contrasting strategies for success illustrates both the power and subtlety of adaptation at even the smallest unit of change, a single amino acid.
Introduction
Adaptation at the molecular level is the fundamental mechanism by which organisms succeed or fail during natural selection. A hallmark of protein evolution is the remarkable plasticity of proteins to change and thereby provide the diversity of structures and functions essential to adaptation. A combination of in vivo and in vitro studies employing experimental evolution has provided a wealth of insights into the molecular basis for adaptation (1–4). In terms of protein evolution, the ability of proteins to maintain function while undergoing adaptation is a key attribute as nonfunctional intermediates will likely reduce the fitness of the organism (5,6). Because function is the key attribute that is acted upon by natural selection, adaptation of proteins to new conditions is expected to be, and is, astoundingly diverse. A central feature to a protein's mutational robustness, and likewise its function, is stability. It has become increasingly apparent that protein stability plays an essential role in circumscribing the accessible thermodynamic landscape for successful protein evolution (7–9). The maintenance of protein stability during adaptation is seen by a wealth of studies documenting the importance of chaperones to fitness and by studies using libraries of molecules generated in vitro that show how changes in stability can buffer against subsequent destabilizing mutations that alter specificity during adaptation (10–17).
To investigate the molecular mechanisms and mutational pathways accessible during protein adaptation, the weak-link method was developed to tightly couple organismal fitness to the performance of a single protein (18). In the weak-link approach, a single chromosomal copy of the adenylate kinase gene (adk) of the thermophile Geobacillus stearothermophilus was replaced by a copy from the mesophile Bacillus subtilis. The recombinant G. stearothermophilus expressing the B. subtilis adk has a temperature-sensitive phenotype that limits its growth to ∼55°C (18,19). At temperatures exceeding 55°C, adenylate homeostasis is disrupted in the recombinant G. stearothermophilus strain due to heat inactivation of the B. subtilis adenylate kinase (AKBSUB). Evolution of a large population (∼1011 cells) to increase their temperature-growth range to that of the original G. stearothermophilus (48–72°C) was performed by a slow ramp of temperature from 55 to 70°C over 30 days. During the course of selection, the population was sampled and functional intermediates of adaptation were observed as mutations to B. subtilis adk.
In our original study, the first mutant to reach fixation was AKBSUB Q199R (glutamine to arginine at residue 199). AKBSUB Q199R has a modestly higher thermostability but significantly improves enzyme function through an increase in kcat (20). AKBSUB Q199R was, in turn, replaced over the temperature range of 62–65°C by five double mutants that arose nearly simultaneously within the population and share AKBSUB Q199R as their progenitor. All five double mutants arose to at least 5% of the population in the temperature range of 62–63°C as the AKBSUB Q199R parent declined (18). Because each of the double mutants has AKBSUB Q199R as its progenitor, the AKBSUB Q199R substitution provides the structural context for all subsequent mutants.
In this study, a combination of x-ray crystallographic, protein-folding, site-directed mutagenesis and biochemical studies were used to elucidate the molecular basis for the conferred thermostability in each AKBSUB mutant. The five AKBSUB double mutants display diverse mechanisms of stabilization that emphasize the plasticity and dynamism underlying protein evolution. Even within the relatively modest selection range of these experiments, molecular mechanisms of adaptation discovered included:
-
1.
Formation of a new electrostatic network on the protein surface.
-
2.
Better nonpolar packing and interactions.
-
3.
A change in protein-folding dynamics consistent with destabilization of the unfolded state.
-
4.
Formation of a new hydrogen bond that stabilizes C-terminal α-helix interactions with the core of the protein (7,21–26).
We also found that each of the double mutants had substantial epistasis (i.e., cooperative mutational effects or intramolecular pleiotropy) with the original AKBSUB Q199R mutant. In turn, we demonstrated that the order of mutations can be a critical determinant for evolutionary success. These results demonstrate a critical role for the versatility of protein evolution as well as its dependence on the history of previous adaptive events that favor specific molecular pathways of adaptation.
Materials and Methods
Protein crystallization and structural analysis
All AK mutants were crystallized near conditions initially determined for the wild-type enzyme. In some cases better crystals were obtained by streak-seeding from crystals of AKBSUB or AKBSUB Q199R. Crystals of mutant AKs were grown over a modest range of concentrations (16–22 mg/mL) by vapor diffusion against 50 mM CHES pH 9.0, 25 mM CaCl2, and 30–34% (w/v) PEG 1500 at 20°C in the presence of inhibitor P1, P5-di(adenosine-5′)pentaphosphate (Ap5A). Statistics for the data and refinement of the structures are summarized in Table S1 in the Supporting Material. With the exception of AKBSUB Q199R/G214R, all the crystals were determined to be in the space group P21, and contained two copies of AK in the asymmetric unit. AK Q199R/G214R crystallized in space group P212121 and contained only one copy in the asymmetric unit. The structure of the mutant AKBSUB proteins were determined by molecular replacement using the structure of wild-type AKBSUB Q199R (PDB 2EU8) as a search model. The data was processed using HKL2000 (27) or d∗trek (28) and the refinement carried out in CNS (29). Accessible polar fraction was calculated using WHAT IF with default parameters (1.4 Å probe radius) (30).
Equilibrium unfolding
GuHCl-induced equilibrium unfolding was performed in 10 mM phosphate buffer, pH 7.0 using far-UV circular dichroism (CD) detection (200–300 nm). Samples were incubated for 2 h before measurements. The equilibrium-unfolding reactions were reversible without any display of protein-concentration dependence. The equilibrium-unfolding curves for AK wild-type and mutant were analyzed using a two-state model to determine ΔGU(H2O) (31). The ΔGU(H2O) values listed in Table 1 are average values from multiple fits. Errors (means ± standard deviations) are derived from multiple experiments, and in all cases are consistent with (i.e., within 10%) the free energy of unfolding in water derived from the rate constants.
Table 1.
Folding and unfolding kinetics of the selected AKBSUB mutants
| Protein | ΔGUnfold(H2O) (kJ/mol) | kF(H2O) (s−1) | kU(H2O) (s−1) | βT |
|---|---|---|---|---|
| AKBSUB | 14 ± 1 | 0.030 ± 0.002 | 8.01 × 10−5 ± 0.5 × 10−5 | 0.45∗ |
| AKBSUB Q199R | 16 ± 1 | 0.015 ± 0.003 | 1.50 × 10−5 ± 0.2 × 10−5 | 0.25∗ |
| AKBSUB Q199R/G213E | 21 ± 1 | 0.13 ± 0.01 | 1.70 × 10−5 ± 0.2 × 10−5 | 0.53 |
| AKBSUB Q199R/T179I | 24 ± 1 | 0.80 ± 0.04 | 2.0 × 10−5 ± 0.3 × 10−5 | 0.58 |
| AKBSUB Q199R/G214R | 25 ± 1 | 0.20 ± 0.02 | 3.0 × 10−6 ± 0.1 × 10−6 | 0.48 |
| AKBSUB Q199R/A193V | 26 ± 1 | 12.2 ± 0.2 | 1.5 × 10−4 ± 0.3 × 10−4 | 0.78 |
| AKBSUB Q199R/Q16L | 28 ± 1 | 0.70 ± 0.04 | 4.40 × 10−6 ± 0.7 × 10−6 | 0.57 |
ΔGUnfold (H2O) values are based on the equilibrium data (Fig. 2a), and kF(H2O) and kU(H2O) values are folding and unfolding rate constants extrapolated to 0 M GuHCl (Fig. 2b). βT (a measure of nativeness/compaction in the transition state for folding) was calculated as mf/(meq) where meq = mf + mu such that mf is the slope of the folding arm and mu is the slope of the unfolding arm in the Chevron plot (Fig. 2b).
Taken from Couñago et al. (20).
Folding dynamics and data analysis
Time-resolved folding and unfolding was monitored by way of far-UV CD, i.e., at 220 nm, using a Pi-Star series spectrometer equipped with a dedicated stopped-flow accessory (Applied Photophysics, Leatherhead, UK). AK mutants were mixed at a 1: 5 ratio with varying concentration of buffered chemical denaturant (GuHCl, 10 mM phosphate buffer, pH 7.0). The observed kinetic traces were free of missing amplitudes, diffusional effects, and protein concentration dependence. For each reaction condition, three-to-six kinetic traces were averaged and fit to a standard single-exponential-decay model equation. In turn, the kinetic data were fit a to a reversible two-state folding model,
where kf and ku are the rate constants for folding and unfolding in water, respectively, and [D] is the concentration of chemical denaturant.
Circular dichroism studies
Thermal denaturation temperatures for AKBSUB mutants were determined by circular dichroism (CD). Denaturation studies were performed at 220 nm using a model No. J-815 chiro-optical spectrometer (JASCO, Easton, MD) in a quartz cuvette (10-mm pathlength) from 20 to 80°C at a rate of 1°C/min. Data was fitted as an apparent two-state transition although, as described previously, thermal denaturation of AK is not completely reversible and limits the interpretation of the data (20). Protein samples were prepared to a final concentration of 20 μM in 10 mM KH2PO4, pH 7.0. All measurements were performed in triplicate.
Results
Crystallographic analysis of AKBSUB mutant structures
The structures of all five double mutants were determined to 1.8 Å resolution in complex with the transition state analog Ap5A (Table S1). The phosphoryl transfer activity of AK depends on a hinge-bending conformational change upon substrate binding that closes the LID domain (residues 128–159) onto the CORE of the protein before catalysis (32–34) (Fig. 1 a). In the presence of Ap5A, the LID domain is closed over the active site. 2Fo-Fc electron density maps made for each AK mutant showed strong and interpretable electron density at the position of each mutation for both copies in the asymmetric unit (Fig. 1) with the exception of AKBSUB Q199R/G214R where there was only one copy of the protein in the asymmetric unit. In addition, electron density difference maps (2Fo-Fc AKBSUB double mutant-2Fo-Fc AKBSUB Q199R) were used to identify changes or rearrangements of nearby atoms (Fig. S1).
Figure 1.
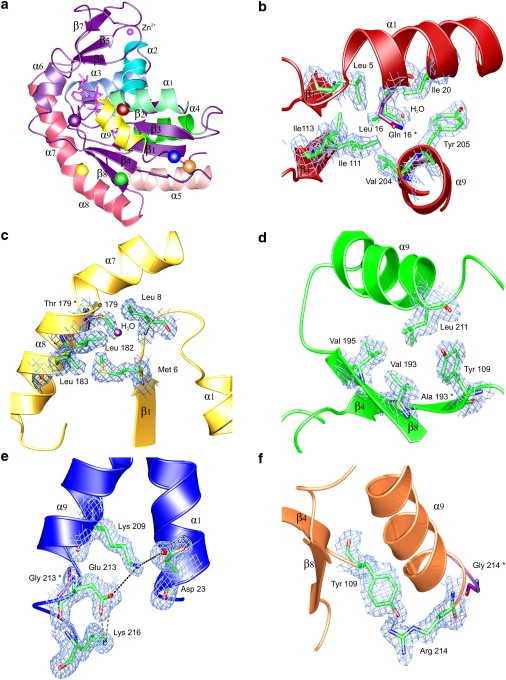
Crystallographic analysis of adaptive AKBSUB mutations at 1.8 Å resolution. (a) Secondary structure of AKBSUB with the positions of mutations indicated as colored spheres: AKBSUBQ199R (purple); AKBSUBQ199R/Q16L (red); AKBSUBQ199R/T179I (yellow); AKBSUBQ199R/A193V (green); AKBSUBQ199R/G213E (blue); and AKBSUBQ199R/G214R (orange). Secondary structure elements are labeled using the convention described by Bae and Phillips (25) with α-helices colored individually. (b–f) 2Fo-Fc σA-weighted electron density maps covering the residues immediately surrounding the sites of each double mutant. The amino acid found originally in AKBSUBQ199R is represented by purple sticks and the residue number indicated with an asterisk. (b) AKBSUBQ199R/Q16L has excellent packing in a nonpolar pocket made up of Leu-5, Ile-20, Ile-111, Ile-113, Val-204, Tyr-205, and Ala-206 (1.2 σ). (c) AKBSUBQ199R/T179I displaces a water and packs into a nonpolar pocket comprised of Met-6, Gly-7, Leu-8, Leu-182, and Leu-183 (0.7 σ). (d) AKBSUBQ199R/A193V is at a largely solvent-exposed position. Leu-211 and Tyr-109 are displaced slightly to form a pocket (1.3 σ). (e) AKBSUBQ199R/G213E is in a highly solvent-exposed position and may form a new electrostatic network made up of Lys-209 ↔ Glu-213 ↔ Lys-216 along the solvent-exposed face of the C-terminal helix-9 as well as Asp-23 from helix-1 (0.9 σ). (f) AKBSUBQ199R/G214R forms a new hydrogen bond to Tyr-109, potentially tethering the C-terminal helix to the core of the protein (1.5 σ).
All structures were analyzed for general trends in the number of electrostatic interactions and hydrogen bonds as well as buried surface area (Table S2). The presence of two copies of AK in the asymmetric unit for all but AKBSUB Q199R/G214R provided an additional level of validation for our structural analyses, because any significant differences correlated to increased thermostability would likely be observed in both copies of the protein. Surprisingly, many changes in potential hydrogen bonding, ion pairs, and accessible surface area number observed between AKBSUB Q199R and the mutants AKBSUB Q199R/Q16L, Q199R/T179I, Q199R/A193V, and Q199R/G213E were of comparable magnitude or number to that seen between their respective copies A and B (Table S2). Although no general trends were apparent, specific interactions at the site of each mutation provided substantial information about the likely mechanisms for stabilization of a particular adenylate kinase mutant in relation to its ancestor, AKBSUB Q199R.
AKBSUB Q199R/Q16L
In the progenitor AKBSUB Q199R, Gln-16 is largely buried with little water-accessible surface area (average accessible polar fraction 0.28). Replacement of Gln-16 with leucine places the nonpolar leucine side chain into a very well-packed nonpolar pocket (Fig. 1 b). In AKBSUB Q199R/Q16L, the Gln-16 Cβ, Cγ, and Cδ atoms are replaced by those of Leu-16, leading to better packing of nonpolar atoms (average accessible polar fraction 0.22) (Fig. 1 b). Two waters are found near the polar Gln-16 Oɛ1/Nɛ2 atoms. In the AKBSUB Q199R/Q16L structure, the positions formally occupied by Oɛ1/Nɛ2 of Gln-16 are replaced with a new third water. This suggests that the stability garnered by the substitution of Gln-16 with leucine is not the result of replacement of the polar Oɛ1/Nɛ2 atoms of Gln-16 with those of the nonpolar Leu-16 side chain.
AKBSUB Q199R/T179I
The structure of AKBSUB Q199R/T179I (Fig. 1 c) shows that a water normally found associated with the polar hydroxyl of Thr-179 in both copies of AKBSUB Q199R as well as AKBSUB Q199R/Q16L, AKBSUB Q199R/G213E, and AKBSUB Q199R/G214R is absent in AKBSUB Q199R/T179I structure. A nonpolar pocket formed by residues Met-6, Leu-8, Leu-182, and Leu-183 in AKBSUB Q199R/T179I had less solvent-accessible surface area than AKBSUB Q199R (34 Å2 vs. 29 Å2). It is consistent with the loss of a bound water and subsequent formation of a well-packed nonpolar interface between helix-7 and the end of β-strand-1 and the loop joining it to helix-1.
AKBSUB Q199R/A193V
Val-193 is in a partially solvent-exposed position of β-strand-8 (accessible polar fraction 0.47). The structure of AKBSUB Q199R/A193V suggests that it may provide better packing leading to stabilization of β-strand-8 to the C-terminal helix-9 (Fig. 1 d). Ala-193 is in a solvent-accessible position that can accommodate a valine and is able to make nonpolar contacts to the side chains of Val-195 and Leu-211 (helix 9) as well as that of Tyr-109 from β-strand-4 (Fig. 1 d). Although Val-193 can be positioned into contact with Leu-211 and Tyr-109, both these residues must displace very slightly to accommodate the additional γ-methyl groups of the valine, suggesting that packing of valine may come at some cost to stability.
AKBSUB Q199R/G213E
Glu-213 is in a highly solvent-accessible position (accessible polar fraction 0.82) near the C-terminus of AKBSUB. Introduction of glutamic acid at position 213 increases the length of the C-terminal helix-9 by one additional turn. The structure of AKBSUB Q199R/G213E allows the formation of a new electrostatic network between the Oɛ1/2 oxygens of Glu-213 and the Nζ nitrogens of Lys-209 (4.5:5.1 Å) and Lys-216 (2.2:3.9 Å) positioned one turn before, and after, position 213 of the C-terminal helix-9 (Fig. 1 e). Structural analysis of AKBSUB Q199R/G213E is made more difficult because the side chain of Glu-213 in copy B of the asymmetric unit may be repositioned by a steric clash with a symmetry-related molecule.
AKBSUB Q199R/G214R
In AKBSUB Q199R/G214R, Arg-214 is positioned to make a new hydrogen bond to the hydroxyl of Tyr-109 with a donor-acceptor distance of NH2 = 2.7 Å and angle of 76°. Although there is a possibility for a bifurcated hydrogen bond from the Nɛ position of Arg-214, there is strong electron density only for the NH2 ↔ OH interaction (Fig. 1 f). Like AKBSUB Q199R/G213E, introduction of an arginine at a position formally held by glycine increases the length of the C-terminal helix-9 by one additional turn.
Protein-folding dynamics
Although high-resolution crystal structures can provide a great deal of information with respect to these final folded structures, changes in protein folding kinetics also provide critical insights into the mechanisms and energetics of stabilization for the observed adaptive mutations. Time-resolved folding and unfolding of the adaptive AK mutants was monitored by far-UV CD using the chemical denaturant guanidine hydrochloride (GuHCl) at 20°C. All traces were consistent with single-exponential processes and free of missing amplitudes within the instrument's dead time (2–4 ms). Thus, the time-resolved folding and unfolding reactions (in addition to chemically induced equilibrium unfolding reactions) of all AK mutants can be considered apparent two-state reactions. Such mechanism is consistent with the observations previously reported for AKBSUB and AKBSUB Q199R (20). Likewise, the logarithms of the observed rate constants versus GuHCl concentration exhibited V-shaped appearances further supporting two-state kinetic behavior (Fig. 2 b). The so-called Chevron plots were used to extrapolate the folding and unfolding rates for a given AK variant (i.e., folding (kF) at low denaturant and unfolding at high denaturant rate (kU) constants). In all cases, the extrapolated rates were compared to nonlinear equilibrium isothermal data by way of independently calculated ΔGU(H2O) to illustrate the confidence-level of the observed rates (Fig. 2, inset).
Figure 2.
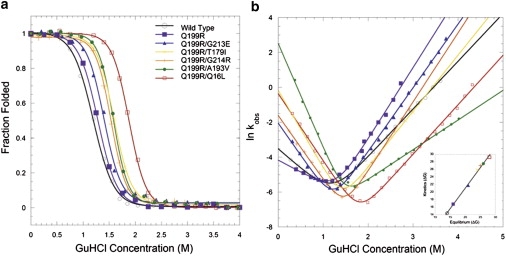
Protein folding dynamics. (a) Equilibrium unfolding and (b) Chevron plots (lnkobs versus [GuHCl]) of folding/unfolding kinetics (pH 7.0, 20°C) for wild-type AKBSUB (open black circles), AKBSUBQ199R (solid purple squares), and the five double mutants: AKBSUBQ199R/Q16L (open red squares), AKBSUBQ199R/T179I (yellow crosses), AKBSUBQ199R/G213E (solid blue triangles), AKBSUBQ199R/G214R (orange plus signs), and AKBSUBQ199R/A19V (solid green circles). All AK double mutants show faster folding and slower unfolding rates relative to their immediate ancestor AKBSUBQ199R with the exception of AKBSUBQ199R/A19V, which displays much faster folding and unfolding rates relative to AKBSUBQ199R.The wild-type AKBSUB, AKBSUBQ199R are taken from Couñago et al. (20). (Inset) Equilibrium isotherms versus kinetic ΔGU(H2O).
In general, all of the AKBSUB species studied exhibit very slow unfolding rates (average kU = 4.1 × 10−5 s−1) whereas the folding rates are substantially faster (average kF = 2.0 s−1). The folding and unfolding rates of the five double mutants were compared to that of AKBSUB Q199R. AKBSUB Q199R exhibits a slower folding rate (0.015 s−1) than wild-type AK, whereas all the adaptive double mutants had rates 9- to 800-fold larger than AKBSUB Q199R (Table 1). With the exception of AKBSUB Q199R/A193V, all of the mutants exhibited only modest changes in unfolding (from 0.8-fold to 5-fold) but like their immediate progenitor, AKBSUB Q199R, unfolding rates were still very slow, ranging from 1.7 × 10−5 to 4.4 × 10−6 s−1. The dynamic behavior of AKBSUB Q199R/A193V is unique in that this variant has faster rates of both folding (by 800-fold) and unfolding (by 10-fold) relative to AKBSUB Q199R. Because the effect on the folding speed is larger than the effect on the unfolding rate, the net effect is stabilizing.
The Tanford β-value is a rough measure of structure and interaction in the transition state for folding (35). All double mutants have higher β-values than AKBSUB Q199R (Table 1), which suggests that their transition states are more ordered (i.e., more nativelike) as compared to that for AKBSUB Q199R. For AKBSUB Q199R/A193V, the β-value is especially high, which may explain the increased unfolding speed. The high β-value indicates structural similarity between the native and the transition states, thus only a few changes or interactions are broken when this variant unfolds. We note that the β-values should be interpreted with caution: although all variants have rather similar folded states based on the crystal structures, the unfolded states may be altered energetically and structurally due to the mutations.
Mutational epistasis and order dependence
To assess the extent to which the original mutant AKBSUB Q199R determines the successful molecular pathways for subsequent mutations, the single mutants AKBSUB Q16L, AKBSUB T179I, AKBSUB A193V, AKBSUB G213E, and AKBSUB G214R were constructed and their thermal unfolding temperatures (Tm) measured by CD (Fig. 3 and Table 2). The AKBSUB double mutants identified previously were also measured under identical solution conditions to provide Tm values for comparison to the single mutants (Fig. 3). The order of thermal stabilities for the double mutants is in excellent agreement with the stabilities measured in GuHCl (Table 1) and by differential scanning calorimetry (36). As shown in Table 2, only AKBSUB Q16L showed substantially higher stability than AKBSUB Q199R (5.9°C). AKBSUB T179I and AKBSUB A193V showed marginally decreased stabilities of −0.6 to −1.7°C, respectively. Strikingly, AKBSUB G213E reduced thermal stability by >25°C. CD of AKBSUB G213E shows little indication of native structure and suggests that the substitution of Gly-213 with glutamic acid had produced a largely unfolded protein at temperatures even at 20°C (Fig. 3 c). This is in marked contrast to their thermal stabilities in the background of the Q199R mutation, where each of the double mutants was 1.9–13.8°C more stable than the single mutant AKBSUB Q199R.
Figure 3.
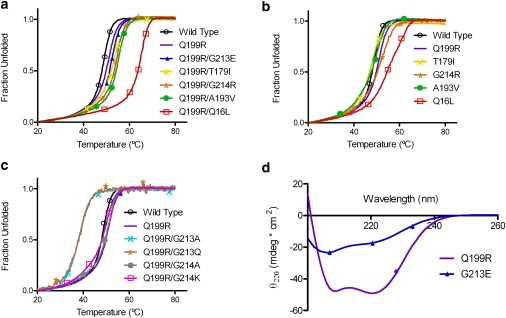
Mutational epistasis determined from a comparison of thermal denaturation temperatures for each adaptive mutant with and without the AKBSUBQ199R background. Circular dichroism was used to monitor unfolding as a function of temperature with data fitted as an apparent two-state transition. (a) Double mutants: AKBSUB (open black circles), AKBSUBQ199R (solid purple squares), AKBSUBQ16L(open red squares), AKBSUBT179I (yellow crosses), AKBSUBA193V (solid green circles), AKBSUBG213E (blue triangles), and AKBSUBG214R (orange stars). (b) AKBSUB (black), AKBSUBQ199R (purple) compared to single mutants: AKBSUB (open black circles), AKBSUBQ199R/Q16L (open red squares); AKBSUBQ199R/T179I (yellow crosses); AKBSUBQ199R/A193V (solid green circles); and AKBSUBQ199R/G214R (orange stars). (c) Point mutation at positions 213 and 214: AKBSUB (open black circles), AKBSUBQ199R (solid purple squares), AKBSUBQ199R/G213A (teal crosses), AKBSUBQ199R/G213Q (brown stars), AKBSUBQ199R/G214A (solid gray circles), and AKBSUBQ199R/G214K (open pink squares). (d) Far-UV wavelength scan of AKBSUBQ199R (purple squares) and AKBSUBG213E (blue triangles) at 20°C.
Table 2.
Thermostability of AKBSUB mutants determined by CD with and without the initial Q199R mutation
| Protein | Tm (°C) | Calculated Tm if mutations had been additive versus actual stability of the double mutants: measured Tm versus calculated (difference)∗ |
|---|---|---|
| AKBSUB | 48.4 ± 0.2 | <31.1 vs. 51.4 (−17.3) |
| AKBSUB Q199R | 49.5 ± 1.2 | |
| AKBSUB Q199R/G213E | 51.4 ± 0.3 | |
| AKBSUB G213E | <30 | |
| AKBSUB Q199R/A193V | 54.6 ± 0.2 | 50.0 vs. 54.6 (−4.6) |
| AKBSUB A193V | 48.9 ± 0.7 | |
| AKBSUB Q199R/T179I | 53.3 ± 0.5 | 48.9 vs. 53.3 (−4.9) |
| AKBSUB T179I | 47.8 ± 0.2 | |
| AKBSUB Q199R/G214R | 53.4 ± 0.6 | 50.9 vs. 53.4 (−2.4) |
| AKBSUB G214R | 49.8 ± 2.3 | |
| AKBSUB Q199R/Q16L | 63.3 ± 1.5 | 56.5 vs. 63.3 (−6.8) |
| AKBSUB Q16L | 55.4 ± 0.4 |
Calculated Tm is estimated by the addition of the Tm values of the individual single mutants.
In the original AKBSUB structure, the C-terminal residues 213–216 are disordered whereas the same residues in the mutant AKBSUB Q199R are well ordered, suggesting that stabilization of the C-terminal helix-9 (residues 201–214) may be an important mechanism of stabilization for AKBSUB (Fig. 1) (25). Approximately midway through helix-9, there is a significant bend in the helix at residues 209–210 that allows good packing to β-strands 4 and 8. Additional mutational studies were performed at Gly-213 and Gly-214 to determine whether mutations at these positions increase helical propensity or make better surface electrostatic interactions which might also provide increases in thermostability. Thermal stabilities for AKBSUB mutants with substitutions at Gly-213 or Gly-214 with and without the initial mutation to Q199R were determined (Tables 2 and 3). Mutations at position Gly-214 to either alanine or lysine were well tolerated but still lowered thermostability with respect to AKBSUB Q199R by 0.4 and 1.1°C, respectively. In contrast, mutations at Gly-213 were poorly tolerated with AKBSUB Q199R/G213A and AKBSUB Q199R/G213Q reducing stability relative to AKBSUB Q199R 11.6 and 10.2°C, respectively. The finding that AKBSUB Q199R/G213Q reduces stability suggests that a negative charge at position 213—although only in combination with Q199R—is able to increase thermal stability significantly.
Table 3.
Thermostability of AKBSUB mutants at Gly-213 and Gly-214 determined by CD
| Protein | Tm (°C) |
|---|---|
| AKBSUB | 48.4 ± 0.2 |
| AKBSUB Q199R | 49.5 ± 1.2 |
| AKBSUB Q199R/G213E | 51.4 ± 0.3 |
| AKBSUB Q199R/G214R | 53.4 ± 0.6 |
| AKBSUB Q199R/G213A | 37.9 ± 0.4 |
| AKBSUB Q199R/G213Q | 39.3 ± 0.9 |
| AKBSUB Q199R/G214A | 49.1 ± 0.1 |
| AKBSUB Q199R/G214K | 48.4 ± 0.6 |
Discussion
The success or failure of organisms within an ever changing environment is critically dependent upon facile and robust adaptation. One of the most important attributes for adaptive protein evolution is the ability for small incremental changes, such as single nucleotide substitutions, to generate molecules with a diverse range of physicochemical properties. In turn, this diverse population will be acted upon by natural selection to produce increasingly more fit individuals. The weak-link method was developed to tightly couple the fitness of a cell to the function of AK, thereby allowing us to explore quantitatively the link between natural selection and the biophysical basis for its outcomes. In the first study, five double mutants arose nearly simultaneously within the population that had at least modest success (making up at least 5% of the population at single temperature) (18). Each of the double mutants has a common parent or progenitor, AKBSUB Q199R and thus the five mutants are essentially siblings competing independently for success within the overall population. Even in this modest number of mutants, a remarkable diversity of mechanistic strategies to increase stability was observed, providing insight into the physical basis for protein evolution.
AKBSUB Q199R/Q16L and AKBSUB Q199R/T179I improve nonpolar contacts and packing
Two of the mutants, AKBSUB Q199R/Q16L and AKBSUB Q199R/T179I, display improved packing within nonpolar pockets that presumably stabilize the core of the protein. In addition, these mutants had among the largest increases in folding rates from 47- to 53-fold over that of AKBSUB Q199R (Table 1) but only very modest decreases in unfolding rates of 0.8–3.4-fold. Thermostability was increased but only in the case of AKBSUB Q199R/Q16L was that change markedly higher than that of the other mutants. Fourier-transform infrared studies showed no substantial changes in exchange rate (Supporting Material). A γ-methyl of Ile-179 in AKBSUB Q199R/T179I displaces a water and fills in a surface cavity leading to better overall packing of the nonpolar atoms in this region. Bae and Phillips (25) compared the structures of AKs from B. subtilis, G. stearothermophilus, and Bacillus globisporus and noted the potential importance of position 179 in mediating thermostability.
AKBSUB Q199R/Q16L appears to work in much same way as the substitution at position Ile-179. The particular side-chain rotamer found for Leu-16 positions the γ- and δ-methyls into a strongly nonpolar pocket made up exclusively of the nonpolar side-chain atoms contributed by Leu-5, Ile-20, Ile-111, Ile-113, Val-204, Tyr-205, and Ala-206. The Leu-16 rotamer in AKBSUB Q199R/Q16L is different than that of Gln-16 in the AKBSUB Q199R structure. In AKBSUB Q199R, the polar Nɛ and Oɛ atoms of Gln-16 are rotated away from the nonpolar pocket filled by AKBSUB Q199R/Q16L. In AKBSUB Q199R/Q16L, a water is found at the position where the Oɛ1/Nɛ2 atoms of Gln-16 would have been, and confirms that the Oɛ1/Nɛ2 positions of Gln-16 are very polar and favor a water or polar moiety. In one copy of AKBSUB Q199R/Q16L, an indirect effect of the loss of the polar atoms of the Gln-16 side chain is a change in rotamer for Arg-19 in helix-1 that, in turn, allows the formation of a potential salt bridge to Asp-23 and repositions Gln-202 away from the core of the protein. Although the change in Arg-19 position was only found in one copy of the AKBSUB Q199R/Q16L asymmetric unit, both copies show the repositioning of Gln-202. The change in Gln-202 position is not observed for any of the other mutants in either copy A or B, suggesting that this may indeed be a distinct contributing factor for stabilization of AKBSUB Q199R/Q16L. Thus, AKBSUB Q199R/Q16L may work to increase nonpolar packing within the core of the protein, and facilitate a new electrostatic network that enables greater stabilization of the C-terminal helix-9.
AKBSUB Q199R/A193V alters protein-folding dynamics
One of the most successful mutants in the in vivo experimental evolution studies was AKBSUB Q199R/A193V (Fig. 1). AKBSUB Q199R/A193V has the largest increase in folding rate (800-fold in the favorable direction) but unexpectedly has an increase in unfolding rate as well (10-fold in the unfavorable direction) relative to AKBSUB Q199R. This is the only double mutant that has an increase in unfolding rate as compared to the parent AKBSUB Q199R (Table 1). To further corroborate the finding that AKBSUB Q199R/A193V has an increase in folding dynamics, Fourier-transform infrared studies on the global hydrogen isotope exchange rate were also performed (Fig. S2 and Table S3). Only AKBSUB Q199R/A193V showed any substantial increase in exchange rate. AKBSUB Q199R/A193V has a twofold change in exchange rate. None of the other mutants had significantly altered exchange rates (Supporting Material).
Structural analysis of this mutant suggested that this partially solvent-exposed position could have altered local nonpolar packing but when taken together with the folding dynamics studies suggests that stabilization of AKBSUB Q199R/A193V may be better understood as a folding dynamics mutant in which both folding and unfolding rates are increased. In other words, the accelerated sampling of the ground states (i.e., the native and denatured states) leads to a faster rate of hydrogen exchange, although at steady state, the magnitude of the folding rate can clearly confer stability under standard laboratory conditions. The substitution of valine at position 193 repositions Val-195 and Leu-211 very slightly away from the γ-methyl groups of the valine, suggesting that there would be a steric clash in accommodating a valine. Valine-193 may also destabilize the unfolded state because it is a solvent-accessible β-branched amino acid that could destabilize the unfolded ensemble (37–43). Together these effects could increase both folding and unfolding rates, yet provide a net increase in stability (if the effect on folding is larger than the effect on unfolding).
AKBSUB Q199R/G213E establishes a new electrostatic network
AKBSUB Q199R/G213E introduces a potential electrostatic network between Lys-209 ↔ Glu-213 ↔ Lys-216 along the solvent-exposed face of the C-terminal helix-9 as well as Asp-23 from helix-1 (4.8:4.1 Å). Taken together with the formation of the original salt bridge from Arg-199 to Asp-207 in the preceding adaptive mutation (AKBSUB Q199R), there is the potential to form an electrostatic network that runs along the entire length of the C-terminal helix to the carboxy terminus. Electrostatic networks have been found to be more common in thermophilic organisms and Glu-213 is a key node within this new network. The distances for these interactions are consistent with the formation of a stabilizing electrostatic network (41). Substitution of Gly-213 with either glutamine or alanine shows that increasing the stability of the helix or increasing the length of the aliphatic side chain is not responsible for increases in global stability. Both AKBSUB Q199R/G213A and AKBSUB Q199R/G213Q dramatically destabilize the protein (Table 3). As noted earlier, helix-9 has a distinct bend that is likely to be facilitated by Gly-213 and 214 in AKBSUB: introduction of residues that favor a more canonical helix and extend it by one additional turn may lead to poorer packing of helix-9 to helix-1 and β-strands 4 and 8 of the CORE domain leading to decreased stability. A similar set of findings were observed for T4 lysozyme where increasing helical propensity led to decreases in stability (44). Helix-9 in AKBSUB Q199R/G213E still has a distinct bend and therefore the electrostatic network introduced by Glu-213 may assist in further stabilizing the C-terminus by facilitating better interactions with the AK CORE. AKBSUB Q199R/G213E leads to a ninefold increase in folding and no appreciable change in unfolding rates (Table 1).
AKBSUB Q199R/G214R establishes a new hydrogen bond that may tether the C terminus to the AK core
AKBSUB Q199R/G214R is largely solvent-exposed, but electron density for the side chain is readily interpretable and shows clear electron density for the Nɛ of Arg-214 to make a hydrogen bond to the hydroxyl of Tyr-109 (Fig. 1 F). We investigated whether changes to the helical propensity in AKBSUB Q199R could increase stability. Mutation AKBSUB Q199R/G214A marginally decreased stability, but not to the same extent that substitution at Gly-213 did (Table 3). AKBSUB Q199R/G214K also reduced stability, suggesting that the positive charge of the Arg-214 mutation alone is not sufficient, and implies a more specific stereochemistry consistent with a hydrogen bond. Introduction of an arginine into helix-9 could have also provided end-capping for the helix dipole, but because introduction of lysine into this position did not substantially increase stability, this cannot be the most important component to stabilization. Folding kinetics data for AKBSUB Q199R/G214R show increased folding and decreased unfolding rates, of 13- and 5-fold, respectively. Although the introduction of a positively charged residue in the C-terminal portion of an α-helix is typically stabilizing, AKBSUB Q199R/G214K is not as stable as AKBSUB Q199R/G214R. Like AKBSUB Q199R/G213E, AKBSUB Q199R/G214R maintains the bend in the C-terminal helix and extends it by one turn. The introduction of a hydrogen bond between Arg-214 and Tyr-109 may tether the C-terminus to the CORE of AK and further stabilize the protein (7).
Conclusions
Protein evolution must be robust to allow facile adaptation to new conditions. An important aspect of protein evolution is the ability to explore diverse molecular pathways to increased function. The fundamental mechanisms underlying adaptation are based in the physical properties of the molecules that carry out functions that contribute to fitness. Although studies of protein adaptation to thermostability have revealed a remarkable diversity of successful physicochemical mechanisms, the extent to which a population undergoing adaptive evolution employs these diverse mechanisms simultaneously in a single protein in vivo remains largely unexplored.
During molecular evolution, adaptive changes in protein structure-function cannot survey all combinations of mutations, but often proceed in a stepwise fashion in which sequential changes build one upon the other. The series of mutational events that lead to an increase in the fitness of the organism is often termed an adaptive walk (45). Because changes that decrease the fitness of the organism will not be fixed within a large population, the adaptive walk is expected to be strongly path-dependent. In excellent agreement with this, AKBSUB G213E decreases AK stability by >25°C and is, by any measure, a disastrous first adaptive step to thermostability compared to AKBSUB Q199R.
Epistasis or intramolecular pleiotropy between the adaptive mutations in AKBSUB was very strong and in the case of the combination of Q199R with G213E was quite dramatic (Table 3). Even in the most modest case of AKBSUB Q199R/G214R, the combination of the single mutations Q199R and G214R is greater-than-twofold more stable than would have been expected if the mutations had been simply additive. Because the AKBSUB variants isolated in the original population were undergoing strong selection, the best adaptive mutations would have had the highest chance for success; this clearly highlights the importance of such strongly synergistic mutations in molecular evolution.
The adaptive changes to AKBSUB Q199R, though highly varied in stereochemistry, strongly influenced refolding reactions as compared to the unfolding reactions, although both phenomena contribute to increases in protein stability. This observation implies that for competitive survival, fast folding is a successful approach (in contrast to a strategy based on slower unfolding, which would increase the protein lifetime but not formation speed) to increasing fitness. This argument also holds true for AKBSUB Q199R/A193V as the effect is largest for the folding speed. Three of the mutants—AKBSUB Q199R/Q16L, AKBSUB Q199R/G213E, and AKBSUB Q199R/G214R—also make changes that are in, or contact, the C-terminal α-helix (Fig. 4), suggesting that adaptive mutations, though varied, can act upon some common aspects of the AK structure. It is important to note that these mutants were isolated from an in vivo selection that had an absolute requirement for function. These mutants are the best 1% of all the potential single amino-acid substitutions, and therefore, small changes in stability conferred by changes in folding rates are a very successful strategy that can produce exceptional advantages during natural selection.
Figure 4.
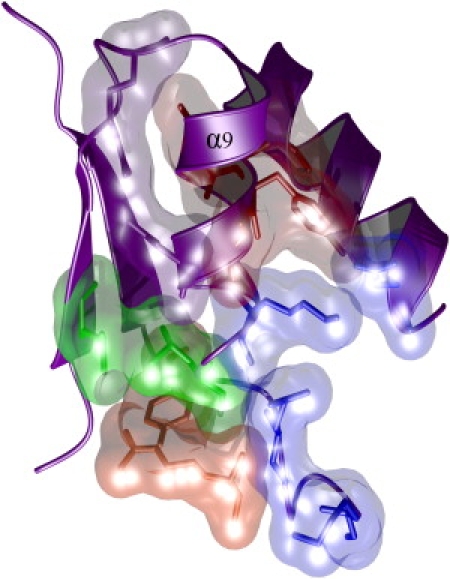
Three of the five adaptive changes to AKBSUB directly contact, or are in, the C-terminal α-helix. Adaptive mutations and the residues in contact with each double mutant are shown as space-filling models AKBSUBQ199R/Q16L (red), AKBSUBQ199R/A193V (green), AKBSUBQ199R/G213E (blue), and AKBSUBQ199R/G214R (orange). Although the stereochemical mechanisms are varied, the net effect is to increase the folding rate (kF) and thereby increase stability. As described previously, the first adaptive step to make AKBSUB Q199R introduced a new electrostatic interaction (purple) that also stabilized the C-terminal helix (20).
We also found that the order of mutational events and the epistasis between them is critical to understanding the particular molecular pathways for success. The first adaptive mutation to produce AKBSUB Q199R provides a unique structural and functional context that enables subsequent mutations to increase the fitness of the organism. The importance of the order of mutational events (i.e., the history of adaptation to earlier events), and the accessible molecular pathways, form the basis for adaptation in all protein evolution. These mechanisms must be clearly understood in order to establish a quantitative understanding of molecular evolution.
Acknowledgments
This work was supported by the National Science Foundation (grant No. 0641792) and The Welch Foundation (grant No. C1584) to Y.S. The Rice University Crystallographic Core Facility is supported by a Kresge Science Initiative endowment grant.
Footnotes
Corey Wilson's present address is Department of Chemical Engineering, Yale University, P.O. Box 208620, New Haven, CT 06520.
Kristopher White's present address is European Molecular Biology Laboratory (EMBL), Grenoble Outstation, 6 rue Jules Horowitz, 38042 Grenoble, France.
Rafael Couñago's present address is Department of Biochemistry, University of Otago, New Zealand.
Gang Wu's present address is Department of Internal Medicine, Health Science Center at Houston, University of Texas, Houston, TX 77030.
Jeffrey Myers' present address is Rigaku Corporation, The Woodlands, Texas 77381.
Accession Numbers
Coordinates and structure factors have been deposited in the Protein DataBank with accession numbers 2osb, 2oo7, 2ori, 2qaj, and 2p3s.
Supporting Material
References
- 1.Dean A.M., Thornton J.W. Mechanistic approaches to the study of evolution: the functional synthesis. Nat. Rev. Genet. 2007;8:675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller S.P., Lunzer M., Dean A.M. Direct demonstration of an adaptive constraint. Science. 2006;314:458–461. doi: 10.1126/science.1133479. [DOI] [PubMed] [Google Scholar]
- 3.Barrick J.E., Yu D.S., Kim J.F. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 4.Bloom J.D., Arnold F.H. In the light of directed evolution: pathways of adaptive protein evolution. Proc. Natl. Acad. Sci. USA. 2009;106(Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patrick W.M., Matsumura I. A study in molecular contingency: glutamine phosphoribosylpyrophosphate amidotransferase is a promiscuous and evolvable phosphoribosylanthranilate isomerase. J. Mol. Biol. 2008;377:323–336. doi: 10.1016/j.jmb.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith J.M. Natural selection and the concept of a protein space. Nature. 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 7.Sterner R., Liebl W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001;36:39–106. doi: 10.1080/20014091074174. [DOI] [PubMed] [Google Scholar]
- 8.Tokuriki N., Tawfik D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 9.DePristo M.A., Weinreich D.M., Hartl D.L. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 10.Tomatis P.E., Fabiane S.M., Vila A.J. Adaptive protein evolution grants organismal fitness by improving catalysis and flexibility. Proc. Natl. Acad. Sci. USA. 2008;105:20605–20610. doi: 10.1073/pnas.0807989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutherford S.L. Between genotype and phenotype: protein chaperones and evolvability. Nat. Rev. Genet. 2003;4:263–274. doi: 10.1038/nrg1041. [DOI] [PubMed] [Google Scholar]
- 12.Bershtein S., Goldin K., Tawfik D.S. Intense neutral drifts yield robust and evolvable consensus proteins. J. Mol. Biol. 2008;379:1029–1044. doi: 10.1016/j.jmb.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 13.Bloom J.D., Lu Z., Arnold F.H. Evolution favors protein mutational robustness in sufficiently large populations. BMC Biol. 2007;5:29. doi: 10.1186/1741-7007-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bloom J.D., Labthavikul S.T., Arnold F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tracewell C.A., Arnold F.H. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol. 2009;13:3–9. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tokuriki N., Tawfik D.S. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459:668–673. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- 17.Fasan R., Meharenna Y.T., Arnold F.H. Evolutionary history of a specialized p450 propane monooxygenase. J. Mol. Biol. 2008;383:1069–1080. doi: 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couñago R., Chen S., Shamoo Y. In vivo molecular evolution reveals biophysical origins of organismal fitness. Mol. Cell. 2006;22:441–449. doi: 10.1016/j.molcel.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 19.Couñago R., Shamoo Y. Gene replacement of adenylate kinase in the Gram-positive thermophile Geobacillus stearothermophilus disrupts adenine nucleotide homeostasis and reduces cell viability. Extremophiles. 2005;9:135–144. doi: 10.1007/s00792-004-0428-x. [DOI] [PubMed] [Google Scholar]
- 20.Couñago R., Wilson C.J., Shamoo Y. An adaptive mutation in adenylate kinase that increases organismal fitness is linked to stability-activity trade-offs. Protein Eng. Des. Sel. 2008;21:19–27. doi: 10.1093/protein/gzm072. [DOI] [PubMed] [Google Scholar]
- 21.Petsko G.A. Structural basis of thermostability in hyperthermophilic proteins, or “there's more than one way to skin a cat”. Methods Enzymol. 2001;334:469–478. doi: 10.1016/s0076-6879(01)34486-5. [DOI] [PubMed] [Google Scholar]
- 22.Zavodszky P., Kardos J., Petsko G.A. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc. Natl. Acad. Sci. USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuler B., Kremer W., Jaenicke R. Role of entropy in protein thermostability: folding kinetics of a hyperthermophilic cold shock protein at high temperatures using 19F NMR. Biochemistry. 2002;41:11670–11680. doi: 10.1021/bi026293l. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S., Tsai C.J., Nussinov R. Factors enhancing protein thermostability. Protein Eng. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- 25.Bae E., Phillips G.N., Jr. Structures and analysis of highly homologous psychrophilic, mesophilic, and thermophilic adenylate kinases. J. Biol. Chem. 2004;279:28202–28208. doi: 10.1074/jbc.M401865200. [DOI] [PubMed] [Google Scholar]
- 26.Wintrode P.L., Zhang D., Goddard W.A., 3rd Protein dynamics in a family of laboratory evolved thermophilic enzymes. J. Mol. Biol. 2003;327:745–757. doi: 10.1016/s0022-2836(03)00147-5. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z., Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1998;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Pflugrath J.W. The finer things in x-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 29.Brünger A.T., Adams P.D., Warren G.L. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 31.Fersht A. W.H. Freeman; New York: 1999. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. [Google Scholar]
- 32.Shapiro Y.E., Sinev M.A., Meirovitch E. Backbone dynamics of Escherichia coli adenylate kinase at the extreme stages of the catalytic cycle studied by 15N NMR relaxation. Biochemistry. 2000;39:6634–6644. doi: 10.1021/bi992076h. [DOI] [PubMed] [Google Scholar]
- 33.Abele U., Schulz G.E. High-resolution structures of adenylate kinase from yeast ligated with inhibitor Ap5A, showing the pathway of phosphoryl transfer. Protein Sci. 1995;4:1262–1271. doi: 10.1002/pro.5560040702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller C.W., Schulz G.E. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 Å resolution. A model for a catalytic transition state. J. Mol. Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- 35.Matouschek A., Fersht A.R. Application of physical organic chemistry to engineered mutants of proteins: Hammond postulate behavior in the transition state of protein folding. Proc. Natl. Acad. Sci. USA. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pena M.I., Davlieva M., Bennett M.R., Olson J.S., Shamoo Y. Evolutionary fates within a microbial population highlight an essential role for protein folding during natural selection. Mol. Syst. Biol. 2010;6:387. doi: 10.1038/msb.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrank T.P., Bolen D.W., Hilser V.J. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc. Natl. Acad. Sci. USA. 2009;106:16984–16989. doi: 10.1073/pnas.0906510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamagata Y., Ogasahara K., Yutani K. Entropic stabilization of the tryptophan synthase α-subunit from a hyperthermophile, Pyrococcus furiosus. X-ray analysis and calorimetry. J. Biol. Chem. 2001;276:11062–11071. doi: 10.1074/jbc.M009987200. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S., Nussinov R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999;293:1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S., Nussinov R. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar S., Nussinov R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 2002;83:1595–1612. doi: 10.1016/S0006-3495(02)73929-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthews B.W., Nicholson H., Becktel W.J. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl. Acad. Sci. USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae E., Bannen R.M., Phillips G.N., Jr. Bioinformatic method for protein thermal stabilization by structural entropy optimization. Proc. Natl. Acad. Sci. USA. 2008;105:9594–9597. doi: 10.1073/pnas.0800938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alber T., Bell J.A., Matthews B.W. Replacements of Pro86 in phage T4 lysozyme extend an α-helix but do not alter protein stability. Science. 1988;239:631–635. doi: 10.1126/science.3277275. [DOI] [PubMed] [Google Scholar]
- 45.Orr H.A. The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.