

Abstract
Affinity purification followed by mass spectrometry (AP-MS) has become a common approach for identifying protein–protein interactions (PPIs) and complexes. However, data analysis and visualization often rely on generic approaches that do not take advantage of the quantitative nature of AP-MS. We present a novel computational method, nested clustering, for biclustering of label-free quantitative AP-MS data. Our approach forms bait clusters based on the similarity of quantitative interaction profiles and identifies submatrices of prey proteins showing consistent quantitative association within bait clusters. In doing so, nested clustering effectively addresses the problem of overrepresentation of interactions involving baits proteins as compared with proteins only identified as preys. The method does not require specification of the number of bait clusters, which is an advantage against existing model-based clustering methods. We illustrate the performance of the algorithm using two published intermediate scale human PPI data sets, which are representative of the AP-MS data generated from mammalian cells. We also discuss general challenges of analyzing and interpreting clustering results in the context of AP-MS data.
Keywords: clustering, mass spectrometry, protein complexes, protein–protein interaction, spectral counts
Introduction
Deciphering how proteins assemble with each other into complexes is key to understanding their biological activity. In recent years, affinity purification coupled to mass spectrometry (AP-MS) has become the technique of choice to recover and identify protein complexes. This has been made possible in part by advances in tandem mass spectrometry (MS/MS) and the development of efficient affinity purification strategies (Gingras et al, 2007). Most often, target proteins (baits) are purified along with their interaction partners (preys) through an epitope tag-based affinity chromatography step. After purification, the protein mixture is digested into peptides that are identified using MS/MS (Aebersold and Mann, 2003). By performing additional AP-MS experiments for interconnected baits, further information can be gained related to the assembly of proteins into interaction networks and delineation of protein complexes involving shared components.
A number of computational approaches were developed for reconstructing protein complexes from ‘binary' representations of AP-MS data (i.e. observation/non-observation of a given interaction; such modeling correctly describes other types of interaction data, such as yeast two-hybrid; Braun et al, 2009). This includes methods based on the socio-affinity index (Gavin et al, 2006; Collins et al, 2007; Pu et al, 2007), as well as a direct application of graph theory based and other advanced approaches (Hart et al, 2007; Zhang et al, 2008; Friedel and Zimmer, 2009). These methods, however, were designed for genome-scale projects, where ideally each prey is also analyzed as a bait (allowing theoretically for scoring the presence–absence of interaction for each protein pair) giving a more complete coverage of the target interactome. At the same time, most current AP-MS data sets are of a relatively small scale where only a subset of proteins are selected for bait purification, especially in human cells (see Sardiu et al (2008) for further discussion). In such cases, potential interactions between non-bait proteins cannot be screened directly, resulting in partial coverage of the interaction network (Scholtens et al, 2005). This renders the methods listed above of limited utility.
Furthermore, most of these methods were also developed for very specific data sets generated in Saccharomyces cerevisiae through tandem affinity purification, which effectively enriches for stable protein complexes that are relatively easily identifiable computationally (Gavin et al, 2006; Krogan et al, 2006). By contrast, more sensitive detection methods, such as those based on single-step affinity purification, typically recover a much higher number of interactors for each bait protein (Chen and Gingras, 2007). In such data sets, clustering of data by computational methods that treat all interactions with equal weight regardless of the quantitative evidence, and where grouping of proteins into protein complexes relies heavily on the topological properties of the network, is no longer effective.
In AP-MS studies, there is additional quantitative information regarding the abundance of proteins in the affinity-purified sample (such as MS/MS spectral counts) that can be extracted from the data (Liu et al, 2004; Powell et al, 2004; Old et al, 2005; Lu et al, 2007; Choi et al, 2008). Recently, hierarchical clustering of a bait–prey matrix data set of normalized spectral abundance factor (NSAF) values (Zybailov et al, 2006) was applied to a human protein–protein interaction (PPI) network. This revealed that clustering quantitative data with continuous distance metrics identifies modules (subnetworks involved in multiple protein complexes) better than clustering binary data (Sardiu et al, 2009). However, this study only investigated commonly used clustering methods that assign proteins to separate or overlapping partitions in single-dimensional space. Such an approach is not ideal because of the aforementioned problem of incomplete interaction data for proteins that appear in the network only as preys.
Here, we present a novel clustering approach for reconstruction of protein complexes that takes advantage of quantitative information and is applicable to incomplete protein interaction data sets that are typically generated in small and intermediate scale AP-MS studies (see Box 1). Our method, nested clustering, essentially performs a two-step sequential clustering (biclustering): creation of bait clusters based on common patterns of label-free quantitative data (spectral counts) across all prey proteins, and identification of nested clusters of preys sharing similar abundance level in each bait cluster. The method effectively addresses the problem of incomplete data, and does not require specification of the number of bait clusters. The performance of the algorithm is illustrated using two published intermediate scale human PPI data sets, which are representative of the AP-MS data generated from mammalian cells.
Summary of the analytical pipeline.

Samples are analyzed by affinity purification and LC-MS/MS. Resulting data are analyzed through a computational pipeline for peptide identification by database searching, and proteins are assembled from peptides. Proteins are quantified by counting the number of MS/MS spectra matched to its constituent peptides (spectral count data). Quantitative data are arranged into the bait–prey matrix, and clustering algorithm is applied to rearrange the data matrix leading to identification of submatrices. This leads to the identification of candidate protein complexes as well as their average abundance level and the associated variance. Green and brown nodes are baits and preys, respectively.
Results and discussion
Overview of the computational method
The input of our clustering approach is AP-MS interaction data that have passed through a processing step to remove contaminants due to non-specific binding, for example by referencing against control purifications or using statistical methods (Sowa et al, 2009; Breitkreutz et al, 2010). The data are represented in a bait–prey matrix, with each column corresponding to purification of a bait protein and each row corresponding to a prey protein: values are spectral counts for each protein. Provided that data are properly normalized to account for the factors affecting spectral count-based quantification (e.g. protein length and the total spectral counts in each AP-MS experiment), relative quantitative measures can be directly compared across purifications, allowing joint statistical modeling of the entire data set (see Materials and methods).
The nested clustering approach is illustrated in Figure 1. The algorithm identifies biclusters by stochastically drawing samples of bait and prey cluster configurations from the appropriate posterior distribution, as well as mean and variance parameters associated with them using the Markov chain Monte Carlo (MCMC) algorithm (see Supplementary information for detail). The biclustering configuration yielding the highest posterior probability is selected as the optimal solution. Figure 1A illustrates a single iteration for drawing bait and prey clusters.
Figure 1.

Overview of the computational method. (A) Nested clustering algorithm. Baits are probabilistically assigned to bait clusters with associated mean and variance. The diameter of circles is proportional to the normalized spectral counts. In bait clusters, mean abundance of each prey is drawn as a square. Mixture modeling is used to group these elements into a small number of abundance levels, completing nested clustering of prey proteins. (B) Resulting biclusters from the algorithm in (A). Each bicluster corresponds to a submatrix consisting of a bait cluster and an associated nested prey cluster. (C) Example of maximum a posteriori estimation. Bait clustering is illustrated in a hypothetical data with two preys. Each dot is a single purification with a different bait. Four unique sets of clustering configurations were generated in 100 samples. The number N is the number of samples sharing the given bait clusters, and maxP is the maximum posterior probability under the fixed bait cluster configuration. The Model 2 is the most frequently sampled configuration with the highest maximum posterior probability, and Model 3 is the second best competing model with similarly high posterior probability. The other two configurations have low posterior probability and low frequency of sampling.
Step I: clustering of baits for anchors of biclusters
The input data are organized into a matrix format with n columns and p rows for baits and preys, respectively. Baits are probabilistically assigned to clusters. Here, each cluster has associated abundance vectors of mean and variance (length p), as shown in vertical arrays of rectangles in the figure. The probability that a bait protein is a member of a bait cluster is proportional to the Gaussian likelihood of the observed spectral counts with the cluster mean and variance times the prior. Without specifying the number of clusters in the data, the algorithm starts with no known bait clusters. First, bait cluster 1 is created to represent bait 1. Then bait cluster 2 is created to explain bait 2 because the abundance vector of cluster 1 is unlikely the data generating distribution for bait 2 (in this hypothetical example shown in Figure 1). Bait 3 is assigned to bait cluster 1 because the bait is similar to bait 1 and thus there is no need to create another bait cluster (number of bait cluster remains at 2). Following this process, baits are sequentially assigned to either existing clusters or a new cluster, and the number of clusters created along the way after assigning all n baits becomes the final number of bait clusters.
Step II: clustering of preys within anchors for final assembly of biclusters
Once baits are organized into bait clusters, preys in each cluster vector are assigned the most likely mean and variance values (Figure 1A, lower panel). In this process, the same mean and variance values are shared across many preys and different bait clusters. Within each bait cluster, the preys sharing the same mean abundance are pooled into nested prey clusters. Combining these preys with the bait cluster results in the assembly of a bicluster (Figure 1B). This completes single iteration of the sampling algorithm.
Sampling steps are iterated many times to ensure sufficient exploration of the search space. Figure 1C shows how bait cluster configuration can change along the iterations using an example of a hypothetical data set. For visualization purposes, the illustration involves only two preys, A and B. The two axes are spectral counts for the two proteins, where each dot represents abundance coordinate of a bait protein. Candidate models are grouped if they share identical bait cluster configurations. In this example, four unique sets (Models 1 through 4) of clustering configurations were created from 100 samples. Among these, a large proportion of models shared bait clusters shown in Models 2 and 3 (50 and 35%, respectively), with high posterior probabilities given the observed data, suggesting that these bait clusters most likely represent the underlying protein complexes. As the assignment process is probabilistic, the total number of clusters can change every time this process is repeated, and the pool of all sampled models automatically generates a posterior distribution of bait clusters as well as the number of bait clusters. This can serve as a reference distribution for the sample with the highest posterior probability (reported clustering result). For final determination of the clustering outcome, the algorithm reports the configuration yielding the highest posterior probability.
Our implementation is based on mixture modeling, which is common in the statistical literature of clustering for high dimensional data (Yeung et al, 2001; Fraley and Raftery, 2002). A critical challenge in the model-based clustering problem is the choice of the number of clusters. Although a number of approaches were proposed to find the optimal number of clusters (Tibshirani et al, 2001; Dudoit and Fridlyand, 2002), our approach is based on Dirichlet process mixture (DPM) models (see Materials and methods), an off-the-shelf framework for Bayesian model-based clustering. The inferential procedure in DPM models surveys a large number of potential candidate models with varying numbers of clusters, and automatically determines the optimal number of bait clusters and prey clusters from the best data fitting model, avoiding the computational burden to fit multiple models with different number of clusters and compare them.
Results from nested clustering can be summarized in several ways. First, the most straightforward outcome is a set of biclusters, submatrices in the bait–prey matrix data, each associated with estimated mean abundance value and variance. A submatrix consists of a bait cluster serving as an anchor and nested prey clusters shared consistently by the baits in the bait cluster (and in a consistent level of abundance). Submatrices can be best illustrated in heatmaps of raw bait–prey matrix, where rows and columns are reorganized using estimated abundance levels (as illustrated in Figure 1B), although such arrangement does not guarantee that nested prey clusters are correctly grouped across all bait clusters in a single plot. Clustering results can also be visualized using tools such as Cytoscape (Shannon et al, 2003): quantitative data can be represented by visual encoding of edge attributes (edge thickness).
Application to TIP49a/b data set
The method was first applied to a human PPI network centered around two AAA+ ATPases Tip49a and Tip49b involved in chromatin remodeling (Sardiu et al, 2008). The network reconstructed in the original study consisted of four major protein complexes SRCAP, hINO80, TRRAP/Tip60, and URI/Prefoldin profiled using 27 bait proteins, with a total of 55 proteins (baits and preys). These protein complexes were established in earlier studies and validated using in vivo coimmunoprecipitation assays (Sardiu et al, 2008, 2009). The authors of the original study applied a variety of existing clustering algorithms to this data set, and provided a detailed performance assessment of each algorithm including parameters affecting the outcome, as well as a useful guideline for using them in practice (Sardiu et al, 2009). Therefore, this data set represents a good choice for evaluating the performance of our approach.
Sample models with associated bait and nested prey clusters were generated from the MCMC algorithm, and the sample leading to the highest posterior probability (maximum a posteriori estimate) was chosen as the final result. Bait clusters recovered major groups of baits belonging to hINO80, URI/Prefoldin, and SRCAP complexes according to the known membership (Sardiu et al, 2008). To further evaluate the clustering solution, we compared the bait cluster configuration with the interbait probability distance matrix (see Materials and methods for computation), and discovered that the separation in probability distance was concordant with the selected bait clusters (Supplementary Figure 1). We also monitored the distribution of the number of bait clusters throughout the sampling, which fluctuated between 9 to 11 clusters with the majority focused on 10 bait clusters. A number of samples showed DPCD and ZNF.HIT2 merged with Prefoldin complex in the case of 9 bait clusters, whereas MRGBP formed an independent bait cluster in the case of 11 bait clusters.
After identifying bait clusters, preys were assigned to mixture component distributions of abundance within each bait cluster, resulting in nested clustering of prey proteins, as represented by colored rectangles in the heatmap (estimated mean abundance in Figure 2B; raw spectral count data in Figure 2A). Notice that, due to the automatic clustering property of DPM model, the estimated mean and variance of all boxes (nested prey clusters) were ‘regularized' toward a small pool of common values, yielding a small number of submatrices. To assemble these boxes into protein complexes, all boxes sharing the same mean abundance value in each bait cluster were combined together.
Figure 2.

Application of nested clustering to TIP49a/b data set. (A) Heatmap of the raw spectral count data organized using estimated mean values. (B) Heatmap of the estimated mean spectral counts. (C) Network visualization of SRCAP, TRRAP, hINO80, and Prefoldin complexes in Sardiu et al (2008). Green and brown nodes are baits and preys, respectively. Baits are shown as circles of larger size to indicate that they are the anchors of protein complexes constructed by nested clustering. Red circles indicate large-protein complexes identified in the form of submatrices.
As indicated on the right-hand side of Figure 2B, nested prey clusters in each bait cluster were nicely separated into the known complex membership reported in Sardiu et al (2009). It also shows a substantial amount of cross-talks involving the members of TRRAP/TIP60 and SRCAP complexes. Four bait proteins, TIP49B, YL1, MRGBP, and H2AZ (marked * in Figure 2B), interact with both members of SRCAP and TRRAP/TIP60 complexes. Although TIP49A does not form a bait cluster with TIP49B due to the heterogeneous quantitative profiles, both proteins show interactions with nearly all the baits in consistently high abundance.
The submatrices in Figure 2B also show groupings of prey proteins according to their association with bait clusters. Several preys were found to be associated with multiple protein complexes. For example, BAF53 shows interactions with two members of SRCAP complex SRCAP and ZINF.HIT1 in medium ‘abundance' (i.e. in AP-MS experiments using baits from this complex, this protein was generally identified with spectral counts in the intermediate abundance range), and with the members of hINO80 complex in low abundance. IES6 provides another such example. Figure 2C shows the same result in Cytoscape. In the graph, baits that serve as anchors for the corresponding protein complexes) are indicated by larger nodes than for the proteins only identified as prey. Note that while the visualization in Figure 2C is a more comprehensive reflection of the multiplicity of protein complexes than the heatmaps in Figure 2A and B, the latter representation allows an easier interpretation of each protein complex, which implies that these different visual formats are complementary.
Analysis of combined data sets in human phosphatase 2A network
To further illustrate several specific challenges of AP-MS data, the method was applied to a data set focusing on the human Protein Phosphatase 2A (PP2A) network. The data set was created by merging two independent data sources, each covering different parts of the target network with a small overlap. The combined data set consisted of 25 purifications of 22 unique baits resulting in a network of 68 proteins including the baits. The first study focused on the characterization of a novel striatin-interacting phosphatase and kinase (STRIPAK) complex with dense interconnections through a catalytic subunit PPP2CA and a scaffolding subunit PPP2R1A (Goudreault et al, 2009). Note that the term ‘complex' here is used in the sense of computationally assembled entity of densely interconnected proteins. The second study covered a comprehensive collection of catalytic, scaffolding, and regulatory subunits in the PP2A network (Glatter et al, 2009). The two studies share three common baits MOBKL3, PPP2CA, and PPP2R1A.
This data set was selected to see whether related data from different studies can be jointly analyzed. As the two data sets share three baits, the coherence of clustering between repeated measurements of the same bait in different studies can be tested. In addition, this data set is interesting in that it poses a unique challenge for clustering because the bait coverage of protein complexes varies widely. After combining the data, STRIPAK complex consisted of almost half the baits leading to an oversampling relative to the remaining PP2A subunits, whereas the catalytic/scaffolding subunits and the regulatory subunits purified in the second study did not show dense interactions among themselves.
Nested clustering of the PP2A data reported seven bait clusters of varying size. However, unlike the TIP49a/b data set discussed above, several clustering solutions with larger number of clusters were found having similarly high posterior probabilities (Supplementary Figure 2 shows the sampling distribution of the number of bait clusters considered). We first note that in all three instances, baits that were used in the two independent studies (PPP2CA, PPP2R1A, and MOBKL3) were assigned to the same bait cluster. This suggests that integrating data from different sources is feasible after proper normalization of spectral counts. As for the bait clustering, the STRIPAK complex formed a standalone cluster (Figure 3A and B), which was also discovered in the interbait probability distance (see Supplementary Figure 2). TRAF3IP3 was an exception that formed an independent cluster, which happened mainly because it was detected as prey in no other purifications but its own (with high counts) as, unlike other baits, it is not expressed endogenously in HEK293 (Goudreault et al, 2009). In addition to the recapitulation of the STRIPAK complex, Figure 3A and B show that PPP2CA and PPP2R1A (PP2A catalytic and scaffolding subunits, respectively) are pivotal links of the PP2A system as they are identified with consistently high abundance in AP-MS runs with all baits, as expected from the literature (Virshup and Shenolikar, 2009).
Figure 3.

Application of nested clustering to PP2A data set. (A) Heatmap of the raw spectral data organized using estimated mean values. (B) Heatmap of the estimated mean spectral counts. (C) Network visualization of the PP2A system along with the STRIPAK and CCT complexes. Green and brown nodes are baits and preys, respectively. Baits are shown as circles of larger size to indicate that they are the anchors of protein complexes constructed by nested clustering. Red circles indicate large-protein complexes identified in the form of submatrices.
At the same time, some of the catalytic, scaffolding, and regulatory subunits formed clusters, despite the lack of interconnections among themselves. These clustering results were found to be in part due to interactions with chaperone-containing TCP1 subunits (CCTs). The eight subunits of CCT complex included in this data are known to interact with each PP2A-family phosphatase catalytic subunit such as PP2A and PP4 (Gingras et al, 2005), as well as with multiple proteins that possess a WD-40 domain, for example each member of the PPP2R2A-2D group, and the striatin molecules (Ho et al, 2002; Valpuesta et al, 2002). However, in these data, the CCT complex components were identified inconsistently and with low spectral counts, possibly due to the common problem of under sampling of low abundance proteins in MS/MS. This inconsistency was sufficiently influential to produce artificial clusters. Still, the use of quantitative spectral count-based abundance data coupled with nested clustering, and with an appropriate choice of the hyperprior (see Supplementary information), was helpful in negating the effect of undersampling of CCT components because the clustering outcome was driven largely by interactions with proteins identified reproducibly and with high abundance. For instance, inconsistent interactions between STRN and STRN3 with CCT complex were suppressed from the output shown in Figure 3B (dashed box in the rows for CCT complex). At the same time, this adjustment also affected the clustering result of other potentially real components of protein complexes (long-dashed boxes in Figure 3B) such as DYNLL1. Overall, our analysis highlighted the importance of improving the robustness of detecting protein interactions (e.g. by means of performing AP-MS with multiple biological replicates of the same bait) to be able to use interaction data involving weak or transient interactions for informative clustering and network analysis.
Comparison to other clustering methods
As observed earlier, nested clustering recovered the known protein complex structure without dependence on the choice of distance metric and data transformation. In Sardiu et al (2009), the authors evaluated several commonly used clustering algorithms on their data, including hierarchical clustering. They pointed out that many of the clustering methods they evaluated failed to recover the full network correctly, and suggested that the availability of various distance metrics and data types (binary/quantitative) have to be explored. The results of our own analysis using hierarchical clustering is in agreement with these observations (see Supplementary Figure 3 and Supplementary information).
In general, these observations reflect the common limitation of methods such as hierarchical clustering, k-means, and fuzzy clustering in that they essentially partition proteins in one dimensional space, that is, separately the rows of the bait–prey matrix. As a biclustering algorithm, nested clustering uses clustering on a single axis (bait side) as an intermediate step only, and as the final output derives submatrices (nested prey clusters) anchored by the bait clusters. In this regard, we have also compared the output of nested clustering with several existing biclustering methods, BiMax (Prelic et al, 2006), Cheng and Church (CC) (Cheng and Church, 2000), and PLAID (Lazzeroni and Owen, 2002). In the two data sets analyzed earlier, BiMax and CC algorithms failed to recover known proteins complexes, whereas the PLAID model was the only method that recovered hINO80 complex and the subcluster shared by SRCAP and TRRAP/TIP60 complexes in the TIP49a/b data set, and also the core hubs PPP2R1A and PPP2CA of the PP2A data set. However, even the PLAID model failed to distinguish a few obvious complexes such as Prefoldin and TRRAP complexes in the TIP49a/b data set and the STRIPAK complex in the PP2A data set (see Supplementary Figures 4 and 5 and Supplementary information).
Practical utility of nested clustering and future extensions
In this study, we addressed the computational challenges of representative AP-MS data sets that are currently being generated, that is data sets containing incomplete interaction data due to purification of a relatively small number of proteins (typically <25–50 baits). This distinguishes our work from most previous computational efforts. As the analysis of protein complexes and interaction networks in the case of mammalian organisms is unlikely to be routinely performed on the global scale as in yeast, methods that can specifically deal with smaller, incomplete interaction data sets are very valuable to the proteomics community. We also note that it is also likely that the use of quantitative information (such as spectral counts used in this work) coupled with methods such as nested clustering will be just as valuable in the analysis of large-scale data sets. However, this question can only be addressed when such data sets become publicly available, along with appropriate benchmarks for evaluating the performance of different computational methods applied to these data.
Our approach has several important technical advantages. Nested clustering groups the prey proteins with similar spectral counts within each bait cluster, and provides an economical expression of abundance levels by identifying a small number of discrete categories (e.g. negligible, low, medium, or high abundance). The outcome is easily interpretable in terms of the participation of each prey in one or multiple protein complexes. Second, the output is reported in the form of a bicluster, which allows individual proteins to belong to multiple complexes either as baits or as preys. Third, the method automatically chooses an optimal number of bait clusters and nested prey clusters by an extensive survey of different clustering models in terms of their likelihood—an important feature often omitted or addressed without proper statistical summaries in generic clustering algorithms. We also note the flexibility of the modeling framework with respect to future extensions. For example, there are alternative label-free quantification strategies (Nesvizhskii et al, 2007), such as those based on total ion current measurement (Wepf et al, 2009). The statistical model can incorporate proper distributional properties of each data such as mean–variance relationship simply by selecting an appropriate likelihood for each data type.
Finally, we remark that the clustering analysis was applied to filtered AP-MS data after removal of non-specific background proteins (common contaminants). The contaminant removal step was performed as a part of the original studies by subtracting all proteins identified in the negative control experiments from the final list of interactors. Furthermore, the PP2A data set (the subset reported in Goudreault et al (2009)) was additionally filtered using a certain minimum spectral count threshold, essentially eliminating all false-positive interactions but at the cost of likely removing some true interactions in the low abundance range. However, the two steps—removal of non-specific background proteins and clustering analysis—can be performed within the same joint statistical framework using spectral count information. We have recently developed a computational model SAINT for model-based assessment of significance of observed individual PPIs using peptide or spectral counts (Breitkreutz et al, 2010). Thus, future work should focus on a combined strategy that marries modeling of spectral count profiles across the entire data set (including control purifications) for computing the confidence in individual interactions simultaneously with the clustering analysis for reconstruction of protein complexes. Such a modeling framework can also be further extended to include additional information, for example to incorporate the knowledge from existing protein interaction databases and/or higher-level information such as gene ontology.
In conclusion, we developed a novel computational method for nested clustering of AP-MS data to identify protein complexes. The method has several key advantages. It uses quantitative information that can be extracted from AP-MS data such as MS/MS spectral counts. It also addresses the problem of incomplete protein interaction data common in many AP-MS data sets. Compared with existing hierarchical and even biclustering methods, nested clustering was able to organize complexes more accurately in both data sets analyzed in this work. As AP-MS approach is being increasingly used for generation of protein interaction networks, novel statistical methods specifically designed for AP-MS data, such as nested clustering presented here, will have an increasingly important function.
Materials and methods
Protein identification and spectral counting
TIP49a/b data set was obtained from Sardiu et al (2009) as a matrix consisting of 55 proteins (rows) and 27 purifications (columns), with elements of the matrix represented by MS/MS spectral counts. The AP-MS data for KIAA bait protein were not used for clustering because only one prey protein was identified as interacting with that bait.
PP2A data set was derived from two publicly available studies (Glatter et al, 2009; Goudreault et al, 2009). To simplify the integration of the data, raw MS data were re-analyzed as a part of this work (see Supplementary information) using X! Tandem, PeptideProphet, and ProteinProphet (Nesvizhskii et al, 2007). ProteinProphet files were parsed, exported into a local MySQL database for further analysis and extraction of spectral count information. Peptides whose sequence is present in multiple proteins cannot be unambiguously assigned to a particular protein or protein group in the protein summary file (Nesvizhskii and Aebersold, 2005). The spectral counts for peptides shared among multiple proteins were weighted when computing the spectral count for each protein. For a peptide identified from n MS/MS spectra and shared between two distinguishable proteins, A and B, its contribution to the adjusted spectral count of protein A was taken as n × NAd/(NAd+NBd). Here, NAd and NBd are the spectral counts of proteins A and B, respectively, determined based on distinct (non-shared) peptides. After this spectral count adjustment procedure, all IPI protein accession numbers were converted to gene names. In the case of multiple proteins mapping to the same gene name, the highest overall spectral counts for the gene were used in the subsequent clustering analysis. Finally, data were exported into Excel files, and manually curated to keep only those proteins that were reported as valid protein interaction partners in the original studies. The final spectral count matrix used for clustering consisted of 22 bait proteins and 25 purifications (including 3 baits that were in common between the two studies).
Data conversion from spectral counts to scaled NSAF values
The conversion of spectral counts to the NSAFs was performed using the definition provided in Zybailov et al (2006). Let Sij denote the spectral count for the interaction between bait j and prey i. Also define Li to be the sequence length of prey i. Then the corresponding NSAF value is defined by
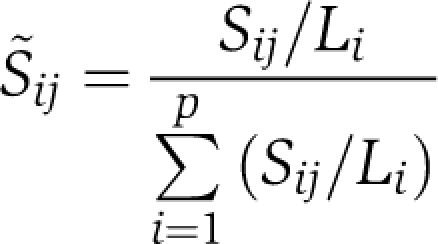 |
where p is the number of preys. To facilitate the modeling in a suitable scale, a natural log transform was applied followed by multiplication by a factor of 100, that is 100 × log(Sij+1) to come up with the final normalized spectral count data.
DPM model
DPM refers to a probability mixture model without a pre-specified number of mixture components, of which the proportions are constructed according to a stochastic process called Dirichlet process. DPM has a clustering property that the stochastic process for mixture proportions almost surely produces a finite number of distributions, resulting in an economical yet flexible expression for the data generating distribution, and therefore an automatic clustering outcome. This property actually renders DPM inappropriate to model the observed data directly, but makes it an attractive choice for specifying a prior distribution for Bayesian statistical inference (e.g. for mean spectral counts, not observed counts). Using DPM as a prior distribution has an inferential advantage that its posterior distribution also follows the same form of DPM with adjusted mixture proportions.
Formally, consider a mixture model of the form 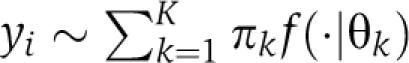 . Each component f(·∣θk) denotes a parametric distribution, for example Gaussian, and K is left unspecified. If it is assumed that the distribution parameters {θk}k=1K are drawn from a common base distribution G0, then the final model can be written as follows. For i=1, 2, ⋯,
. Each component f(·∣θk) denotes a parametric distribution, for example Gaussian, and K is left unspecified. If it is assumed that the distribution parameters {θk}k=1K are drawn from a common base distribution G0, then the final model can be written as follows. For i=1, 2, ⋯,
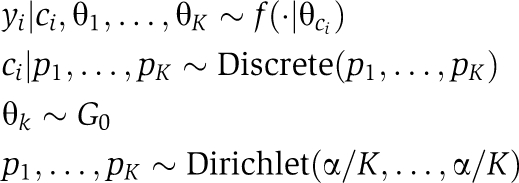 |
where ci is the cluster membership for yi into one of the K groups. A shorthand expression for this model is yi∣θi∼f (θi), θi∼G, and G∼DP(G0, α), where α is a concentration parameter. For a more detailed description of DPMs, see Antoniak (1974). From this basic framework, DPM models have been extended to a more sophisticated form that allows dependence between components of {θk}k=1K, as in the hierarchical Dirichlet process (Teh et al, 2006) and the nested Dirichlet process (Rodriguez et al, 2008), of which the former was used in this work. In particular, a hierarchical DPM is used to model the distribution of abundance of individual preys with the same set of a finite number of abundance levels shared across all proteins but with distinct probabilities of taking on those values.
Nested mixture model
Let X={xji} denote the bait–prey matrix data with n baits and p preys, indexed by j and i, respectively. Nested clustering of the bait–prey matrix data can be built upon the model proposed in Kim et al (2006), which carries out one-way partition clustering for gene expression data using the original DPM prior above. Following the approach, let xj denote a p-dimensional vector of spectral counts in p preys for bait j, then the bait clustering DPM can be written as
where f is a multivariate Gaussian distribution with mean and variance parameters 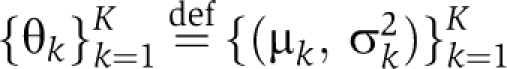 and G0 denotes the base distributions for p-dimensional vectors {μk}k=1K and {σk2}k=1K. It is further assumed that the base distribution can be decomposed as
and G0 denotes the base distributions for p-dimensional vectors {μk}k=1K and {σk2}k=1K. It is further assumed that the base distribution can be decomposed as 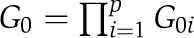 and the individual protein-level DPM G0i follows a hierarchical Dirichlet process such that G0i∼DP(H, γ) for i=1,⋯, p and H∼DP(H0, ρ), where γ and ρ are individual prey level and entire data level concentration parameters, respectively. See Supplementary information for the prior elicitation for each data set and the inference procedure based on MCMC sampling algorithm.
and the individual protein-level DPM G0i follows a hierarchical Dirichlet process such that G0i∼DP(H, γ) for i=1,⋯, p and H∼DP(H0, ρ), where γ and ρ are individual prey level and entire data level concentration parameters, respectively. See Supplementary information for the prior elicitation for each data set and the inference procedure based on MCMC sampling algorithm.
Determination of clusters
The outcome is an ordinary mixture model of the following form. A mixture model is first obtained for bait clustering
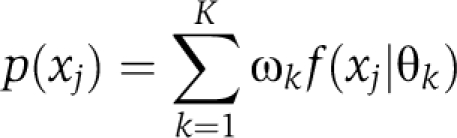 |
with a fixed number K. Here, bait j is assigned to the cluster that gives the maximum posterior probability, that is
Given the bait clusters {ĉj}j=1n, θki=(μki, σki2) denotes the mean (abundance level) and the variance of prey i in bait cluster k. As a result of hierarchical Dirichlet process prior, a mixture model for prey clustering is obtained as follows
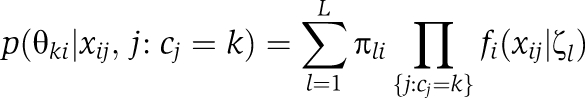 |
for all preys i=1,…,p and bait clusters k=1,…,K. Here, fi denotes the marginal distribution of xij for all j=1,…,n. The nested prey clusters are determined as
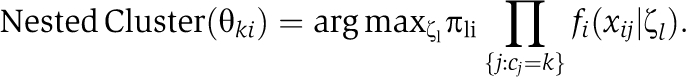 |
Note that the finite set of mean and variance values (ζ1,⋯, ζL) are shared in the posterior distribution of all preys i=1,…,p, with different weights π·i=(π1i,⋯, πLi) in each prey. In the notation above, L is an unknown number of prey clusters. Note that this number was also automatically determined by the clustering property of DPM.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH grant R01 CA-126239 to AIN. ACG is supported by a grant from the Canadian Institutes of Health Research (MOP-84314) and holds a Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics. We are grateful to Damian Fermin for technical assistance, Mihaela Sardiu and Michael Washburn for providing the raw spectral count data for the TIP49a/b network, and to Timo Glatter, Matthias Gstaiger, and Ruedi Aebersold for making their PP2A data set publicly available through the Tranche data distribution system.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Antoniak CE (1974) Mixtures of Dirichlet processes with applications to Bayesian nonparametric problems. Ann Stat 2: 1152–1174 [Google Scholar]
- Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M (2009) An experimentally derived confidence score for binary protein-protein interactions. Nat Methods 6: 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom J, Boucher L, Neduva V, Larsen B, Lin Z-Y, Breitkreutz B-J, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras A-C, Nesvizhskii AI, Tyers M (2010) Global architecture of the yeast protein kinase and phosphatase interaction network. Science 328: 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GI, Gingras AC (2007) Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 42: 298–305 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Church GM (2000) Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol 8: 93–103 [PubMed] [Google Scholar]
- Choi H, Fermin D, Nesvizhskii AI (2008) Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics 7: 2373–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Kemmeren P, Zhao XC, Greenblatt JF, Spencer F, Holstege FC, Weissman JS, Krogan NJ (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics 6: 439–450 [DOI] [PubMed] [Google Scholar]
- Dudoit S, Fridlyand J (2002) A prediction-based resampling method for estimating the number of clusters in a dataset. Genome Biol 3: RESEARCH0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, Raftery AE (2002) Model-based clustering, discriminant analysis, and density estimation. J Am Stat Assoc 97: 611–631 [Google Scholar]
- Friedel CC, Zimmer R (2009) Identifying the topology of protein complexes from affinity purification assays. Bioinformatics 25: 2140–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M et al. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636 [DOI] [PubMed] [Google Scholar]
- Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B, Aebersold R (2005) A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol Cell Proteomics 4: 1725–1740 [DOI] [PubMed] [Google Scholar]
- Gingras AC, Gstaiger M, Raught B, Aebersold R (2007) Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol 8: 645–654 [DOI] [PubMed] [Google Scholar]
- Glatter T, Wepf A, Aebersold R, Gstaiger M (2009) An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol Syst Biol 5: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreault M, D'Ambrosio LM, Kean MJ, Mullin MJ, Larsen BG, Sanchez A, Chaudhry S, Chen GI, Sicheri F, Nesvizhskii AI, Aebersold R, Raught B, Gingras AC (2009) A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics 8: 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GT, Lee I, Marcotte ER (2007) A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183 [DOI] [PubMed] [Google Scholar]
- Kim S, Tadesse MG, Vannucci M (2006) Variable selection in clustering via Dirichlet process mixture models. Biometrika 93: 877–893 [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643 [DOI] [PubMed] [Google Scholar]
- Lazzeroni L, Owen A (2002) Plaid models for gene expression data. Stat Sinica 12: 61–86 [Google Scholar]
- Liu H, Sadygov RG, Yates JR III (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76: 4193–4201 [DOI] [PubMed] [Google Scholar]
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol 25: 117–124 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4: 1419–1440 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Vitek O, Aebersold R (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods 4: 787–797 [DOI] [PubMed] [Google Scholar]
- Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics 4: 1487–1502 [DOI] [PubMed] [Google Scholar]
- Powell DW, Weaver CM, Jennings JL, McAfee KJ, He Y, Weil PA, Link AJ (2004) Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol Cell Biol 24: 7249–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelic A, Bleuler S, Zimmermann P, Wille A, Buhlmann P, Gruissem W, Hennig L, Thiele L, Zitzler E (2006) A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics 22: 1122–1129 [DOI] [PubMed] [Google Scholar]
- Pu SY, Vlasblom J, Emili A, Greenblatt J, Wodak SJ (2007) Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 7: 944–960 [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Dunson DB, Gelfand AE (2008) The nested Dirichlet process. J Am Stat Assoc 103: 1131–1154 [Google Scholar]
- Sardiu ME, Cai Y, Jin J, Swanson SK, Conaway RC, Conaway JW, Florens L, Washburn MP (2008) Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci USA 105: 1454–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiu ME, Florens L, Washburn MP (2009) Evaluation of clustering algorithms for protein complex and protein interaction network assembly. J Proteome Res 8: 2944–2952 [DOI] [PubMed] [Google Scholar]
- Scholtens D, Vidal M, Gentleman R (2005) Local modeling of global interactome networks. Bioinformatics 21: 3548–3557 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh YW, Jordan MI, Beal MJ, Blei DM (2006) Hierarchical Dirichlet processes. J Am Stat Assoc 101: 1566–1581 [Google Scholar]
- Tibshirani R, Walther G, Hastie T (2001) Estimating the number of clusters in a dataset via the Gap statistic. J Roy Stat Soc B 63: 411–423 [Google Scholar]
- Valpuesta JM, Martin-Benito J, Gomez-Puertas P, Carrascosa JL, Willison KR (2002) Structure and function of a protein folding machine: the eukaryotic cytosolic chaperonin CCT. FEBS Lett 529: 11–16 [DOI] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S (2009) From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 33: 537–545 [DOI] [PubMed] [Google Scholar]
- Wepf A, Glatter T, Schmidt A, Aebersold R, Gstaiger M (2009) Quantitative interaction proteomics using mass spectrometry. Nat Methods 6: 203–205 [DOI] [PubMed] [Google Scholar]
- Yeung K, Fraley C, Murua A, Raftery A, Ruzzo W (2001) Model-based clustering and data transformation for gene expression data. Bioinformatics 17: 977–987 [DOI] [PubMed] [Google Scholar]
- Zhang B, Park BH, Karpinets T, Samatova NF (2008) From pull-down data to protein interaction networks and complexes with biological relevance. Bioinformatics 24: 979–986 [DOI] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5: 2339–2347 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.