

Abstract
Background
In utero exposure to ethanol can result in severe fetal brain defects. Previous studies showed that ethanol induces apoptosis in differentiated cortical neurons. However, we know little about ethanol’s effects on proliferating embryonic cortical progenitors. This study investigated the impact of ethanol exposure on the Fas/Apo-1/CD95 suicide receptor pathway, and on the survival of proliferating cortical neuroepithelial progenitors.
Methods
Murine embryonic-derived primary cortical neuroepithelial cells were maintained as neurosphere cultures and exposed to a dose-range of ethanol for periods ranging from 1 to 5 days. Programmed cell death was measured by four independent means (Annexin-V staining, caspase activation, DNA fragmentation and autophagic vacuole formation). Surface Fas/Apo-1 suicide receptor expression was measured by flow cytometry. Expression of Fas/Apo-1-associated DISC-complex genes was measured by quantitative polymerase chain reaction.
Results
Ethanol exposure did not substantially increase apoptosis, necrosis, or surface Fas/Apo-1 expression. Moreover, ethanol significantly decreased caspase activation and autophagic activity. Finally, ethanol exposure induced mRNA expression of genes that constitute the death receptor complex.
Conclusions
This study provides surprising evidence that ethanol does not induce either programmed cell death or necrosis of immature progenitors during neurogenesis, though ethanol may render neural progenitors susceptible to future apoptotic insults. Furthermore, our novel observation that ethanol suppresses autophagy is consistent with a hypothesis that ethanol promotes premature neural progenitor maturation. Taken together with our previous data regarding the role of the Fas/Apo-1 receptor in neural development, we conclude that ethanol disrupts basic proliferation and differentiation machinery rather than initiating cell death per se.
Keywords: Fetal Alcohol Syndrome, Neurogenesis, Apoptosis, Fas/Apo-1/CD95, Differentiation
INTRODUCTION
Heavy prenatal ethanol exposure can result in a spectrum of physical abnormalities and growth deficiencies, collectively known as the Fetal Alcohol Spectrum Disorder (FASD). Defects associated with FASD include mental retardation, developmental delay and attention-deficit hyperactivity disorder (Mattson et al., 1996; Coles et al., 1997; Mattson and Riley, 1998; Roebuck et al., 1998; Thomas et al., 1998b; Kodituwakku et al., 2001; Schonfeld et al., 2001). The developing cortex is an important target of ethanol-induced damage. For example, it has been demonstrated that prenatal ethanol exposure results in a disproportionate reduction in frontal cortex size in the absence of global microcephaly (Wass et al., 2001).
Much of the research on gestational exposure to ethanol has focused on cell loss as a mechanism of ethanol-induced damage. Prenatal ethanol exposure during a crucial developmental period in the rat (gestational day [GD] 6 to birth) leads to a significant decrease in the number of neurons generated during the peak period of neurogenesis (GD 12–19) (Miller, 1988), and consequently, a decreased neuron number in the mature cortex (Miller and Potempa, 1990). This cell loss has largely been attributed to the ability of ethanol to induce apoptosis (Bhave and Hoffman, 1997; Cartwright et al., 1998; Bhave et al., 1999; Cheema et al., 1999; McAlhany et al., 2000; Ramachandran et al., 2003; Young et al., 2003; Takadera and Ohyashiki, 2004; Dikranian et al., 2005). Although we know that ethanol can induce apoptosis of fetal cortical neurons in late gestation (Ramachandran et al., 2003), relatively few studies (Kentroti and Vernadakis, 1991; Hao et al., 2003; Santillano et al., 2005) have focused on the apoptotic effects of ethanol on embryonic neural progenitors during early neurogenesis. Paradoxically, the onset of neurogenesis is accompanied by a wave of apoptotic cell death, similar to the death observed later in development as neurons seek appropriate synaptic targets (Blaschke et al.,1996; Thomaidou et al., 1997; Blaschke et al., 1998; Cheema et al., 1999; Bähr, 2000; De la Rosa and De Pablo, 2000). Indeed, apoptosis occurring during neurogenesis is as crucial to normal development as is the proliferation competency of progenitors. In fact, disruption of apoptosis via overexpression of Bcl-2 results in abnormal neurogenesis and neuronal determination in Xenopus embryos (Yeo and Gautier, 2003).
Our laboratory has been interested in the role of apoptosis during the period of neurogenesis, and in particular the extent to which the suicide receptor Fas/Apo [Apoptosis]-1/CD95 is involved in the deletion of neural precursor cells. We have previously shown the transient expression of this receptor in cells of the developing cortex during the peak period of naturally occurring apoptosis, in close proximity to FasL expressing cells (Cheema et al., 1999). In this same study, we illustrated that administration of FasL or anti-Fas/Apo-1 antibody induced caspase-dependent cell death in primary embryonic cortical neuroblast cultures, indicating that a functional Fas/Apo-1 death pathway is present in these neural progenitors. We demonstrated that p53 activation is followed by the induction of Fas/Apo-1 expression (Cheema et al., 2004). Ethanol, in turn, induces p53 expression (Kuhn and Miller, 1998; de-la-Monte et al., 2000) and p53-dependent suicide factors including Bax (Moore et al., 1999). Furthermore, we observed an increase in Fas/Apo-1 mRNA expression in postnatal organotypic cortical explants treated with ethanol, coupled with a dose-dependent increase in apoptosis (Cheema et al., 2000). This led us to hypothesize that the Fas/Apo-1 receptor might be involved in mediating the apoptotic consequences of ethanol exposure in immature progenitors as well.
In the present study we examined the vulnerability of neural progenitors to ethanol-induced apoptosis during the early period of neurogenesis in the mouse, an intensely proliferative phase and a time when crucial cell fate decisions are being made. Given the evidence illustrating that ethanol causes apoptosis of cortical neurons in both late gestation and the early postnatal period, and also considering the effects of this teratogen in ultimately decreasing the size of the cortex, we hypothesized that ethanol would result in a dose-dependent increase in apoptosis of proliferating cortical progenitors. Our previous data suggested that the Fas/Apo-1 receptor was a candidate ethanol target (Cheema et al., 2000), therefore we also hypothesized that ethanol would cause an increase in surface Fas/Apo-1 expression in neural progenitor cells along with an increase in downstream adaptor proteins of the extrinsic apoptotic pathway.
Contrary to our hypothesis, our data show that murine neural progenitors obtained during the early period of neurogenesis are relatively resistant to the apoptosis-inducing effects of ethanol. Interestingly, ethanol dramatically decreases both caspase activation and the formation of autophagic vacuoles. Ethanol exposure failed to cause an overall induction of surface Fas/Apo-1 expression. However, downstream adaptor proteins containing death effector domains were up-regulated following ethanol treatment. New evidence regarding the role of the DISC-complex adaptor proteins and autophagy in differentiation suggests that ethanol exposure during early neurogenesis may alter cortical progenitor maturation. These data suggest that ethanol exposure early in cortical development may be detrimental in ways unrelated to apoptosis---perhaps involving non-apoptotic functions of the extrinsic suicide pathway.
METHODS
Isolation and Culture of Cortical Progenitors
Timed pregnant C57BL6 mice were bred in-house, from breeder pairs initially purchased from Harlan (Harlan, TX). Gestational day (G.D.) 0.5 was the day that a vaginal mucus plug was detected. Pregnant females (G.D. 12.5) were anesthetized with a mixture of ketamine (0.09mg/gram body weight) and xylazine (0.106mg/gram body weight) by intramuscular injection. All animal procedures were approved by the Texas A&M University Laboratory Animal Care Committee. Fetuses were removed under aseptic conditions and rinsed in chilled phosphate buffered saline (PBS). Fetal brains were dissected, meningeal tissue removed and the structural precursor of the neocortex isolated. Fetal cortical tissue from 3–4 litters per experiment was combined into a single collection tube, dispersed by trypsinization (0.05%, Invitrogen #25300-054) and cells counted using a hemocytometer. Cells were grown in serum free mitogenic media (DMEM-F12 Invitrogen #11039-24) supplemented with 20ng/mL human recombinant Basic Fibroblast Growth Factor (bFGF, BD Biosciences #354060), 20ng/mL human Epidermal Growth Factor (hEGF, BD Biosciences #356052), ITS (Insulin, Transferrin, Selenium)-X supplement (Invitrogen #51500-056), 5ug/mL heparin (Sigma #4784), 0.15ng/mL Leukemia Inhibitory Factor (LIF, Alomone #L-200) and 20nM progesterone (Sigma #P7556). Cells were allowed to proliferate as neurospheres until cultures achieved a density of 2 × 106 cells per T25 flask (approximately 6–8 passages), and then used for experiments.
Ethanol Treatment
Neurosphere cultures at a density of 2 × 106 cells per T25 flask were randomly assigned to control or ethanol treatment groups representing low (60–95mg/dL (~13mM–21mM) based on gas chromatographic analyses), moderate (130–218mg/dL (~28mM–47mM)), and high doses (270–350mg/dL (~58–76mM)) of ethanol. Ethanol containing medium was prepared freshly before use, from 95% ethanol (Sigma), and ethanol concentrations in fresh and culture-exposed medium were measured for each experiment, by gas chromatography. Each flask was defined as a single sample. Culture medium was changed every 2 days. Control, and ethanol-treated flasks were capped tightly with phenolic caps, and sealed with parafilm to limit the loss of ethanol, and ethanol concentrations in the culture medium remained stable through the course of the experiment. These measured doses range in equivalence to consumption levels that can be attained by social drinkers to those attained by chronic alcoholics (Perper et al., 1986; Adachi et al., 1991). All values reported on graphs and tables are averages of actual gas chromatography measures of ethanol content taken from six experimental samples per dosage condition.
Flow Cytometry
Measurement of surface antigen levels was conducted according to our previously published protocols (Santillano et al., 2005) on a flow-cytometer (FACSCalibur, Beckton Dickinson). Excitation wavelength was set at 488nm (argon laser) and emission spectra for fluorescein and propidium iodide were 525nm and 630 nm respectively. Analyses were generated using Cell Quest software for Macintosh, (V. 3.5).
Labeling for Surface Fas/Apo-1/CD95 Receptor Expression
Cells were harvested, washed twice in PBS and triturated to dissociate neurosphere structures into individual cells. Cells were then immediately fixed in 1% paraformaldehyde buffered in PBS for either 30 minutes or overnight at 4ºC. Subsequently cells were washed twice in PBS and 106 cells were labeled with a fluorescein-conjugated antibody against the surface Fas/Apo-1/CD95 antigen (Pharmingen #554257). An equal amount of fluorescein-conjugated isotype-matched IgG antibody was added to samples not receiving Fas/Apo-1/CD95 antibody to account for nonspecific staining. Fixed, unlabeled cells served to measure background fluorescence. The proportion of labeled cells was analyzed via flow cytometry from each sample (N=104–105 cells per sample).
Annexin-V Staining for Apoptosis Detection
Annexin-V labeling was conducted as per the Annexin-V-Fluorescein Staining Kit (Roche #1858777) protocol, and per our previously published protocols (Cheema et al., 1999). Briefly, 106 cells were washed in PBS, resuspended in labeling solution containing Annexin-V-Fluorescein and propidium iodide and incubated for 10–15 minutes at room temperature. The proportion of labeled cells was analyzed via flow cytometry from each sample (N=104–105 cells per sample).
ELISA for Apoptosis Detection
We utilized a photometric enzyme-immunoassay for the in vitro determination of cytoplasmic histone-associated-DNA-fragments (Cell Death Detection ELISAPLUS Roche #1774425). Briefly, 104 cells were incubated in lysis buffer for 30 minutes at room temperature. The lysate was centrifuged at 200× g for 10 minutes, and 20μL from the supernatant was added to a streptavidin coated microplate for analysis. This solution was incubated for 2 hours at room temperature with an antibody mixture of anti-histone-biotin (which binds the histone component of the nucleosomes and captures the immuno-complex via biotin to the coated microplate) and anti-DNA-peroxidase (which binds the DNA components of the nucleosomes and is conjugated to horseradish peroxidase to provide a color reaction). After incubation the plate is washed three times with incubation buffer and ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid)) solution is added to provide a color reaction. The color product was measured by photometric analysis at a wavelength of 405nm, with a reference wavelength of 490nm. All samples were analyzed in triplicate, and in order to account for possible differences in the amounts of cells lysed per sample, all values were normalized to total protein levels.
Homogenous Caspase Assay
We utilized a fluorimetric assay for the quantification of activated caspases (Homogenous Caspase Assay, Roche #3005372). This assay is sensitive for caspases 2, 3, 6, 7, 8, 9, and 10. Briefly, 104 cells were simultaneously lysed and incubated in caspase substrate (DEVD-Rho110 [Asp-Glu-Val-Asp-Rhodamine 110]) at 37ºC for 2 hours. Activated caspases recognize and cleave at DEVD sites, and free R110 is determined fluorimetrically at 521nm. The developed fluorochrome is proportional to the concentration of activated caspases.
Fluorimetric Assay for Detection of Autophagic Vacuoles
The presence of autophagic vacuoles was detected with the fluorescent dye monodansylcadaverine (MDC, Sigma #30432) according to a previously published report (Biederbick et al., 1995). Cells were treated with ethanol for 72 hours, then harvested by centrifugation and resuspended in a .05mM solution of monodansylcadaverine dye in warm serum free mitogenic media (from 0.5M stock solution in DMSO), and incubated at 37ºC for 1 hour. Intracellular MDC fluorescence levels were measured by fluorescence photometry in a FLX-800 microplate reader (excitation wavelength 380nm, emission wavelength 525nm). An aliquot of cells was harvested and starved in warm Hank’s Balanced Salt Solution (Invitrogen) for one hour to induce autophagy and serve as a positive control. All samples were analyzed in triplicate, and in order to account for possible differences in the amounts of cells lysed per sample, all values were normalized to total protein levels.
BCA Protein Assay
Protein concentrations of each lysate utilized for the Cell Death Detection ELISA and autophagy assay were obtained using a BCA Protein Assay Kit (Pierce #23225).
RNA Extraction and cDNA Synthesis
Total RNA was extracted from ethanol-treated cultures using TRIZOLTM reagent (Invitrogen #15596), followed by DNAse treatment and column purification using the SV Total RNA Isolation kit (Promega, #23100). The quality and quantity of RNA was assessed by micro-capillary electrophoresis on an Agilent 2100 Bioanalyzer. Five micrograms of total RNA per ethanol-treated sample was reverse transcribed with Superscript III (Invitrogen #18080-093), utilizing the manufacturer’s protocol.
Real-Time Polymerase Chain Reaction
Real-time polymerase chain reactions were conducted on a Biorad MyIQTM ICycler Real-time machine using 96-well pcr plates (iCycler 96 well, Biorad #2239441). Reactions utilized iQ SYBR Green Supermix (Biorad #170-8880), and a starting primer concentration of 10μM. Standard curves were created for each primer set using various concentrations (ranging from 200ng/μL to 20ng/μL) of whole embryonic RNA from untreated GD 12.5 cultures. All target genes examined were compared to β-actin, a standard housekeeping reference gene. No-template controls were included for each primer set. We used a modification of the method outlined by M.W. Pfaffl (Pfaffl, 2001) to quantify gene expression (see Table 1 for primer characteristics), which takes into account the primer efficiency and the mean cycle threshold value for the control group as per the equation below.
Table 1.
PCR Primers
| Name | Accession # | E% | C.C. | Forward Primer Sequence (5′--3′) | Reverse Primer Sequence (5′--3′) |
|---|---|---|---|---|---|
| Dedd | NM_011615 | 102 | .99 | ATC TGG AGG AAA CAT CAA TTC G | GCA GCA CAC CAC AGG ATA G |
| Fadd | NM_010175 | 90 | .97 | CGC CGA CAC GAT CTA CTG | TTC TTC TTC TCA GCA TTC TTC C |
| Madd | BC003255 | 131 | .99 | TTC AAC TCT GCT AAC G | GGC CTT GTC ACC AAT AAG G |
| Cradd | NM_009950 | 167 | .98 | CTT TAT GGT ACA GGT TCC C | TGT CCA GCA ACA GCA TTG TC |
| β-Actin | XO3672 | 84 | .99 | CAT CCG TAA AGA CCT CTA TGC | ACT CCT GCT TGC TGA TCC |
E%= Primer Efficiency (%)
C.C.= Correlation Coefficient for Primer Amplification
CT = cycle threshold
target = gene of interest
reference gene = β-Actin
Data Analysis
Analysis of Variance (ANOVA), Multivariate Analysis of Variance (MANOVA) and post-hoc Fischer’s Least Significant Difference (Fisher’s LSD) tests were computed using a standard statistical package, SPSS (V. 11). Statistical significance was set at p<0.05. When necessary single outlying values greater than 3 standard deviations from the mean were eliminated from analysis.
RESULTS
Cortical Progenitors are Resistant to Ethanol Induced Apoptosis
In the present study, we sought to determine if immature neural progenitors, obtained from the second trimester equivalent period of gestation, were vulnerable to ethanol-induced apoptosis. We chose to assess levels of apoptosis after ethanol exposure by measuring Annexin-V staining (phosphatidylserine translocation to the outer leaflet of the cell membrane) by flow cytometry. Ethanol treatment did not result in a statistically significant difference in the percentage of cells gated in the apoptotic (Figure 1A, B, C) or in the necrotic fraction (Figure 2A, B, C) regardless of dosage or temporal exposure. However, the possibility remained that by measuring an early event in apoptosis, we failed to detect apoptosis that had already progressed to a later stage. Therefore, we measured DNA fragmentation (cytoplasmic oligonucleosomal fragments), a late event in the apoptotic cascade and one upon which a variety of caspase-dependent and independent pathways converge (Slagsvold et al., 2003; Chu et al., 2005; Culmsee et al., 2005). An overall ANOVA indicated a statistically significant interaction between ethanol concentration and time in culture (p<0.027). This interaction was mainly due to exposure to low and moderate doses of ethanol. Exposure to a low dose of ethanol (average ethanol concentration, 63mg/dL) led to a small decrease in DNA fragmentation as measured by the detection of (Figure 3, overall Fisher’s LSD p<0.004). This was due to a statistically significant reduction in apoptosis for cells exposed to ethanol for 1 or 2 days (p<0.027 and 0.001 respectively), but not 5 days. The moderate dosage (average ethanol concentration, 167mg/dL) in contrast, resulted in a small but significant increase in DNA fragmentation (overall Fisher’s LSD p<0.002). This was due to either a return to baseline control levels (at 1 day treatment) or a slight, but statistically significant increase in DNA fragmentation after 2 and 5-day exposures (all p<0.05). Overall, the highest dose (average ethanol concentration, 306mg/dL) did not result in a significant change in DNA fragmentation. In the aggregate, a modest 0.2-fold increase in apoptosis, as measured by DNA fragmentation, was observed only at the moderate ethanol dose. Caspases mediate both the extrinsic and intrinsic apoptotic pathways, and therefore we next assessed overall caspase activation in response to ethanol treatment as a measure of participation of either arm of the apoptotic program. We found that ethanol administration resulted in a dramatic and statistically significant (ANOVA, p=0.013) decrease in caspase activation that was observable even at the lowest dosage of ethanol (Figure 4).
Fig. 1.
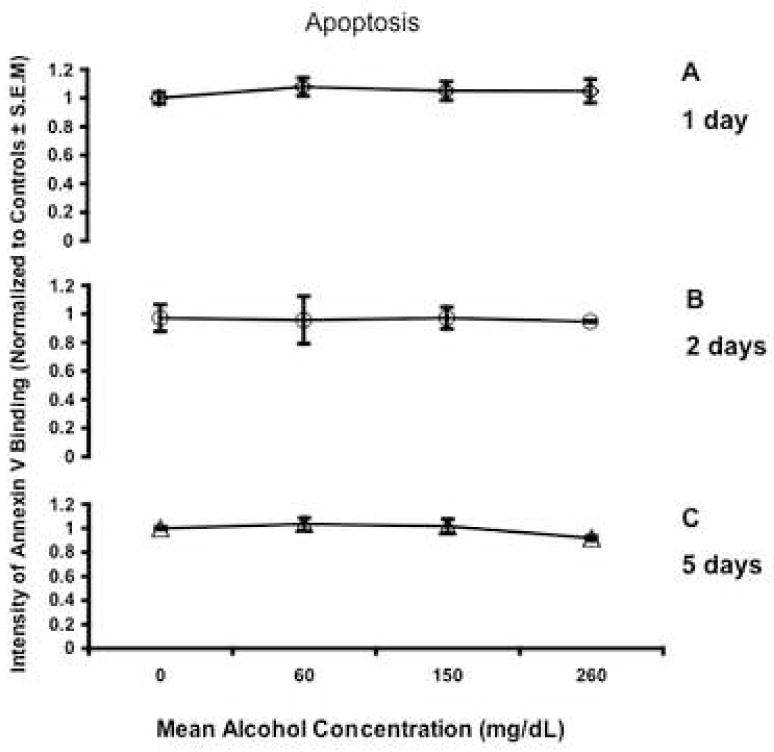
Line graphs depict intensity of Annexin V binding normalized to control cultures. There was no significant change in apoptosis, as detected by Annexin-V staining after a 1, 2 or 5 days of ethanol exposure. Apoptotic cells were defined as those that expressed Annexin-V but not propidium iodide binding. All error bars represent S.E.M.
Fig. 2.
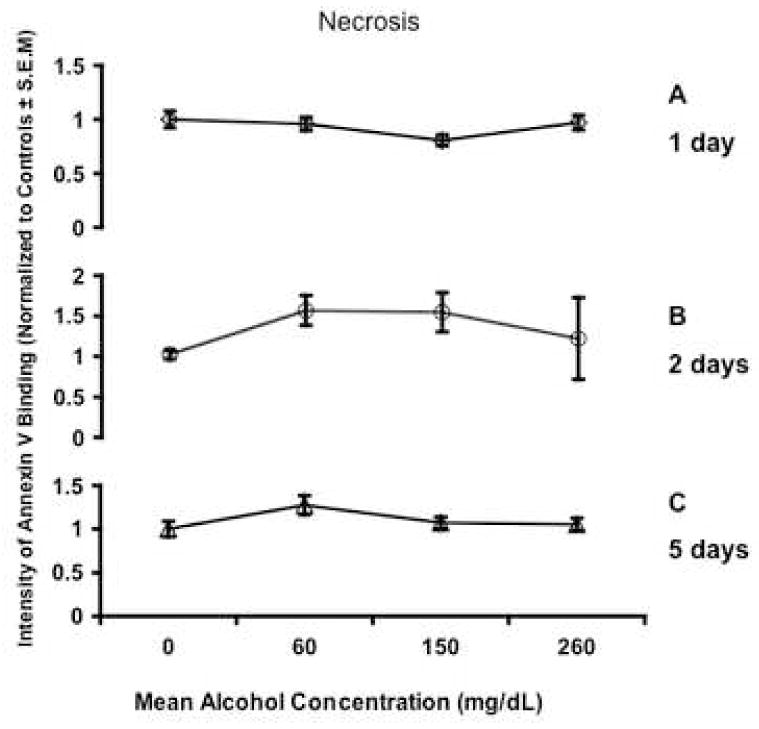
Line graphs depict intensity of Annexin V binding normalized to control cultures. There was no significant change in necrosis as detected by Annexin-V staining after a 1, 2, or 5 days of ethanol exposure. Necrotic cells were defined as expressing both Annexin-V and propidium iodide binding. All error bars represent S.E.M.
Fig. 3.
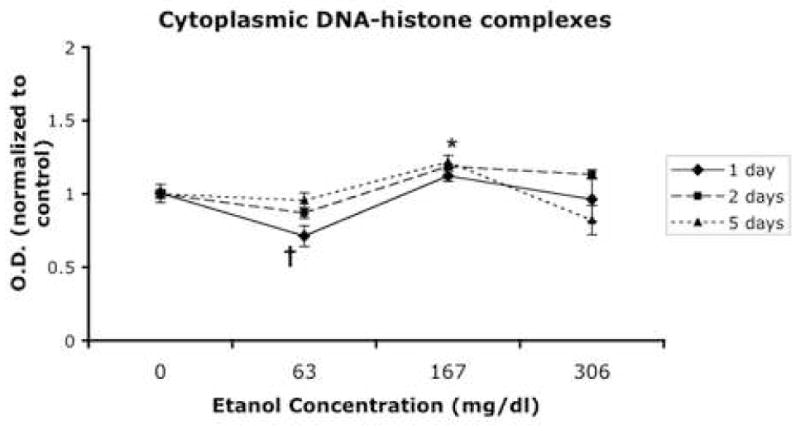
DNA fragmentation as measured by enzyme-linked immunosorbent assay (ELISA) observed after a 1, 2, and 5 days exposure. The y axis represents optical density normalized to controls. The † symbol indicates an overall statistically significant suppression compared to controls (LSD post-hoc p<0.004) at the low dose of ethanol, while * indicates an overall statistically significant induction compared to controls (LSD post-hoc p<0.002) at the moderate ethanol dose. The high ethanol dose was not statistically different from controls (LSD post-hoc p<0.581). All error bars represent S.E.M.
Fig. 4.
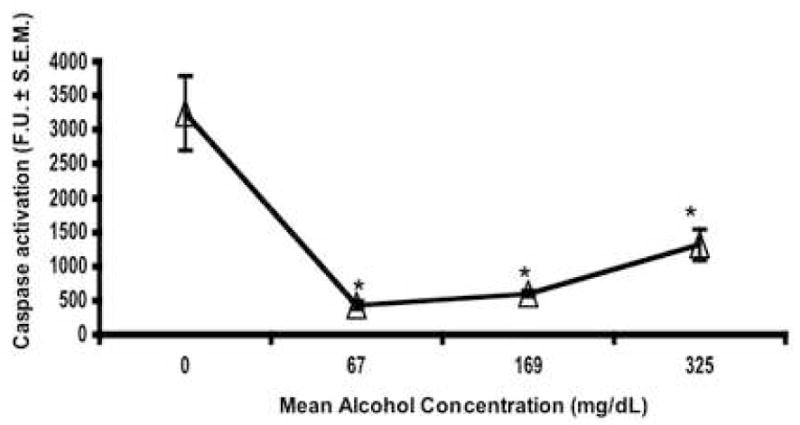
Ethanol decreased total caspase activation as measured by a fluorometric enzyme assay. F.U. indicates fluorescence units normalized to total protein. * indicates a significant difference when compared to controls (ANOVA p<0.013). All error bars represent S.E.M.
Ethanol Suppresses Autophagy in Proliferating Cortical Neuroepithelial Precursors
Autophagy is classified as a type II cell death mechanism characterized by the engulfment of cytoplasmic contents and organelles into membrane-bound autophagic vesicles, which are targeted for destruction to lysosomes within the same cell (Clarke, 1990). Unlike apoptosis, autophagic death does not result in pronounced DNA degradation (Gozuacik and Kimchi, 2004). Thus, we chose to examine whether or not ethanol might induce cell death via autophagy, using the uptake of monodansylcadaverine (MDC), a fluorescent dye that specifically marks the acidic autophagosomes/autolysosomes and not other endosomal structures (Biederbick et al., 1995). We exposed GD 12.5-derived neural progenitor cultures to ethanol for 72 hours and then measured the incorporation of MDC dye (Figure 5). Interestingly, we observed a statistically significant decrease in autophagic vacuole formation as indicated by a decrease in fluorescence after treatment with moderate and high doses of ethanol (ANOVA, p=0.021).
Fig. 5.
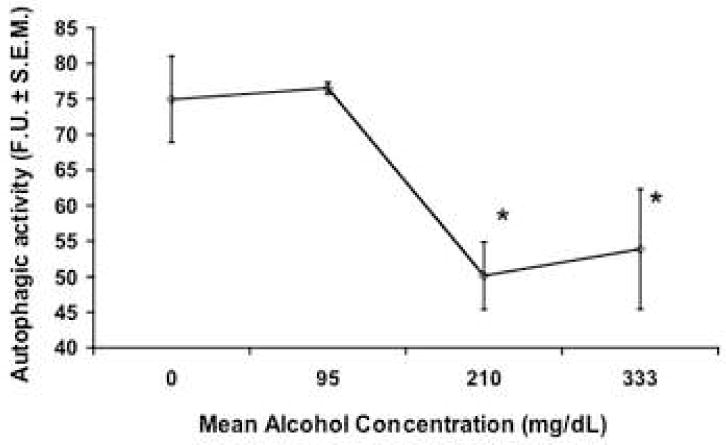
Ethanol decreased autophagy, as measured by MDC (monodansylcadaverine) fluorescent dye incorporation, after a 3 day exposure. F.U. is fluorescence units normalized to total protein. * indicates a significant difference when compared to controls (ANOVA p<0.021) All error bars represent S.E.M.
Ethanol Treatment Does Not Alter Surface Fas/Apo-1 CD95 Receptor Expression
The developing brain expresses and is sensitive to the Fas/Apo-1 suicide receptor (Cheema et al., 1999; Le-Niculescu et al., 1999; Cheema et al., 2000; Felderhoff-Mueser et al., 2000; Cheema et al., 2004). Even though ethanol did not induce substantial apoptosis, ethanol may increase sensitivity to apoptosis signals from the environment by up-regulating Fas/Apo-1 receptor expression. Neurosphere cultures were labeled with FITC-conjugated antibodies to the Fas/Apo-1 receptor and analyzed via flow cytometry. After subtraction of background fluorescence, cells expressing above 101 units of fluorescence were defined as Fas/Apo-1 expressing cells (FasTotal, Figure 6A), and cells gated in the fraction above 102 as expressing a high level of the surface receptor (FasHi, Figure 6A) as per our previously published protocols (Cheema et al., 2004). Ethanol exposure did not result in a change in the percentage of cells gated in the FasTotal or FasHi (Figure 6B and C) populations. However, we did observe an inherent heterogeneity of Fas/Apo-1 expression amongst cortical progenitors that was unrelated to ethanol exposure. By day 2, approximately 40–50% of cells sampled expressed some level of Fas/Apo-1 (FasTotal), and of these 1–3% express a high amount of surface Fas/Apo-1 (FasHi). Moreover, as the time spent in culture increases there is a pronounced and statistically significant (ANOVA, p= 0.001) decrease in overall expression levels (Figure 6B,C), so that expression was close to the limit of detection for control, low and moderate alcohol doses, and undetectable at the high dose.
Fig. 6.
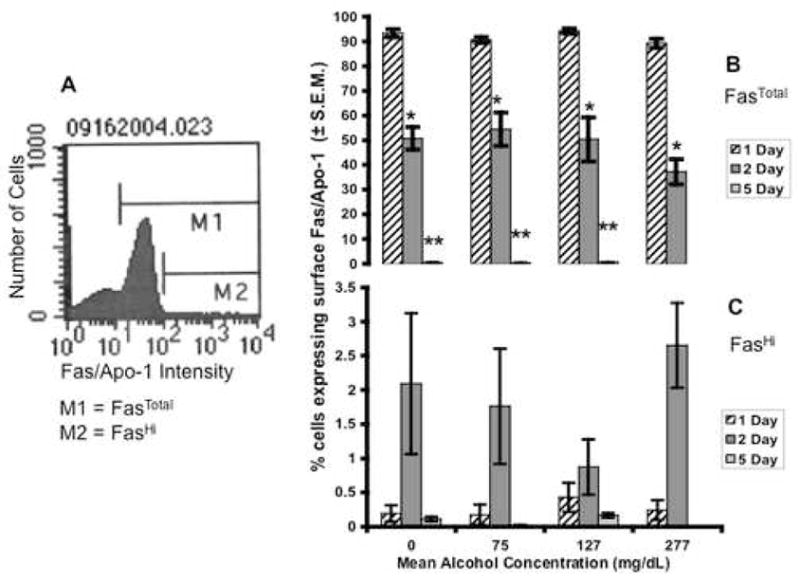
(A) Flow cytometry frequency histogram depicting the number of cells (Y axis) expressing cell-surface immunofluorescence for the Fas/Apo-1 suicide receptor (X-axis). M1 denotes all cells expressing cell-surface Fas/Apo-1 above isotype background control (FasTotal). M2 denotes cells expressing high levels of Fas/Apo-1 on their cell surface (FasHi). (B) Ethanol did not significantly change FasTotal at either 1, 2 or 5 days of exposure. A decrease in surface Fas receptor expression was observed across time, * indicates significance with respect to 1 day of ethanol, ** indicates significance with respect to 2 days of ethanol (ANOVA, p<0.001 for both). (B) Ethanol did not change the proportion of cells in the FasHi population, although at this developmental stage cortical progenitors maintained in vitro for 2 days have a higher basal level of Fas expression. All error bars represent S.E.M.
The Expression of DISC-complex Genes Is Upregulated By Ethanol Exposure
We examined the expression of genes that code for proteins involved in the extrinsic apoptotic pathway, namely Fadd (Fas/Apo-1/CD95 associated adaptor protein), Dedd (Death effector domain-containing protein), Madd (Map-kinase activating death domain) and Cradd/Raidd (caspase and RIP adaptor with death domain). These specific adaptor proteins associate with ligand-bound receptor complexes, recruiting procaspases and other components to form the DISC complex (Alcivar et al., 2003; Peter and Krammer, 2003; Jabado et al., 2004; Wang et al., 2006). Multivariate analysis of variance (MANOVA) resulted in a Wilks’ Lambda value of 0.034, indicating that overall a strong relationship exists between ethanol administration and expression of this group of death-receptor-associated genes. After two days of exposure to ethanol, we observed a general (though not statistically significant) upward dose-related trend in Fadd mRNA expression (Figure 7A). We did however observe a significant dose-related increase in the expression of Dedd (Figure 7B, * indicates ANOVA p=0.001) and Cradd/Raidd (Figure 7C, ** indicates ANOVA p=0.038) mRNA expression when compared to controls. Madd expression was virtually undetectable in our population, with control group cycle threshold (CT) values outside of the range of reliable detection (CT>37).
Fig. 7.
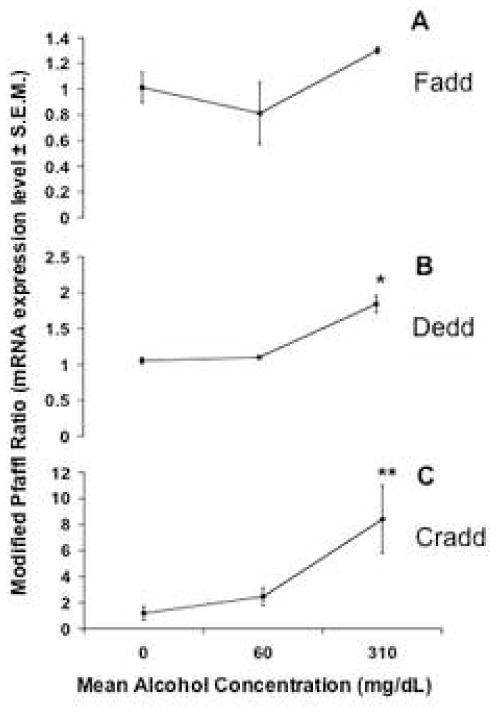
mRNA expression levels for death-receptor-associated genes Fadd, Dedd and Cradd after exposure to ethanol for 48 hours. The y axis represents mRNA expression levels as a modified Pfaffl ratio (see Methods). * indicates significant difference (ANOVA, p=0.000) for Dedd, ** indicates significant difference for Cradd (ANOVA, p=0.038) when compared to controls. All error bars represent S.E.M.
DISCUSSION
The ventricular neuroepithelium of the anterior neural tube expands rapidly during the second trimester-equivalent period of human gestation to generate telencephalic structures like the cerebral cortex. Deletion of relatively few neural progenitors from the neuroepithelium during the critical developmental period of neuroepithelial proliferation is likely to lead to a disproportionate reduction in the growth of telencephalic structures. We isolated neuroepithelial cells from gestational day 12.5 mouse cerebral cortex to capture neuroepithelial progenitors that would give rise to the earliest (i.e., deepest) laminae of the developing cerebral cortical plate (Angevine and Sidman, 1961; Caviness, 1982; Takahashi et al., 1995) as well as progenitors that give rise to later-developing laminae, since evidence from several laboratories suggests that initial fate specification occurs within the ventricular zone itself (Donoghue and Rakic, 1999; Desai and McConnell, 2000).
At the start of the reported experiments, we hypothesized that ethanol would induce apoptosis in cortical progenitors similar to our observed induction of apoptosis in a more differentiated neural tissue (Cheema et al., 2000). We measured a variety of features characteristic of the apoptotic process, and our data show that cortical progenitors maintained as neurosphere cultures are remarkably resistant to ethanol-induced apoptosis, even at doses typically attained only by chronic alcoholics. Interestingly, total caspase activity was suppressed by ethanol. The lack of caspase activation, together with the fact that ethanol did not also induce necrosis, indicates that the alternate calpain pathway (Neumar et al., 2003) is also unlikely to be a target of ethanol. We therefore investigated the effect of ethanol on autophagy, a lysozome-mediated cellular mechanism that can result in programmed cell death. To the best of our knowledge this is the first study examining the effect of ethanol on autophagy, and surprisingly, we found that ethanol exposure significantly reduced the formation of autophagic lysozomes. The suppression of autophagy indicates that ethanol also did not lead to a significant destruction of cellular organelles. These experiments confirm initial studies in our laboratory demonstrating that ventricular zone progenitors isolated from a somewhat later developmental stage (i.e., the peak period of cortical plate neurogenesis) exhibited a similar resistance to apoptosis (Santillano et al., 2005).
Despite some evidence suggesting that ethanol can kill immature neuroblasts under specific circumstances (Kentroti and Vernadakis, 1991; Hao et al., 2003), ethanol-induced apoptosis may be a differentiation state-specific phenomenon. For example, the pro-apoptotic protein Bax, a necessary mediator of ethanol-induced apoptosis in differentiating neurons (Young et al., 2003), is not expressed in proliferating cortical neuroepithelial cells, and its expression is induced only following neuronal differentiation (Wade et al., 1999). The concept of developmental stages conferring either vulnerability or resistance to ethanol is certainly a documented phenomenon in the post-natal period, and in other brain regions such as the cerebellum (Thomas et al., 1998a; Heaton et al., 2003). Cerebellar granule cells for example, exhibit delayed apoptosis in response to ethanol exposure that follows a decline in their proliferation rate (Li et al., 2001), suggesting that cell cycle status may be linked to ethanol sensitivity. Given the inverse relationship between the expression of pro-apoptotic factors like p53 and cell cycle (Wade et al., 1999), along with our observations that ethanol induces cell cycle (Santillano et al., 2005), it is likely that the early proliferative stage of neurogenesis is a period of relative resistance to ethanol-induced apoptosis. It is also possible that sensitivity to ethanol-induced cell death is model system-dependent rather than differentiation state-dependent, i.e., that neural progenitors are more resistant to apoptosis simply because of the specific local milieu that emerges with an aggregate of cells maintained as neurospheres. However, the neurosphere model more closely mimics the natural, cell-dense environment of the second trimester neuroepithelium, consisting of high-density aggregates of stem and progenitor cells within ventricular and emerging sub-ventricular zones, than do more typically used, dissociated culture models. Nevertheless, apoptosis-resistance is not absolute, since recent work in our laboratory (unpublished observations) indicates that disruption of c-kit signaling, or exit from cell cycle, does render progenitor cells vulnerable to ethanol-induced apoptosis.
Dis-regulation of the Fas/Apo-1 Receptor System
Our previous work showed that the Fas/Apo-1 is expressed in the developing cerebral cortex during the perinatal period, both by immature ventricular zone progenitors, and later, by maturing cortical plate neurons (Cheema et al., 1999). Additionally, our lab showed that ethanol induced a dose-dependent increase in Fas/Apo-1 mRNA in differentiated postnatal cortical explant cultures (Cheema et al., 2000). We subsequently found that high levels of cell-surface Fas/Apo-1 expression are associated with entry into G2 and M phases of the cell cycle (Cheema et al., 2004), suggesting that this suicide receptor may modulate progression through cell cycle checkpoints. Furthermore, the expression of Fas/Apo-1 is associated with the activation of cell cycle regulatory proteins like p53, further supporting a role for the Fas/Apo-1 pathway in cell cycle regulation. Cortical progenitors maintained in neurosphere cultures exhibit increased cell cycle activity in response to ethanol exposure (Santillano et al., 2005). We therefore hypothesized that if ethanol induced cell cycle activity, it would also increase the expression of the Fas/Apo-1 in proliferating neural progenitors. The present study demonstrates that neuroepithelial progenitors do exhibit a nascent heterogeneity with respect to cell surface Fas/Apo-1 expression. However, ethanol did not alter the numbers of cells expressing either moderate or high levels of surface Fas/Apo-1. Because cell-surface Fas/Apo-1 expression in embryonic cortical progenitors is tied to cell cycle and because ethanol induced cell cycle, our data may be interpreted to suggest that ethanol induces dissociation between cell cycle and sensitivity to receptor-mediated apoptosis. This dissociation may permit the survival of cell populations that would otherwise be eliminated. One prediction that arises from these data is that ethanol exposure may permit cells with defects in DNA replication to proceed through cell cycle checkpoints, leading to an increased accumulation of defective neural progenitors, and ultimately, the accumulation of aberrant neurons within the cortical plate. These neurons may not behave appropriately during development, and may contribute to lamination defects and the formation of heterotopias (Komatsu et al., 2001; Mooney et al., 2004).
Though ethanol did not alter cell-surface Fas/Apo-1 expression, multivariate analyses indicate that ethanol has a general dose-related inductive effect on the expression of mRNA transcripts for several DED (death-effector-domain)-containing genes that constitute part of the DISC (death-inducing signaling complex), the intracellular initiating component of the extrinsic apoptotic cascade. These data predict that while ethanol does not directly induce apoptosis in immature neuroepithelial cells, exposure to ethanol during the mitogenic period may render daughter neuroblasts susceptible to apoptotic stimuli as they differentiate into neurons.
Evidence for an Alternate Hypothesis of Ethanol-Induced Neuroepithelial Maturation
An alternate hypothesis, also collectively supported by our data is that ethanol promotes premature neuroepithelial differentiation. While autophagy is often associated with cell death, it also represents a major strategy for cellular adaptation. Autophagic degradation of long-lived proteins and organelles enables a cell to adapt to stressful conditions like amino acid starvation (Gozuacik and Kimchi, 2004). Autophagy is also associated with cellular differentiation. For example, undifferentiated colon adenocarcinoma cells express high levels of autophagic activity, and this activity is repressed following differentiation (Houri et al., 1995). Similarly, while Fas/Apo-1 was originally characterized as an obligatory cell-death receptor (Nagata and Goldstein, 1995), more recent evidence from a variety of laboratories including ours, have shown that this receptor also regulates cell cycle (Cheema et al., 2004) and differentiation (Cheema et al., 1999; Desbarats et al., 2003; Ceccatelli et al., 2004; Tamm et al., 2004) signaling cascades. Additionally, DISC-complex adaptor proteins also exhibit non-apoptotic functions (Park et al., 2005). Consequently, in the absence of ethanol-induced apoptosis, the observed suppression of autophagy and induction of DISC mRNA transcripts are consistent with a model of ethanol-induced neuroepithelial maturation.
Other data from our group also support this ‘premature differentiation’ hypothesis. For example, we have shown that ethanol suppresses the expression of stem cell markers like Sca-1, ABCG2, CD117 and CD133 and induces asymmetric cell division (Santillano et al., 2005). Asymmetric division, resulting in two dissimilar daughter cells, promotes the emergence of neuronal lineage-committed precursors from the cortical ventricular zone that then proceed to populate the sub-ventricular zone (Noctor et al., 2004), before differentiating into cortical plate neurons. Collectively, we interpret these data to indicate that ethanol promotes stem-cell to blast-cell differentiation in embryonic cortical neuroepithelial-derived cells, and our current data are certainly compatible with that hypothesis.
Conclusion
Our data collectively support an unexpected and surprising conclusion---that ethanol is not obviously or immediately cytotoxic to cerebral cortical neuroepithelial-derived progenitors. Rather, the effects of ethanol appear to be more complex and pervasive. Ethanol appears to influence cortical neuroepithelial maturation programs, and collectively our data predict that episodes of maternal ethanol consumption during the period of fetal neuroepithelial proliferation are likely to have long-term consequences for subsequent neuronal differentiation. Furthermore, disrupting the timing of neuroepithelial maturation during the second trimester-equivalent period of brain development can lead to significant disorganization of the laminar pattern of the mature cortical plate, without the need to invoke cyto-toxicity as ethanol’s mechanism of action.
Acknowledgments
Special thanks to Jane Miller for her assistance with the flow cytometry and Henry Rudy Thomas for experiment preparation. The authors thank Danielle Lewis, and Drs. Wei-Jung Chen, Leena Kumar, and Cynthia Camarillo for a critical evaluation of the manuscript. This work was supported by an NIAAA predoctoral F31 fellowship to T.L.P. (#AA015232) and NIAAA grant #AA13440 to R.C.M.
List of Abbreviations
- Caspase
cysteine, aspartic-acid proteases
- CRADD/RAIDD
caspase and RIP adaptor with death domain
- DEDD
Death effector domain-containing protein
- DISC
Death Inducing Signaling Complex
- FADD
Fas/Apo-1/CD95 associated adaptor protein
- Fas/Apo-1
Fas/Apo(Apoptosis)-1, CD95 suicide receptor
- FASD
Fetal Alcohol Spectrum Disorder
- FasL
Fas/Apo-1 ligand
- GD
Gestational day
- MADD
MAPk (Mitogen-activated protein kinase) activating death domain
Contributor Information
Terasa L. Prock, Email: prock@medicine.tamuhsc.edu.
Rajesh C. Miranda, Email: miranda@medicine.tamuhsc.edu.
References
- Adachi J, Mizoi Y, Fukunaga T, Ogawa Y, Ueno Y, Imamichi H. Degrees of alcohol intoxication in 117 hospitalized cases. Journal of Studies on Alcohol. 1991;52:448–453. doi: 10.15288/jsa.1991.52.448. [DOI] [PubMed] [Google Scholar]
- Alcivar A, Hu S, Tang J, Yang X. DEDD and DEDD2 associate with caspase 8/10 and signal cell death. Oncogene. 2003;22:291–297. doi: 10.1038/sj.onc.1206099. [DOI] [PubMed] [Google Scholar]
- Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Bähr M. Live or let die---retinal ganglion cell death and survival during development and in lesioned CNS. Trends Neurosci. 2000;23:483–490. doi: 10.1016/s0166-2236(00)01637-4. [DOI] [PubMed] [Google Scholar]
- Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: Signal transduction cascades and the site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave SV, Hoffman PL. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J Neurochem. 1997;68:578–586. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66(1):3–14. [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122(4):1165–74. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396(1):39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Tessmer LL, Smith SM. Ethanol-induced neural crest cell apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol Clin Expt Res. 1998;22:142–149. [PubMed] [Google Scholar]
- Caviness VS. Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Research, Developmental Brain Research. 1982;4:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Ceccatelli S, Tamm C, Sleeper E, Orrenius S. Neural stem cells and cell death. Toxicol Lett. 2004;149(1–3):59–66. doi: 10.1016/j.toxlet.2003.12.060. [DOI] [PubMed] [Google Scholar]
- Cheema ZF, Santillano DR, Wade SB, Newman JM, Miranda RC. The extracellular matrix, p53 and estrogen compete to regulate cell-surface Fas/Apo-1 suicide receptor expression in proliferating embryonic cerebral cortical precursors, and reciprocally, Fas- ligand modifies estrogen control of cell-cycle proteins. BMC Neurosci. 2004;5(1):11. doi: 10.1186/1471-2202-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema ZF, Wade SB, Sata M, Walsh K, Sohrabji F, Miranda RC. Fas/Apo[Apoptosis]-1 and Associated Proteins in the Differentiating Cerebral Cortex: Induction of Caspase-Dependent Cell Death and Activation of NF-KB. J Neurosci. 1999;19(5):1754–1770. doi: 10.1523/JNEUROSCI.19-05-01754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema ZF, West JR, Miranda RC. Ethanol Induces Fas/Apo[Apoptosis]-1 mRNA and Cell Suicide in the Developing Cerebral Cortex. Alcohol Clin Expt Res. 2000;24(4):535–543. [PubMed] [Google Scholar]
- Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94(6):1685–95. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181(3):195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- Coles CD, Platzman KA, Raskind-Hood CL, Brown RT, Falek A, Smith IE. A comparison of children affected by prenatal alcohol exposure and attention deficit-hyperactivity disorder. Alcohol Clin Expt Res. 1997;21:150–161. [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-Ribose) polymerase and bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25(44):10262–72. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Rosa EJ, De Pablo F. Cell death in early neuronal development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454–458. doi: 10.1016/s0166-2236(00)01628-3. [DOI] [PubMed] [Google Scholar]
- de-la-Monte SM, Gangu N, Banerjee K, Brown NV, Luong T, Wands JR. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol Clin Expt Res. 2000;24:716–726. [PubMed] [Google Scholar]
- Desai AR, McConnell SK. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development. 2000;127(13):2863–72. doi: 10.1242/dev.127.13.2863. [DOI] [PubMed] [Google Scholar]
- Desbarats J, Birge RB, Mimouni-Rongy M, Weinstein DE, Palerme JS, Newell MK. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol. 2003;5(2):118–25. doi: 10.1038/ncb916. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res Dev Brain Res. 2005;22:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Donoghue MJ, Rakic P. Molecular gradients and compartments in the embryonic primate cerebral cortex. Cereb Cortex. 1999;9(6):586–600. doi: 10.1093/cercor/9.6.586. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi UC, Edwards AD, Mehmet H. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 2000;10(1):17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Hao H, Parker GC, Zhao J, Barami K, Lyman W. Human neural stem cells are more sensitive than astrocytes to ethanol exposure. Alcohol Clin Expt Res. 2003;27(8):1310–7. doi: 10.1097/01.ALC.0000080671.56559.EF. [DOI] [PubMed] [Google Scholar]
- Heaton MW, Paiva M, Madorsky I, Shaw G. Ethanol effects on neonatal rat cortex: comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Developmental Brain Research. 2003;145:249–262. doi: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Houri JJ, Ogier-Denis E, De Stefanis D, Bauvy C, Baccino FM, Isidoro C, Codogno P. Differentiation-dependent autophagy controls the fate of newly synthesized N-linked glycoproteins in the colon adenocarcinoma HT-29 cell line. Biochem J. 1995;309 ( Pt 2):521–7. doi: 10.1042/bj3090521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabado O, Wang Q, Rideout HJ, Yeasmin M, Guo KX, Vekrellis K, Papantonis S, Angelastro JM, Troy CM, Stefanis L. RAIDD aggregation facilitates apoptotic death of PC12 cells and sympathetic neurons. Cell Death Differ. 2004;11(6):618–30. doi: 10.1038/sj.cdd.4401397. [DOI] [PubMed] [Google Scholar]
- Kentroti S, Vernadakis A. Survival and proliferation in developing neuroblasts in cultures derived from embryos treated with ethanol during early neuroembryogenesis: effects attenuated by somatostatin. J Neurosci Res. 1991;30(4):641–8. doi: 10.1002/jnr.490300407. [DOI] [PubMed] [Google Scholar]
- Kodituwakku PW, May PA, Clericuzio CL, Weers D. Emotion-related learning in individuals prenatally exposed to alcohol: an investigation of the relation between set shifting, extinction of responses, and behavior. Neuropsycholgia. 2001;39(7):699–780. doi: 10.1016/s0028-3932(01)00002-1. [DOI] [PubMed] [Google Scholar]
- Komatsu S, Sakata-Haga H, Sawada K, Hisano S, Fukui Y. Prenatal exposure to ethanol induces leptomeningeal heterotopia in the cerebral cortex of the rat fetus. Acta Neuropathol (Berl) 2001;101:22–26. doi: 10.1007/s004010000257. [DOI] [PubMed] [Google Scholar]
- Kuhn PE, Miller MW. Expression of p53 and Alz-50 immunoreactivity in rat cortex: effect of prenatal exposure to ethanol. Exp Neurol. 1998;154:418–429. doi: 10.1006/exnr.1998.6907. [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H, Bonfoco E, Kasuya Y, Claret F-X, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas Ligand induction and cell death. Molecular and Cellular Biology. 1999;19(1):751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lin H, Zhu Y, Wang M, Luo J. Disruption of cell cycle kinetics and cyclin-dependent kinase system by ethanol in cultured cerebellar granule progenitors. Brain Res Dev Brain Res. 2001;132(1):47–58. doi: 10.1016/s0165-3806(01)00294-2. [DOI] [PubMed] [Google Scholar]
- Mattson S, Riley EP, Delis DC, Stern C, Jones KL. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol Clin Expt Res. 1996;20(5):810–6. doi: 10.1111/j.1530-0277.1996.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Expt Res. 1998;22:279–294. doi: 10.1111/j.1530-0277.1998.tb03651.x. [DOI] [PubMed] [Google Scholar]
- McAlhany RE, West JR, Miranda RC. Glial-derived neurotrophic factor (GDNF) prevents ethanol-induced apoptosis and JUN kinase phosphorylation. Brain Res Dev Brain Res. 2000;119(2):209–16. doi: 10.1016/s0165-3806(99)00171-6. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of Prenatal Exposure to Ethanol on the Development of Cerebral Cortex: I. Neuronal Generation. Alcohol Clin Expt Res. 1988;12:440–448. doi: 10.1111/j.1530-0277.1988.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Miller MW, Potempa G. Number of neurons and glia in mature rat somatosensory cortex. J Comp Neurol. 1990;293:92–102. doi: 10.1002/cne.902930108. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Siegenthaler JA, Miller MW. Ethanol induces heterotopias in organotypic cultures of rat cerebral cortex. Cereb Cortex. 2004;10:1071–80. doi: 10.1093/cercor/bhh066. [DOI] [PubMed] [Google Scholar]
- Moore DB, Walker DW, Heaton MB. Neonatal ethanol exposure alters bcl-2 family mRNA levels in the rat cerebellar vermis. Alcohol Clin Expt Res. 1999;23:1251–1261. doi: 10.1111/j.1530-0277.1999.tb04286.x. [DOI] [PubMed] [Google Scholar]
- Nagata S, Goldstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278(16):14162–7. doi: 10.1074/jbc.M212255200. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nature Neurosci. 2004;7(2):136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Park SM, Schickel R, Peter ME. Nonapoptotic functions of FADD-binding death receptors and their signaling molecules. Curr Opin Cell Biol. 2005;17(6):610–6. doi: 10.1016/j.ceb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Perper JA, Twerski A, Wienand JW. Tolerance at High Blood Alcohol Concentration: A Study of 110 Cases and Review of the Literature. Journal of Forensic Sciences. 1986;31:212–221. [PubMed] [Google Scholar]
- Peter ME, Krammer PH. The CD95(Apo-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neruons. J Neurosci Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Roebuck TM, Mattson SN, Riley E. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Expt Res. 1998;22:339–344. doi: 10.1111/j.1530-0277.1998.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Santillano D, Kumar L, Prock T, Tingling J, Miranda RC. Ethanol induces cell-cycle activity and reduces stem cell heterogeneity in cerebral cortical neuroepithelial precursors. Biomed Central. 2005;6:59. doi: 10.1186/1471-2202-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld AM, Lang MS, Delis DC, Riley EP. Verbal and nonverbal fluency in children with heavy prenatal alcohol exposure. J Studies Alcohol. 2001;62(2):239–46. doi: 10.15288/jsa.2001.62.239. [DOI] [PubMed] [Google Scholar]
- Slagsvold HH, Rosseland CM, Jacobs C, Khuong E, Kristofferson N, Gaarder M, Fallgren AB, Huitfeld HS, Paulsen RE. High molecular weight DNA fragments are processed by caspase sensitive or caspase independent pathways in cultures of cerebellar granule neurons. Brain Res. 2003;984(1–2):111–21. doi: 10.1016/s0006-8993(03)03119-6. [DOI] [PubMed] [Google Scholar]
- Takadera T, Ohyashiki T. Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur J Pharmacol. 2004;24:239–245. doi: 10.1016/j.ejphar.2004.07.115. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15(9):6046–57. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm C, Robertson JD, Sleeper E, Enoksson M, Emgard M, Orrenius S, Ceccatelli S. Differential regulation of the mitochondrial and death receptor pathways in neural stem cells. European Journal of Neuroscience. 2004;19:2613–2621. doi: 10.1111/j.0953-816X.2004.03391.x. [DOI] [PubMed] [Google Scholar]
- Thomaidou D, Mione MC, Cavanagh JF, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17(3):1075–85. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Goodlett CR, West JR. Alcohol-induced Purkinje cell loss depends on developmental timing of alcohol exposure and correlates with motor performance. Brain Res Dev Brain Res. 1998a;105(2):159–66. doi: 10.1016/s0165-3806(97)00164-8. [DOI] [PubMed] [Google Scholar]
- Thomas SE, Mattson SN, Riley EP. Comparison of social abilities of children with fetal alcohol syndrome to those of children with similar IQ scores and normal controls. Alcohol Clin Expt Res. 1998b;22(2):528–33. [PubMed] [Google Scholar]
- Wade SB, Oommen P, Conner WC, Earnest DJ, Miranda RC. Overlapping and divergent actions of estrogen and the neurotrophins on cell fate and p53-dependent signal transduction in conditionally immortalized cerebral cortical neuroblasts. J Neurosci. 1999;15(19):6994–7006. doi: 10.1523/JNEUROSCI.19-16-06994.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Maniati M, Jabado O, Pavlaki M, Troy CM, Greene LA, Stefanis L. RAIDD is required for apoptosis of PC12 cells and sympathetic neurons induced by trophic factor withdrawal. Cell Death Differ. 2006;13(1):75–83. doi: 10.1038/sj.cdd.4401690. [DOI] [PubMed] [Google Scholar]
- Wass TS, Persutte WH, Hobbins JC. The impact of prenatal alcohol exposure on frontal cortex development in utero. Am J Obstet Gynecol. 2001;185:737–742. doi: 10.1067/mob.2001.117656. [DOI] [PubMed] [Google Scholar]
- Yeo W, Gautier J. A role for programmed cell death during early neurogenesis in Xenopus. Developmental Biology. 2003;260:31–45. doi: 10.1016/s0012-1606(03)00222-7. [DOI] [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin Y-Q, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death and Differentiation. 2003;10:1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]