

Abstract
DNA mismatch repair (MMR) corrects mismatched base pairs mainly caused by DNA replication errors. The fundamental mechanisms and proteins involved in the early reactions of MMR are highly conserved in almost all organisms ranging from bacteria to human. The significance of this repair system is also indicated by the fact that defects in MMR cause human hereditary nonpolyposis colon cancers as well as sporadic tumors. To date, 2 types of MMRs are known: the human type and Escherichia coli type. The basic features of the former system are expected to be universal among the vast majority of organisms including most bacteria. Here, I review the molecular mechanisms of eukaryotic and bacterial MMR, emphasizing on the similarities between them.
1. Introduction
DNA mismatch repair (MMR) is a highly conserved DNA repair system (Table 1) that greatly contributes to maintain genome stability through the correction of mismatched base pairs. The major source of mismatched base pairs is replication error, although it can arise also from other biological processes [1]. In Escherichia coli, MMR increases the accuracy of DNA replication by 20–400-fold [2]. Mutations and epigenetic silencing in MMR genes have been implicated in up to 90% of human hereditary nonpolyposis colon cancers [3–8], indicating the significance of this repair system. Postreplicative MMR is performed by the long-patch MMR mechanism in which multiple proteins are involved and a relatively long tract of the oligonucleotide is excised during the repair reaction [9, 10]. In contrast, particular kinds of mismatched base pairs are repaired through very short-patch MMR in which a short oligonucleotide tract is excised to remove the lesion [11–13]. In this paper, I refer to long-patch MMR as MMR.
Table 1.
Distribution of MMR proteins.
| Molecular function | Thermus thermophilus | Escherichia coli | Saccharomyces cerevisiae | Homo sapiens |
|---|---|---|---|---|
| Mismatch recognition | MutS | MutS | MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) | MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) |
| Strand incision | β-clamp∗1 | — | PCNA | PCNA |
| clamp-loader∗1 | RFC | RFC | ||
| MutL | MutLα (MLH1/PMS1) MutLγ ∗2 (MLH1/MLH3) | MutLα (MLH1/PMS2) MutLγ ∗2 (MLH1/MLH3) | ||
| Strand incision | — | MutH | — | — |
| Match making | MutL | MutL | MutLα (MLH1/PMS1) MutLβ (MLH1/MLH2) MutLγ (MLH1/MLH3) | MutLα (MLH1/PMS2) MutLβ (MLH1/PMS1) MutLγ (MLH1/MLH3) |
| Strand excision (single-stranded DNA-binding) | SSB | SSB | RPA | RPA |
| Strand excision (exonuclease) |
RecJ ExoI |
RecJ ExoI ExoVII ExoX |
EXO1∗3 | EXO1∗3 |
| Strand excision (helicase) | UvrD |
UvrD |
— | — |
| Repair synthesis | DNA polymerase III | DNA polymerase III | DNA polymerase δ | DNA polymerase δ |
∗1The involvement of bacterial clamp and clamp-loader in the strand incision reaction has not yet been confirmed. ∗2It is demonstrated that the endonuclease motif in MLH3 is responsible for in vivo MMR [83]; however, the endonuclease activity of MutLγ has not yet been confirmed biochemically. ∗3In yeast and human, EXO1 has the 5′-flap endonuclease activity in addition to 5′–3′ exonuclease activity.
Currently, 2 types of MMR mechanisms have been elucidated: one is expected to be employed by eukaryotes and the majority of bacteria, and the other is specific to E. coli and closely related bacteria. As shown in Figures 1(a) and 1(b), MMR in eukaryotes and most bacteria directs the repair to the error-containing strand of the mismatched duplex by recognizing the strand discontinuities. On the other hand, as shown in Figure 1(c), E. coli MMR reads the absence of methylation as a strand discrimination signal. In both MMR systems, strand discrimination is conducted by nicking endonucleases. MutL homologues from eukaryotes and most bacteria incise the discontinuous strand to introduce the entry or termination point for the excision reaction. In E. coli, MutH nicks the unmethylated strand of the duplex to generate the entry point of excision.
Figure 1.

A schematic representation of MMR pathway models. (a) Eukaryotic MMR. A DNA mismatch is generated by the misincorporation of a base during DNA replication. MutSα recognizes base-base mismatches and MutLα nicks the 3′- or 5′-side of the mismatched base on the discontinuous strand. The resulting DNA segment is excised by the EXO1 exonuclease, in cooperation with the single-stranded DNA-binding protein RPA. The DNA strand is resynthesized by DNA polymerase δ and DNA ligase 1. (b) MMR in mutH-less bacteria. Mismatched bases are recognized by MutS. After the incision of discontinuous strand by MutL, the error-containing DNA strand is removed by the cooperative functions of DNA helicases, such as UvrD, the exonucleases RecJ and ExoI, and the single-stranded DNA-binding protein SSB. DNA polymerase III and DNA ligase fill the gap to complete the repair. (c) E. coli MMR. MutS recognizes mismatched bases, and MutL interacts with and stabilizes the complex. Then, MutH endonuclease is activated to incise the unmethylated GATC site to create an entry point for the excision reaction. DNA helicase, a single-stranded DNA-binding protein, and several exonucleases are involved in the excision reaction. PDB IDs of crystal structures in this figure are 2O8B (human MutSα), 1H7S (human MutLα), 1L1O (human RPA), 3IAY (human DNA polymerase δ), 1X9N (human DNA ligase 1), 1E3M (bacterial MutS), 1B63 (bacterial MutL), 2AZO (E. coli MutH), 2ISI (bacterial UvrD), 2ZXO (bacterial RecJ), 3C95 (bacterial ExoI), 2CWA (bacterial SSB), 2HQA (bacterial DNA polymerase III), and 2OWO (bacterial DNA ligase).
Here, I first review the overviews of MMR systems in E. coli, eukaryotes, and the majority of bacteria. Second, I refer to the molecular features of MutS and MutL homologues, the key enzymes in MMR. Third, molecular mechanisms for strand-discrimination and excision reaction are discussed. Finally, I introduce the cellular functions of MMR other than postreplication mismatch correction.
2. Overview of Methyl-Directed MMR in E. coli
The E. coli MMR system has been well characterized and reconstituted using recombinant proteins (Figure 1(c)) [14]. In this system, a mismatched base is recognized by a MutS homodimer. A MutL homodimer interacts with the MutS-DNA complex, and then a MutH restriction endonuclease is activated by MutL. The MMR system needs to discriminate the newly-synthesized/error-containing strand; however, a mismatched base itself contains no such signal. The E. coli MMR system utilizes the absence of methylation at the restriction site to direct the repair to the newly synthesized strand. MutH nicks the unmethylated strand at the hemimethylated GATC site to introduce an entry point for the excision reaction [15–17]. The error-containing strand is removed by helicases [18] and exonucleases [19–21], and a new strand is synthesized by DNA polymerase III and ligase. The absence of methylation serves as a signal for the discrimination of the error-containing strand, and hence, E. coli MMR is called methyl-directed MMR. Although homologues of E. coli MutS and MutL exist in almost all organisms, no homologue of E. coli MutH has been identified in the majority of organisms including eukaryotes and most bacteria (Table 1) [22].
3. Overview of Eukaryotic MMR
In eukaryotes, strand discontinuity serves as a signal that directs MMR to the discontinuous strand of a mismatched duplex (Figure 1(a)). In newly synthesized strands, discontinuities can exist as 3′-ends or termini of Okazaki fragments. These termini of the DNA strand may function as strand discrimination signals in vivo. For the biochemical characterization of eukaryotic MMR, nicked plasmid DNAs have been used as a model substrate containing a strand discontinuity, on which several reviews elaborated [10, 23, 24]. For this assay system, the shorter path from a nick to the mismatch is removed by the excision reaction, indicating that 5′- and 3′-directed MMRs are distinct (Figure 2) [25–27]. Intriguingly, the 5′–3′ exonuclease activity of exonuclease 1 (EXO1) is required for both 5′- and 3′-directed strand removals [28, 29]. The reason for the apparently contradictory requirement of 5′–3′ exonuclease activity for the 3′-discontinuity-directed excision reaction had been unknown because it was thought that strand discontinuity was also the entry point for excision reaction. This issue was resolved by the discovery that the human MutL homologue MutLα (MLH1-PMS2 heterodimer) and yeast MutLα (MLH1-PMS1 heterodimer) harbor latent endonuclease activity that nicks the discontinuous strand of the mismatched duplex in a MutSα-, PCNA-, RFC-, and ATP-dependent manner (Figure 3) [10, 30, 31]. In eukaryotic 5′- and 3′-directed MMR, the 3′- and 5′-sides of a mismatch are incised, respectively, by MutLα, and the 5′–3′ exonuclease activity of EXO1 removes the DNA segment spanning the mismatch, that is, in eukaryotic 3′-directed MMR, the preexisting strand discontinuity does not serve as an entry point for the excision reaction, and the entry point is introduced by the endonuclease activity of MutLα (Figure 3(b)).
Figure 2.

Bidirectionality of eukaryotic MMR. The 5′-nicked (a) and 3′-nicked (b) heteroduplexes are used as model substrate. The shorter path is chosen to remove the mismatched base. The 5′–3′ exonuclease activity of EXO1 is required for excision reaction in both 5′- and 3′-nicked heteroduplexes.
Figure 3.

The 5′-nick-(a) and 3′-nick-(b) directed eukaryotic MMR. After recognition of a mismatched base by MutSα, MutLα incises the discontinuous strand of the heteroduplex in a mismatch-MutSα-, PCNA-, RFC-, and ATP-dependent manner [10, 30, 31]. Incisions by MutLα occur dominantly on the distal side of the mismatched base relative to the pre-existed strand break although it can also occur proximal to the mismatch [30].
Intriguingly, MutLα is essential for 3′-nick-directed MMR but it is dispensable for 5′-nick-directed MMR, although MutLα possesses an enhancing effect on 5′-nick-directed MMR [32, 33]. This result indicates that there is an alternative pathway for 5′-nick-directed MMR. In vitro experiments suggest that MutLα-independent 5′-nick-directed human MMR requires at least 3 proteins, MutSα, EXO1, and replication protein A (RPA) [32].
4. Overview of MMR in mutH-Less Bacteria
The DQHA(X)2E(X)4 motif in the C-terminal domain of the PMS2 subunit of human MutLα comprises a metal-binding site that is essential for the endonuclease activity of MutLα and MMR activity of nuclear extract [30]. Except for E. coli and closely related bacteria, most mutH-lacking bacteria possess MutL homologues that contain this metal-binding motif [30, 34]. Therefore, the molecular mechanism established on the basis of the results obtained from eukaryotic MMR systems is expected to be universal for organisms lacking mutH. In agreement with this prediction, several studies demonstrated that MutL homologues from mutH-less bacteria, for example, Thermus thermophilus, Aquifex aeolicus, and Neisseria gonorrhoeae, possess endonuclease activity that is dependent on the DQHA(X)2E(X)4 motif [34–36]. Furthermore, in T. thermophilus, it is clarified that the endonuclease activity of MutL is essential for in vivo DNA repair activity [34]. Thus, the molecular mechanism of MMR in mutH-less bacteria is expected to resemble that of eukaryotic one (Figure 1(b)).
5. Molecular Functions of MutS Homologues
The amino acid sequence of MutS is highly conserved among bacteria regardless of the presence and absence of mutH [37–39]. Bacterial MutS forms a homodimer and recognizes mispaired bases and short insertion/deletion loops [9, 40, 41]. Eukaryotes possess several mismatch-recognizing MutS homologues: MSH2, MSH3, and MSH6. These 3 homologues contain amino acid sequences homologous to bacterial MutS and form 2 heterodimers, namely, MutSα (MSH2/MSH6) and MutSβ (MSH2/MSH3). MutSα recognizes single base-base mismatches and short insertion/deletion loops while MutSβ is responsible for the repair of relatively large insertion/deletion loops that contain ~16-mer excess nucleotides [42].
The crystal structures of C-terminal dimerization domain-deleted bacterial MutS [37, 38] and human MutSα [43] were solved in complex with mismatched DNA (Figure 4). The structures revealed that the mismatch-recognition mechanisms of bacterial MutS and eukaryotic MutSα fundamentally resemble each other. The bacterial MutS homodimer/DNA complex shows a functionally asymmetric protein dimer in which only 1 of the 2 subunits recognizes the mismatched or unpaired base. Thus, although bacterial MutS binds to double-stranded DNA as a homodimer, its functionality is heterodimeric.
Figure 4.

Crystal structures of MutS-mismatch complex. (a) Crystal structure of E. coli MutS bound to a G:T-mismatched heteroduplex (PDB ID: 1E3M). One of the 2 subunits of E. coli MutS is shown in color. DNA is shown in salmon. Domains I, II, III, IV, and V are shown in red, pink, violet, purple blue, and blue, respectively. Domains I, IV, and V are responsible for mismatch recognition, double-stranded DNA binding, and dimerization/ATP binding, respectively. (b) The mismatch-recognition site in E. coli MutS-G:T mismatch complex. The mismatch-recognizing phenylalanine residue (Phe36) and G:T mismatch are shown in red and blue, respectively. (c) Crystal structure of human MutSα (PDB ID: 2O8B), which is comprised of full-length MSH2 and a protease-resistant fragment of MSH6 lacking the first 340 amino acid residues. MSH2 is shown in white and MSH6 is in color. Mismatch-binding, Connector, Levers, Clamp, and ATPase domains are colored in red, pink, magenta, purple, and blue, respectively. (d) The mismatch-recognition site in human MutSα-G:T mismatch complex. Phe432 and G:T mismatch is shown in red and blue, respectively.
Bacterial MutS and human MutSα recognize the heteroduplex by stacking the mismatched base with a phenylalanine residue (Phe36 and Phe432 in E. coli MutS and human MSH6, resp.) that is intercalated from the minor groove side [38] (Figure 4). This phenylalanine residue is perfectly conserved among bacterial MutS homologues and eukaryotic MSH6 [37], and the alteration of this residue to an alanine results in a drastic decrease in the mismatch-recognition ability of Thermus aquaticus MutS [49]. The adjacent glutamate residue (Glu38 and Glu434 in E. coli MutS and human MSH6, resp.) also plays a central role in mismatch recognition by forming a hydrogen bond with the mismatched base (Figures 5(a)–3(c)) [38]. This glutamate residue is also conserved among bacterial MutS and eukaryotic MSH6 [37], and the mutation of this glutamate into an alanine or a glutamine abolishes in vivo MMR activity [50–52]. Elimination of this hydrogen bond by replacing the thymidine with 2,4-difluorotoluene, which lacks the N3, resulted in the decrease in mismatch selectivities of E. coli MutS and yeast MutSα [50, 52].
Figure 5.

Mismatch recognition mode of MutS. (a) G:T mismatch bound to E. coli MutS (PDB ID: 1E3M). (b) C:A mismatch bound to E. coli MutS (PDB ID: 1OH5). Cytosine residue is in a syn conformation. (c) G:T mismatch bound to human MutSα (PDB ID: 2O8B). (d) Side view of the E. coli MutS-mismatch complex (PDB ID: 1E3M). The mismatched duplex is sharply kinked in the complex with MutS. MutS and mismatched DNA are colored grey and red, respectively. The mismatched G and T are shown in the sphere model.
The excellent crystallographic analyses of E. coli MutS complexes with various kinds of mismatched heteroduplexes remarkably enhanced our understanding of the mismatch recognition mechanism at an atomic resolution [53]. MutS recognizes a wide range of mismatches in a common manner. Heteroduplexes bound by MutS homologues are sharply kinked with the minor groove wide-opened (Figure 5(d)), which allows the mismatched base to be in close contact with the phenylalanine and the glutamate residues of the mismatch-recognition domain. The carbonyl oxygen (OE2) of Glu38 in E. coli MutS forms a hydrogen bond with N3 of mismatched pyrimidine or N7 of mismatched purine base [53]. It is expected that N7 of mismatched purine or OE2 of the glutamate is protonated. As previously discussed, this might be the reason why the mutation of Glu38 to Gln in E. coli MutS eliminates MMR activity though Gln also can form hydrogen bonds [53]. The acidity of the glutamate but not glutamine might be suitable for the required pK a shift. Interestingly, E38A mutant of E. coli MutS exhibited the enhanced DNA-binding activity to a perfectly matched homoduplex DNA [50]. Schofield et al. proposed the idea that Glu38 would take part in the promotion of DNA kinking by destabilizing the unkinked DNA-MutS complex through the electrostatic repulsion of the Glu side-chain with phosphate backbone [50].
The lack of a crystal structure for MutS homologues with a homoduplex has prevented us from further understanding the mechanism; however, atomic force microscopy is a powerful tool that can be used to investigate the events that occur during the initial substrate binding by MutS homologues. Observations of the E. coli MutS-DNA complex using atomic force microscopy revealed that the DNA is bent at the perfectly matched site while it is bent and unbent at the mismatched site [54]. Taken together with previous biochemical and crystallographic data, it is proposed that MutS nonspecifically binds to DNA and bends it in order to search for a mismatch, when it encounters a mismatch it kinks the DNA at the mismatched site and finally forms an ultimate recognition complex in which the DNA is unbent [54]. In order to explain why an unbent conformation is more stable at a mismatch than at a homoduplex site, Wang et al. speculated the possibility that a mismatched base is flipped out upon mismatch recognition by MutS [54]. Base flipping is one of the major base-recognition mechanisms observed among base-processing enzymes such as DNA glycosylases or methyltransferases [55]. Although further experimental evidence should be required, this idea may be attractive especially when we remember the recent report that a nonenzymatic protein, alkylguanine alkyltransferase-like protein, also flips out the methylated base upon substrate recognition [56, 57].
The mismatch-recognizing property of MutS homologues is closely related to their ATP-binding/hydrolyzing activity [58–63]. MutS homologues contain a Walker's ATP-binding motif in each subunit that is formed through the association of the subunits. It has been shown that E. coli MutS exchanges ADP to ATP upon mismatch binding then undergoes a conformational change to form a sliding clamp [61, 63]. The ATP-binding-dependent formation of the sliding clamp was also confirmed in eukaryotic MutS homologues [63–66]. Several studies revealed that the ATPase activity of the E. coli MutS homodimer is also heterodimeric [67, 68]. One of the 2 nucleotide-binding sites exhibits a high affinity to ADP, and the other shows a high affinity to ATP [60, 69, 70], therefore, we should discriminate between the different adenine nucleotide-binding forms of MutS homologues in MMR. Recently, a computational study using normal-mode analysis was applied to assess the conformational dynamics of E. coli MutS and human MutSα [71]. Normal-mode analysis is one of the simulation methodologies which can deal with the dynamics of large molecules [72]. The analyses suggested the existence of the distinct conformational states which are expected to reflect the functional cycles including DNA scanning, mismatch recognition, repair initiation, and sliding along DNA after mismatch recognition [71]. These computational studies can provide start points for further experiments.
It is thought that the sliding clamp of MutS leaves the mismatch and diffuses along the DNA to activate the downstream reactions, which is called the “moving” or “cis” model [23, 74]. The “moving” model is supported by the result that blockages between the mismatch and the nick prevent in vitro E. coli MMR [75]. In contrast, another mechanism, which is called the “stationary” or “trans” model, is also proposed to account for the activation of downstream reactions [23, 74]. In the “stationary” model, MutS remains bound at the mismatch after recognition, and the ATPase activity of MutS is required for verification of mismatch recognition. The “stationary” model is supported by the observation that MutS-mismatch complex can activate MutH on separate homoduplex molecule [67].
The dynamism and transiency of the ternary complex of MutS (MutSα)-MutL (MutLα)-mismatch had prevented us from characterizing their physical interactions. Hydrogen/deuterium exchange mass spectrometry (DXMS) is suitable to study the interactions in large and transient complexes. DXMS analysis of the formation of E. coli MutS-MutL-mismatch complex revealed that a relatively small region in domain II (Figure 4) of MutS serves as an interface for binding MutL [76]. On the basis of the structural analyses of the MutS and MutL N-terminal domains, Wei Yang and coworkers also predicted this region to be a MutL-interacting site [37]. Although the sequence similarity of this region is limited, a structurally conserved region in the MSH2 subunit of Saccharomyces cerevisiae MutSα is also essential for interaction with MutLα [76].
E. coli MutS proteins exist not only in a dimeric form but also in a tetrameric form at high concentrations [41, 77]. The C-terminal domain (the last 53 amino acids in E. coli MutS) is responsible for the tetramerization of full-length MutS. Although the crystallographic analyses were achieved by using C-terminal truncations, it has been pointed out that the deletion of the C-terminal domain causes defects in mismatch recognition, MutH stimulation, and in vivo MMR activity [77–79]. On the basis of the crystallographic analysis of the C-terminal domain (Figure 6), tetramer-disrupting mutants of E. coli MutS were prepared [44]. Examination of the effects of those mutations revealed that dimerization but not tetramerization of the MutS C-terminal domain is essential for in vivo mismatch repair [44].
Figure 6.
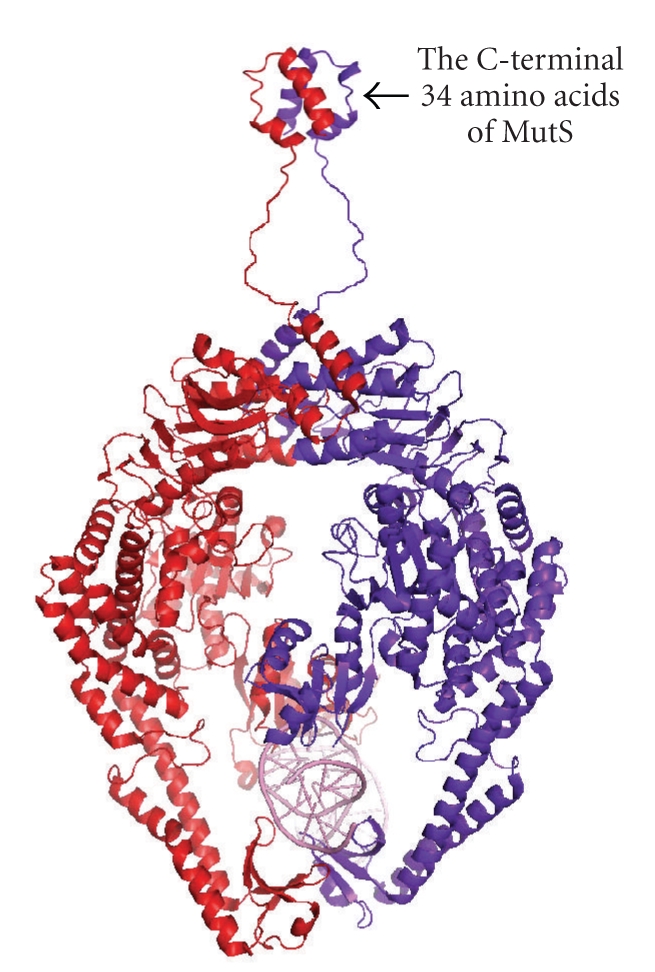
The model for full length of E. coli MutS dimer. The crystal structure of the C-terminal 34 amino acids of E. coli MutS (2OK2) [44] was connected to the C-termini of E. coli MutS (residues 2-800) structure (1E3M).
6. Molecular Functions of MutL Homologues
Bacterial MutL homologues exist as homodimers [47] while eukaryotic MutL homologues form the heterodimers MutLα, MutLβ, and MutLγ [80–83]. It is suggested that a large portion of eukaryotic MMR is performed by MutLα. In contrast, there is only a slight effect of inactivation of MutLβ or MutLγ on in vivo MMR activity. Crystallographic and biochemical analyses revealed that the bacterial MutL homodimer is comprised of an N-terminal ATPase/DNA-binding domain and a C-terminal dimerization/DNA-binding domain (Figure 7) [47, 84]. Eukaryotic MutL homologues are expected to have the same domain organization as bacterial MutL, except for the prolonged interdomain linker (Figure 7) [45, 85].
Figure 7.

(a) A schematic representation of the domain structure of MutL homologues. ATPase, nuclease, and dimer indicate the ATPase, endonuclease, and dimerization domains, respectively. The crystal structures of N-terminal ATPase domain of human PMS2 (PDB ID: 1EA6) [45], ATPase domain of E. coli MutL (PDB ID: 1B63) [46], and C-terminal dimerization domain of E. coli MutL (PDB ID: 1X9Z) [47, 48] are shown.
The N-terminal ATPase domain contains a single ATP-binding motif per subunit. MutL homologues belong to the GHKL ATPase superfamily that includes homologues of DNA gyrase, Hsp90, and histidine kinase in addition to MutL homologues [46, 86]. The GHKL superfamily proteins undergo large conformational changes upon ATP binding and/or hydrolysis. X-ray crystallographic analysis clarified the ATP-binding-dependent conformational change of the N-terminal domain of E. coli MutL (Figure 8(a)) [46]. In addition, study using atomic force microscopy revealed that full-length S. cerevisiae MutLα can exist in several ATP-binding species with specific conformations in a solution [73]. The conformational change of the full-length MutL homologue seems to involve the interaction between the N-terminal and C-terminal domains (Figure 8(c)). ATP-dependent conformational changes are also implicated for bacterial MutL endonucleases [34, 87]. Such ATPase-cycle-dependent conformational changes in MutL homologue should be necessary to perform the MMR reaction [88–90].
Figure 8.

Crystal structures of the apo form ((a) PDB ID: 1BKN) and ADPNP-bound form ((b) PDB ID: 1B63) of the E. coli MutL N-terminal domain. (c) A schematic representation of a model for the ATP-dependent conformational change of full-length MutLα [73]. NTD and CTD indicate the N-terminal ATPase domain and the C-terminal endonuclease domain, respectively. In the apo form of MutLα, the PMS2 and MLH1 subunits dimerize via their C-terminal domains. ATP binding induces the dimerization of the N-terminal domain and condensation of the molecule.
The C-terminal domain of MutL homologues is the endonuclease domain in mutH-less organisms [30, 31]. The inactivation of the metal-binding motif in the C-terminal domain of Homo sapiens PMS2, S. cerevisiae PMS1, T. thermophilus MutL, A. aeolicus MutL, and N. gonorrhoeae MutL diminishes their endonuclease activity [34–36]. Eukaryotic MutLα and bacterial MutL show apparently nonspecific endonuclease activity against lesionless DNA, indicating that MMR requires the sequence- or structure-nonspecific endonuclease activity to introduce excision entry point wherever it is needed [30, 34]. The regulatory mechanism of this apparently nonspecific endonuclease activity has been argued [10, 91].
The endonuclease activity of MutL homologues is affected by the binding of ATP to their N-terminal domain. Isolated T. thermophilus MutL tightly binds ATP in the absence of the MutS-mismatch complex and the ATP-binding form of MutL has decreased endonuclease activity against perfectly matched substrates in vitro [34]. The in vitro endonuclease activities of A. aeolicus and N. gonorrhoeae MutL are also suppressed by the addition of ATP [34, 35]. In addition, T. thermophilus MutL is stably associated with a MutS-mismatch complex in the presence of ATP [34]. Since it has been known that the ATPase activity of MutL is activated by its interaction with MutS, formation of MutS-mismatch-MutL complex is expected to stimulate the endonuclease activity of MutL by canceling the ATP-dependent suppression (Figure 9). In other words, ATP may be utilized to suppress the apparently nonspecific endonuclease activity of MutL until it is required. However, it remains to be determined whether the regulatory mechanism is used by other MutL homologues, including eukaryotic MutLα, because the endonuclease activity of MutLα is enhanced by the addition of ATP instead of being suppressed [30, 31]. Interestingly, it is clarified that ATP stimulates the endonuclease activity of a relatively high concentration of A. aeolicus MutL in the absence of a MutS-mismatch complex [36], suggesting that the effect of ATP on the MutL endonuclease activity depends on the concentration of MutL. This finding also strongly indicates that the ATPase activity of MutL is required for its endonuclease activity, that is, ATP is utilized not only to suppress the nonspecific endonuclease activity of MutL but also to actively enhance its activity.
Figure 9.

A model for the ATPase-cycle-dependent regulation of bacterial MutL endonuclease activity. Free MutL exists as an ATP-bound form whose endonuclease activity is inactive, but preferably binds to a MutS-mismatch complex. The interaction with the MutS-mismatch complex induces the ATP hydrolysis of MutL, resulting in the stimulation of its endonuclease activity. Adapted from the work of Fukui et al. [34].
The C-terminal endonuclease domain of MutL homologues contains the highly conserved motif, CPHGRP. Mutations in this motif result in the deficiency of in vivo and in vitro MMR activities [34, 85]. Bioinformatic analysis indicated that this motif takes part in the formation of the metal-binding motif and it resembles a metal-dependent transcriptional regulator [85]. Using biochemical procedures, the C-terminal domain of the human PMS2 subunit of MutLα was demonstrated to bind a zinc ion [85]. Although the detailed functions of this zinc ion remain unknown, the involvement of the CPHGRP motif in the ATP-dependent conformational change in T. thermophilus MutL is suggested [34].
As mentioned above, recent biochemical studies on the endonuclease activity of MutL homologues have been achieved by using homologues from thermophilic bacteria, such as T. thermophilus, A. aeolicus, and Thermotoga maritima [34, 36, 87, 92]. Proteins from these bacteria are extremely stable and suitable for physicochemical examinations including crystallographic analysis. In addition, a variety of gene manipulating procedures are established in T. thermophilus. T. thermophilus may be one of the ideal model organisms for the study of nick-directed MMR.
7. Strand Discrimination in Nick-Directed MMR
Accumulating evidence indicates that a pre-existing strand break can serve as a signal to discriminate the error-containing strand in eukaryotic MMR [27, 30]. Since newly synthesized strands always contain strand break as 3′-ends or 5′-termini of Okazaki fragments, these ends can be utilized as strand discrimination signals in vivo. This is consistent with the observation that MutSα-dependent yeast MMR corrects mismatches more efficiently in the lagging strand than in the leading strand [93]. As mentioned above, MutLα is responsible for the strand-discrimination by nicking the discontinuous strand of the mismatched duplex (Figures 1 and 3). Interestingly, it is demonstrated that MutLα incises the discontinuous strand at a distal site from the pre-existing strand break. How does MutLα discriminate the discontinuous strand at a site distant from the strand-discrimination signal? One possible explanation is that MutSα (or MutSβ) and MutLα are loaded onto the substrate DNA through their interactions with the replication machinery such as proliferating cell nuclear antigen (PCNA), replication factor C (RFC), and/or DNA polymerase δ to render the newly synthesized strand in the catalytic site of the MutLα endonuclease domain [23, 94, 95]. It is known that MSH3 and MSH6 contain the PCNA-interacting motif QX2(L/I)X2FF [96]. In fact, several studies have demonstrated the associations of MutSα and MutSβ with PCNA [95, 97, 98]. In addition, PCNA and RFC are necessary not only for repair synthesis [99] but also for the mismatch-provoked incision and excision reactions [30, 100]. Inhibitors of PCNA abolish 3′-nick-directed excision and 40–50% of 5′-nick-directed excision [99–101]. The excision reaction of MutLα-independent 5′-nick-directed MMR may be performed independently of PCNA. In mutH-less bacteria, the C-terminal region of MutS contains the putative β-clamp-binding motif QLSFF [102]. The deletion of this region abolishes the in vitro interaction of MutS with the β-clamp and in vivo MMR activity [103]; furthermore, this interaction is necessary for the in vivo localization of MutS and MutL in response to mismatches [103]. These interactions may also be responsible for the strand discrimination in bacterial nick-directed MMR.
8. Downstream Events in Nick-Directed MMR
The excision reaction of in vitro eukaryotic MMR is performed by the 5′–3′ single-stranded DNA-specific exonuclease EXO1. To date, EXO1 is a unique exonuclease that is involved in eukaryotic MMR [30, 104]. In addition, no reports have identified the MMR-related eukaryotic DNA helicase. The exonuclease activity of EXO1 is enhanced by its direct interaction with MutSα [99]. As mentioned above, MutSα forms a sliding clamp and diffuses along the DNA after mismatch recognition. The purpose of the diffusion of MutSα from the mismatch may be to activate EXO1 at the 5′-terminus. In mutH-less bacteria, A. aeolicus MutL enhances the DNA helicase activity of UvrD [105] whose amino acid sequence is ubiquitous among bacteria. Furthermore, a genetic study implied the simultaneous involvements of the 5′–3′ exonuclease RecJ and a 3′–5′ exonuclease ExoI in T. thermophilus MMR [106]. In mutH-less bacteria, the error-containing DNA segment generated by the endonuclease activity of MutL may be removed bi-directionally by the cooperative function of multiple exonucleases and helicases (Figure 10).
Figure 10.

Two parallel pathways of excision reaction in T. thermophilus. RecJ (red) and ExoI (purple) are thought to be responsible for the 5′- and 3′-directed excision, respectively. UvrD helicase (magenta) functions in cooperation with RecJ. DNA helicase (blue) which translocates 5′ to 3′ direction has been unknown.
The termination of the EXO1-dependent excision reaction in eukaryotic 3′-nick-directed and MutLα-dependent 5′-nick-directed MMR can be directed by 3′-termini that are pre-existing and newly introduced by MutLα, respectively (Figure 3). In contrast, excision termination in MutLα-independent 5′-nick-directed MMR appears to employ a relatively complicated mechanism, because the termination of the excision reaction is not directed by the terminus of the DNA strand. Excision termination in MutLα-independent 5′-nick-directed MMR is conducted by the inhibitory function of RPA, a single-stranded DNA-binding protein [32]. In mutH-less bacteria, the mechanism for terminating the excision reaction remains unknown. The implication of the involvements of 5′–3′ and 3′–5′ exonucleases [106] raises the possibility that the excision termination of 5′- and 3′-nick-directed MMR in mutH-less bacteria is directed by the 3′- and 5′-termini of the DNA strand that are introduced by MutL (Figure 10). Biochemical and structural studies on the exonucleases are required to further understand the excision reaction. Structural analyses of the RecJ 5′–3′ exonuclease from T. thermophilus were successfully performed [20, 107]; thus, proteins from T. thermophilus are known to be suitable for physicochemical examinations [108, 109]. The structure of RecJ consists of 4 domains that form a ring-like structure with the catalytic site in the center of the ring (Figure 11). Based on this structure, the molecular basis for the high processivity and substrate specificity of this enzyme was discussed [107]. In addition, a detailed biochemical study on the T. thermophilus ExoI was also performed [106]. It is expected that, unlike other model organisms, T. thermophilus possesses only a single set of 5′–3′ and 3′–5′ exonucleases [106]. Hence, in vitro and in vivo experiments concerning the excision reaction would be relatively straightforward in this bacterium. It will be intriguing to examine whether MutS can stimulate the exonuclease activities of RecJ and ExoI.
Figure 11.

Crystal structure of T. thermophilus RecJ (PDB ID: 2ZXR). Full-length T. thermophilus RecJ is comprised of domains I–IV and forms a ring-like structure. The catalytic active site is located in the cavity between domains I and II as indicated by an arrow. Domain III shows a structural similarity to the oligonucleotide/oligosaccharide-binding fold that is often found in single-stranded DNA-binding proteins. The ring-like structure and oligonucleotide/oligosaccharide-binding fold will ensure the high processivity and strict specificity for single-stranded DNA.
9. Other Functions
Mismatch base pairs can arise not only from replication error but also from other biological processes including homeologous recombination, oxidation, and methylation of bases. Long-patch MMR also has a role in the repair machinery for those mismatches.
The involvement of MMR proteins in the suppression of homeologous recombination, that is, the strand exchange between nonidentical DNA molecules, has been reported [110, 111]. Inhibition of homeologous recombination contributes to genome integrity by limiting the invasion of foreign replicons and the excessive intracellular rearrangement of genome. Although the requirement of MutS and MutL homologues for this inhibitory function has been demonstrated, the downstream reactions following the recognition of the mismatch had not been described. Recently, it is suggested that the endonuclease activity of MutLα is required for this system [112].
Oxidative damage is one of the major spontaneously arising forms of DNA damage. Aerobic cells yield reactive oxygen species via respiration events that attack biomolecules such as proteins, lipids, and DNAs [113]. The attack of reactive oxygen species on DNA bases generates oxidized bases including 8-oxoguanine (8OG) (Figure 12) [114]. An 8OG base can pair not only with cytosine but also with adenine [115]. An 8OG:A pair can be converted to a T:A pair through replication, forming a G:C-T:A transversion mutation. To prevent this mutational process, base-excision repair employs MutM (OGG1 in eukaryotes) and MutY (MYH in eukaryotes) glycosylases that excise 8OG and adenine from 8OG:C and 8OG:A pairs, respectively [115]. In vitro and in vivo studies indicated that bacterial and eukaryotic MMR can recognize an 8OG:A pair as a substrate and remove the adenine residue because the mismatched adenine residue exists in the newly synthesized strand [116, 117], that is, MMR can perform the same role as the MutY (MYH)-dependent base-excision repair pathway (Figure 12).
Figure 12.

Prevention of 8OG-induced G:C-T:A transversion mutations. The 8OG base is one of the major forms of oxidative DNA damage and can be generated by reactive oxygen species. Since 8OG can pair not only with C but also with A, it causes a G:C-T:A transversion through DNA replication. MutM (OGG1)- and MutY (MYH)-dependent base-excision repair pathways are known to remove the 8OG and A from 8OG:C and 8OG:A pairs, respectively. MMR is also responsible for the removal of A from 8OG.
O 6-methylguanine (O 6MeG) is generated by the action of S N1-alkylating agents, such as N-methyl-N-nitrosourea and N-methyl-N′-nitro-N-nitrosoguanidine that are used in cancer chemotherapy [118]. O 6MeG can pair with thymine, resulting in a G:C-A:T transition mutation through replication. Although the major repair activity for O 6MeG is derived from O 6MeG methyltransferase and/or its homologues [56, 119], MutSα can also bind to an O 6MeG:T mismatch [43, 120]. However, if the O 6MeG is in the template strand of the duplex, MMR does not remove the lesion. The accumulation of the complex of unrepaired O 6MeG with MMR proteins is thought to result in the induction of apoptosis [121] through the crosstalk between MMR proteins and check point kinases [120].
10. Conclusions
In this paper, the molecular mechanism of widely conserved human-type MMR has been described. Since the accumulating evidence indicates the similarities of basic features among bacterial and eukaryotic MMR, mutH-less bacteria may serve as a model organism for biochemical and structural studies of MMR proteins.
The structural analyses on initial recognition complex should be required for further understanding of mismatch-recognition mechanism of MutS homologues. It would be beneficial to identify the amino acid residue which is responsible for the kinking of heteroduplex by MutS homologues. Such mutant might be utilized for the crystallographic analysis of the initial recognition complex. In order to understand the ATP-dependent functional cycles of MutL endonucleases, structural analyses on the full-length MutL homolgoues should be necessary. Since MutL endonucleases from several thermophilic bacteria have relatively short linker region between N-terminal and C-terminal domains [34, 36, 92, 105], these proteins are expected to be suitable for crystallographic analyses. However, we should not underestimate the importance of this interdomain linker region. This region might be responsible for the protein-protein interaction or domain-domain interaction just like intrinsically disordered proteins [122]. It also remains to be clarified how MutL endonucleases discriminate the discontinuous strand of mismatched heteroduplex. Further experiments using in vitro reconstituted system may provide the key findings to understand the mechanism. Discrimination between the “moving” and “stationary” models should also be argued in this context.
Acknowledgments
The author thanks Drs. Seiki Kuramitsu and Taisuke Wakamatsu for critical reading of this paper.
References
- 1.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. 2nd edition. Washington, DC, USA: American Society for Microbiology; 2005. [Google Scholar]
- 2.Schaaper RM. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli . Journal of Biological Chemistry. 1993;268(32):23762–23765. [PubMed] [Google Scholar]
- 3.Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Current Opinion in Genetics and Development. 1995;5(3):382–395. doi: 10.1016/0959-437x(95)80055-7. [DOI] [PubMed] [Google Scholar]
- 4.Suter CM, Martin DIK, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nature Genetics. 2004;36(5):497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
- 5.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Research. 1997;57(5):808–811. [PubMed] [Google Scholar]
- 6.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annual Review of Biochemistry. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 7.Fishel R, Lescoe MK, Rao MRS, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75(5):1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 8.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75(6):1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 9.Lyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chemical Reviews. 2006;106(2):302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 10.Modrich P. Mechanisms in eukaryotic mismatch repair. Journal of Biological Chemistry. 2006;281(41):30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hennecke F, Kolmar H, Brundl K, Fritz H-J. The vsr gene product of E. coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature. 1991;353(6346):776–778. doi: 10.1038/353776a0. [DOI] [PubMed] [Google Scholar]
- 12.Tsutakawa SE, Jingami H, Morikawa K. Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell. 1999;99(6):615–623. doi: 10.1016/s0092-8674(00)81550-0. [DOI] [PubMed] [Google Scholar]
- 13.Tsutakawa SE, Muto T, Kawate T, et al. Crystallographic and functional studies of very short patch repair endonuclease. Molecular Cell. 1999;3(5):621–628. doi: 10.1016/s1097-2765(00)80355-x. [DOI] [PubMed] [Google Scholar]
- 14.Lahue RS, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245(4914):160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 15.Modrich P. Methyl-directed DNA mismatch correction. Journal of Biological Chemistry. 1989;264(12):6597–6600. [PubMed] [Google Scholar]
- 16.Längle-Rouault F, Maenhaut-Michel G, Radman M. GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO Journal. 1987;6(4):1121–1127. doi: 10.1002/j.1460-2075.1987.tb04867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lahue RS, Modrich P. Methyl-directed DNA mismatch repair in Escherichia coli . Mutation Research. 1988;198(1):37–43. doi: 10.1016/0027-5107(88)90037-1. [DOI] [PubMed] [Google Scholar]
- 18.Mechanic LE, Frankel BA, Matson SW. Escherichia coli MutL loads DNA helicase II onto DNA. Journal of Biological Chemistry. 2000;275(49):38337–38346. doi: 10.1074/jbc.M006268200. [DOI] [PubMed] [Google Scholar]
- 19.Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(12):6765–6770. doi: 10.1073/pnas.121183298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamagata A, Kakuta Y, Masui R, Fukuyama K. The crystal structure of exonuclease RecJ bound to Mn2+ ion suggests how its characteristic motifs are involved in exonuclease activity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(9):5908–5912. doi: 10.1073/pnas.092547099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamagata A, Masui R, Kakuta Y, Kuramitsu S, Fukuyama K. Overexpression, purification and characterization of RecJ protein from Thermus thermophilus HB8 and its core domain. Nucleic Acids Research. 2001;29(22):4617–4624. doi: 10.1093/nar/29.22.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisen JA. A phylogenomic study of the MutS family of proteins. Nucleic Acids Research. 1998;26(18):4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel TA, Erie DA. DNA mismatch repair. Annual Review of Biochemistry. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 24.Spampinato CP, Gomez RL, Galles C, Lario LD. From bacteria to plants: a compendium of mismatch repair assays. Mutation Research. 2009:110–128. doi: 10.1016/j.mrrev.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Fang W-H, Modrich P. Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. Journal of Biological Chemistry. 1993;268(16):11838–11844. [PubMed] [Google Scholar]
- 26.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Molecular Cell. 2004;15(1):31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. Journal of Biological Chemistry. 2005;280(48):39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. Journal of Biological Chemistry. 2002;277(15):13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- 29.Wei K, Clark AB, Wong E, et al. Inactivation of exonuclease I in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes and Development. 2003;17(5):603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of mutLα in human mismatch repair. Cell. 2006;126(2):297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 31.Kadyrov FA, Holmes SF, Arana ME, et al. Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. Journal of Biological Chemistry. 2007;282(51):37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genschel J, Modrich P. Functions of MutLα, replication protein A (RPA), and HMGB1 in 5′-directed mismatch repair. Journal of Biological Chemistry. 2009;284(32):21536–21544. doi: 10.1074/jbc.M109.021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drummond JT, Anthoney A, Brown R, Modrich P. Cisplatin and adriamycin resistance are associated with MutLα and mismatch repair deficiency in an ovarian tumor cell line. Journal of Biological Chemistry. 1996;271(33):19645–19648. doi: 10.1074/jbc.271.33.19645. [DOI] [PubMed] [Google Scholar]
- 34.Fukui K, Nishida M, Nakagawa N, Masui R, Kuramitsu S. Bound nucleotide controls the endonuclease activity of mismatch repair enzyme MutL. Journal of Biological Chemistry. 2008;283(18):12136–12145. doi: 10.1074/jbc.M800110200. [DOI] [PubMed] [Google Scholar]
- 35.Duppatla V, Bodda C, Urbanke C, Friedhoff P, Rao DN. The C-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochemical Journal. 2009;423(2):265–277. doi: 10.1042/BJ20090626. [DOI] [PubMed] [Google Scholar]
- 36.Mauris J, Evans TC., Jr. Adenosine triphosphate stimulates Aquifex aeolicus MutL endonuclease activity. PLoS ONE. 2009;4(9, article no. e7175) doi: 10.1371/journal.pone.0007175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407(6805):703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 38.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, De Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G·T mismatch. Nature. 2000;407(6805):711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 39.Tachiki H, Kato R, Masui R, et al. Domain organization and functional analysis of Thermus thermophilus MutS protein. Nucleic Acids Research. 1998;26(18):4153–4159. doi: 10.1093/nar/26.18.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tachiki H, Kato R, Kuramitsu S. DNA binding and protein-protein interaction sites in MutS, a mismatched DNA recognition protein from Thermus thermophilus HB8. Journal of Biological Chemistry. 2000;275(52):40703–40709. doi: 10.1074/jbc.M007124200. [DOI] [PubMed] [Google Scholar]
- 41.Takamatsu S, Kato R, Kuramitsu S. Mismatch DNA recognition protein from an extremely thermophilic bacterium, Thermus thermophilus HB8. Nucleic Acids Research. 1996;24(4):640–647. doi: 10.1093/nar/24.4.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCulloch SD, Gu L, Li G-M. Nick-dependent and -independent processing of large DNA loops in human cells. Journal of Biological Chemistry. 2003;278(50):50803–50809. doi: 10.1074/jbc.M309025200. [DOI] [PubMed] [Google Scholar]
- 43.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese L. Structure of the human MutSα DNA lesion recognition complex. Molecular Cell. 2007;26(4):579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 44.Mendillo ML, Putnam CD, Kolodner RD. Escherichia coli MutS tetramerization domain structure reveals that stable dimers but not tetramers are essential for DNA mismatch repair in vivo . Journal of Biological Chemistry. 2007;282(22):16345–16354. doi: 10.1074/jbc.M700858200. [DOI] [PubMed] [Google Scholar]
- 45.Guarné A, Junop MS, Yang W. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATpase. EMBO Journal. 2001;20(19):5521–5531. doi: 10.1093/emboj/20.19.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97(1):85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 47.Guarné A, Ramon-Maiques S, Wolff EM, et al. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO Journal. 2004;23(21):4134–4145. doi: 10.1038/sj.emboj.7600412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kosinski J, Steindorf I, Bujnicki JM, Giron-Monzon L, Friedhoff P. Analysis of the quaternary structure of the MutL C-terminal domain. Journal of Molecular Biology. 2005;351(4):895–909. doi: 10.1016/j.jmb.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 49.Malkov VA, Biswas I, Camerini-Otero RD, Hsieh P. Photocross-linking of the NH2-terminal region of Taq MutS protein to the major groove of a heteroduplex DNA. Journal of Biological Chemistry. 1997;272(38):23811–23817. doi: 10.1074/jbc.272.38.23811. [DOI] [PubMed] [Google Scholar]
- 50.Schofield MJ, Brownewell FE, Nayak S, Du C, Kool ET, Hsieh P. The Phe-X-Glu DNA binding motif of MutS. The role of hydrogen bonding in mismatch recognition. Journal of Biological Chemistry. 2001;276(49):45505–45508. doi: 10.1074/jbc.C100449200. [DOI] [PubMed] [Google Scholar]
- 51.Junop MS, Yang W, Funchain P, Clendenin W, Miller JH. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair. 2003;2(4):387–405. doi: 10.1016/s1568-7864(02)00245-8. [DOI] [PubMed] [Google Scholar]
- 52.Drotschmann K, Yang W, Brownewell FE, Kool ET, Kunkel TA. Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2-Msh6. Journal of Biological Chemistry. 2001;276(49):46225–46229. doi: 10.1074/jbc.C100450200. [DOI] [PubMed] [Google Scholar]
- 53.Natrajan G, Lamers MH, Enzlin JH, Winterwerp HHK, Perrakis A, Sixma TK. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Research. 2003;31(16):4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Yang Y, Schofield MJ, et al. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts RJ, Cheng X. Base flipping. Annual Review of Biochemistry. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 56.Morita R, Nakagawa N, Kuramitsu S, Masui R. An O 6-methylguanine-DNA methyltransferase-like protein from Thermus thermophilus interacts with a nucleotide excision repair protein. Journal of Biochemistry. 2008;144(2):267–277. doi: 10.1093/jb/mvn065. [DOI] [PubMed] [Google Scholar]
- 57.Tubbs JL, Latypov V, Kanugula S, et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459(7248):808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blackwell LJ, Martik D, Bjornson KP, Bjornson ES, Modrich P. Nucleotide-promoted release of hMutSα from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. Journal of Biological Chemistry. 1999;273(48):32055–32062. doi: 10.1074/jbc.273.48.32055. [DOI] [PubMed] [Google Scholar]
- 59.Blackwell LJ, Bjornson KP, Allen DJ, Modrich P. Distinct MutS DNA-binding modes that are differentially modulated by ATP binding and hydrolysis. Journal of Biological Chemistry. 2001;276(36):34339–34347. doi: 10.1074/jbc.M104256200. [DOI] [PubMed] [Google Scholar]
- 60.Bjornson KP, Modrich P. Differential and simultaneous adenosine di- and triphosphate binding by MutS. Journal of Biological Chemistry. 2003;278(20):18557–18562. doi: 10.1074/jbc.M301101200. [DOI] [PubMed] [Google Scholar]
- 61.Acharya S, Foster PL, Brooks P, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Molecular Cell. 2003;12(1):233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 62.Kato R, Kataoka M, Kamikubo H, Kuramitsu S. Direct observation of three conformations of MutS protein regulated by adenine nucleotides. Journal of Molecular Biology. 2001;309(1):227–238. doi: 10.1006/jmbi.2001.4752. [DOI] [PubMed] [Google Scholar]
- 63.Mendillo ML, Putnam CD, Mo AO, et al. Probing DNA- and ATP-mediated conformational changes in the MutS family of mispair recognition proteins using deuterium exchange mass spectrometry. Journal of Biological Chemistry. 2010;285(17):13170–13182. doi: 10.1074/jbc.M110.108894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Molecular Cell. 1999;3(2):255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 65.Iaccarino I, Marra G, Dufner P, Jiricny J. Mutation in the magnesium binding site of hMSH6 disables the hMutSα sliding clamp from translocating along DNA. Journal of Biological Chemistry. 2000;275(3):2080–2086. doi: 10.1074/jbc.275.3.2080. [DOI] [PubMed] [Google Scholar]
- 66.Jiang J, Bai L, Surtees JA, Gemici Z, Wang MD, Alani E. Detection of high-affinity and sliding clamp modes for MSH2-MSH6 by single-molecule unzipping force analysis. Molecular Cell. 2005;20(5):771–781. doi: 10.1016/j.molcel.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 67.Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Molecular Cell. 2001;7(1):1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 68.Lamers MH, Winterwerp HHK, Sixma TK. The alternating ATPase domains of MutS control DNA mismatch repair. EMBO Journal. 2003;22(3):746–756. doi: 10.1093/emboj/cdg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martik D, Baitinger C, Modrich P. Differential specificities and simultaneous occupancy of human MutSα nucleotide binding sites. Journal of Biological Chemistry. 2004;279(27):28402–28410. doi: 10.1074/jbc.M312108200. [DOI] [PubMed] [Google Scholar]
- 70.Antony E, Hingorani MM. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry. 2003;42(25):7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mukherjee S, Law SM, Feig M. Deciphering the mismatch recognition cycle in MutS and MSH2-MSH6 using normal-mode analysis. Biophysical Journal. 2009;96(5):1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tama F, Valle M, Frankt J, Brooks CL., III Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9319–9323. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine nucleotide-induced conformational changes in MutLα . Molecular Cell. 2008;29(1):112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Research. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 75.Pluciennik A, Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(31):12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mendillo ML, Hargreaves VV, Jamison JW, et al. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22223–22228. doi: 10.1073/pnas.0912250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bjornson KP, Blackwell LJ, Sage H, Baitinger C, Allen D, Modrich P. Assembly and molecular activities of the MutS tetramer. Journal of Biological Chemistry. 2003;278(36):34667–34673. doi: 10.1074/jbc.M305513200. [DOI] [PubMed] [Google Scholar]
- 78.Calmann MA, Nowosielska A, Marinus MG. Separation of mutation avoidance and antirecombination functions in an Escherichia coli mutS mutant. Nucleic Acids Research. 2005;33(4):1193–1200. doi: 10.1093/nar/gki263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calmann MA, Nowosielska A, Marinus MG. The MutS C terminus is essential for mismatch repair activity in vivo . Journal of Bacteriology. 2005;187(18):6577–6579. doi: 10.1128/JB.187.18.6577-6579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flores-Rozas H, Kolodner RD. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(21):12404–12409. doi: 10.1073/pnas.95.21.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lipkin SM, Wang V, Jacoby R, et al. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nature Genetics. 2000;24(1):27–35. doi: 10.1038/71643. [DOI] [PubMed] [Google Scholar]
- 82.Harfe BD, Minesinger BK, Jinks-Robertson S. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Current Biology. 2000;10(3):145–148. doi: 10.1016/s0960-9822(00)00314-6. [DOI] [PubMed] [Google Scholar]
- 83.Nishant KT, Plys AJ, Alani E. A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae . Genetics. 2008;179(2):747–755. doi: 10.1534/genetics.108.086645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95(4):541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 85.Kosinski J, Plotz G, Guarné A, Bujnicki JM, Friedhoff P. The PMS2 subunit of human MutLα contains a metal ion binding domain of the iron-dependent repressor protein family. Journal of Molecular Biology. 2008;382(3):610–627. doi: 10.1016/j.jmb.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 86.Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends in Biochemical Sciences. 2000;25(1):24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 87.Kim TG, Cha HJ, Lee HJ, et al. Structural insights of the nucleotide-dependent conformational changes of Thermotoga maritima Mutl using small-angle X-ray scattering analysis. Journal of Biochemistry. 2009;145(2):199–206. doi: 10.1093/jb/mvn157. [DOI] [PubMed] [Google Scholar]
- 88.Spampinato C, Modrich P. The MutL ATPase is required for mismatch repair. Journal of Biological Chemistry. 2000;275(13):9863–9869. doi: 10.1074/jbc.275.13.9863. [DOI] [PubMed] [Google Scholar]
- 89.Räschle M, Dufner P, Marra G, Jiricny J. Mutations within the hMLH1 and hPMS2 subunits of the human MutLα mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSα . Journal of Biological Chemistry. 2002;277(24):21810–21820. doi: 10.1074/jbc.M108787200. [DOI] [PubMed] [Google Scholar]
- 90.Hall MC, Shcherbakova PV, Kunkel TA. Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and Pms1 proteins. Journal of Biological Chemistry. 2002;277(5):3673–3679. doi: 10.1074/jbc.M106120200. [DOI] [PubMed] [Google Scholar]
- 91.Yang W. Human MutLα: the jack of all trades in MMR is also an endonuclease. DNA Repair. 2007;6(1):135–139. doi: 10.1016/j.dnarep.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim TG, Heo S-D, Ku JK, Ban C. Functional properties of the thermostable mutL from Thermotoga maritima . BMB Reports. 2009;42(1):53–58. doi: 10.5483/bmbrep.2009.42.1.053. [DOI] [PubMed] [Google Scholar]
- 93.Pavlov YI, Mian IM, Kunkel TA. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Current Biology. 2003;13(9):744–748. doi: 10.1016/s0960-9822(03)00284-7. [DOI] [PubMed] [Google Scholar]
- 94.Masih PJ, Kunnev D, Melendy T. Mismatch repair proteins are recruited to replicating DNA through interaction with Proliferating Cell Nuclear Antigen (PCNA) Nucleic Acids Research. 2008;36(1):67–75. doi: 10.1093/nar/gkm943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iyer RR, Pohlhaus TJ, Chen S, et al. The MutSα-proliferating cell nuclear antigen interaction in human DNA mismatch repair. Journal of Biological Chemistry. 2008;283(19):13310–13319. doi: 10.1074/jbc.M800606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moldovan G-L, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129(4):665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 97.Shell SS, Putnam CD, Kolodner RD. The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Molecular Cell. 2007;26(4):565–578. doi: 10.1016/j.molcel.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lau PJ, Kolodner RD. Transfer of the MSH2·MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. Journal of Biological Chemistry. 2003;278(1):14–17. doi: 10.1074/jbc.C200627200. [DOI] [PubMed] [Google Scholar]
- 99.Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Molecular Cell. 2003;12(5):1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- 100.Umar A, Buermeyer AB, Simon JA, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87(1):65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 101.Guo S, Presnell SR, Yuan F, Zhang Y, Gu L, Li G-M. Differential requirement for proliferating cell nuclear antigen in 5′ and 3′ nick-directed excision in human mismatch repair. Journal of Biological Chemistry. 2004;279(17):16912–16917. doi: 10.1074/jbc.M313213200. [DOI] [PubMed] [Google Scholar]
- 102.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simmons LA, Davies BW, Grossman AD, Walker GC. β clamp directs localization of mismatch repair in Bacillus subtilis . Molecular Cell. 2008;29(3):291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Genschel J, Modrich P. Analysis of the excision step in human DNA mismatch repair. Methods in Enzymology. 2006;408:273–284. doi: 10.1016/S0076-6879(06)08017-7. [DOI] [PubMed] [Google Scholar]
- 105.Mauris J, Evans TC., Jr. A human PMS2 homologue from Aquifex aeolicus stimulates an ATP-dependent DNA helicase. Journal of Biological Chemistry. 2010;285(15):11087–11092. doi: 10.1074/jbc.M109.050955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shimada A, Masui R, Nakagawa N. A novel single-stranded DNA-specific 3′–5′ exonuclease, Thermus thermophilus exonuclease I, is involved in several DNA repair pathways. doi: 10.1093/nar/gkq350. Nucleic Acids Research. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wakamatsu T, Kitamura Y, Kotera Y, Nakagawa N, Kuramitsu S, Masui R. Structure of RecJ exonuclease defines its specificity for single-stranded DNA. The Journal of Biological Chemistry. 2010;285:9762–9769. doi: 10.1074/jbc.M109.096487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iino H, Naitow H, Nakamura Y, et al. Crystallization screening test for the whole-cell project on Thermus thermophilus HB8. Acta Crystallographica Section F. 2008;64(6):487–491. doi: 10.1107/S1744309108013572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yokoyama S, Hirota H, Kigawa T, et al. Structural genomics projects in Japan. Nature Structural Biology. 2000;7, supplement:943–945. doi: 10.1038/80712. [DOI] [PubMed] [Google Scholar]
- 110.Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annual Review of Genetics. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- 111.Harfe BD, Jinks-Robertson S. Mismatch repair proteins and mitotic genome stability. Mutation Research. 2000;451(1-2):151–167. doi: 10.1016/s0027-5107(00)00047-6. [DOI] [PubMed] [Google Scholar]
- 112.Erdeniz N, Nguyen M, Deschênes SM, Liskay RM. Mutations affecting a putative MutLα endonuclease motif impact multiple mismatch repair functions. DNA Repair. 2007;6(10):1463–1470. doi: 10.1016/j.dnarep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cadet J, Douki T, Gasparutto D, Ravanat J-L. Oxidative damage to DNA: formation, measurement and biochemical features. Mutation Research. 2003;531(1-2):5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 114.Jovanovic SV, Simic MG. The DNA guanyl radical: kinetics and mechanisms of generation and repair. Biochimica et Biophysica Acta. 1989;1008(1):39–44. doi: 10.1016/0167-4781(89)90167-x. [DOI] [PubMed] [Google Scholar]
- 115.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Macpherson P, Barone F, Maga G, Mazzei F, Karran P, Bignami M. 8-Oxoguanine incorporation into DNA repeats in vitro and mismatch recognition by MutSα . Nucleic Acids Research. 2005;33(16):5094–5105. doi: 10.1093/nar/gki813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mazurek A, Berardini M, Fishel R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. Journal of Biological Chemistry. 2002;277(10):8260–8266. doi: 10.1074/jbc.M111269200. [DOI] [PubMed] [Google Scholar]
- 118.Roth RB, Samson LD. Gene transfer to suppress bone marrow alkylation sensitivity. Mutation Research. 2000;462(2-3):107–120. doi: 10.1016/s1383-5742(00)00021-1. [DOI] [PubMed] [Google Scholar]
- 119.Pegg AE. Mammalian O 6 -alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Research. 1990;50(19):6119–6129. [PubMed] [Google Scholar]
- 120.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSa and MutLa in response to cytotoxic O 6-methylguanine adducts. Molecular Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSα and hMutLa-dependent phosphorylation of p53 in response to DNA methylator damage. Proceedings of the National Academy of Sciences of the Unites States of America. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Uversky VN, Dunker AK. Understanding protein non-folding. Biochimica et Biophysica Acta. 2010;1804(6):1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]