

Abstract
Australia was probably settled soon after modern humans left Africa, but details of this ancient migration are not well understood. Debate centers on whether the Pleistocene Sahul continent (composed of New Guinea, Australia, and Tasmania) was first settled by a single wave followed by regional divergence into Aboriginal Australian and New Guinean populations (common origin) or whether different parts of the continent were initially populated independently. Australia has been the subject of relatively few DNA studies even though understanding regional variation in genomic structure and diversity will be important if disease-association mapping methods are to be successfully evaluated and applied across populations. We report on a genome-wide investigation of Australian Aboriginal SNP diversity in a sample of participants from the Riverine region. The phylogenetic relationship of these Aboriginal Australians to a range of other global populations demonstrates a deep common origin with Papuan New Guineans and Melanesians, with little evidence of substantial later migration until the very recent arrival of European colonists. The study provides valuable and robust insights into an early and important phase of human colonization of the globe. A broader survey of Australia, including diverse geographic sample populations, will be required to fully appreciate the continent's unique population history and consequent genetic heritage, as well as the importance of both to the understanding of health issues.
Main Text
There is strong fossil and genetic evidence that modern humans arose in Africa ∼200,000 years ago, with a subset departing the continent much later (∼40,000–80,000 years ago) to populate the rest of the world.1 Mitochondrial DNA (mtDNA) suggests that these migrants exited Africa by the “southern route,” across the Red Sea to Arabia, moving relatively rapidly along the coast to reach Southeast Asia and Australia.2 Indeed, despite its distance from Africa, Australia has some of the earliest reliable evidence of human habitation outside Africa, dating to at least ∼46,000 and probably ∼60,000 years ago.3–5 Archeological evidence suggests that New Guinea and Melanesia, the islands immediately north and northeast of Australia, collectively termed Near Oceania, were also settled by ∼40,000 years ago.6 During this late Pleistocene period, sea levels were lower and the first humans entered the region when present day Australia, Tasmania, and New Guinea were part of a single landmass known as the Sahul. However, details of dispersal routes and timing of the settlement remain debated. The common origin hypothesis proposes a single major migration from Eurasia to the Sahul followed by divergence into separate geographic populations. The independent origin model, by contrast, posits a multiwave early settlement of the Sahul with largely independent migrations to present-day New Guinea and mainland Australia. There is also debate around whether the first settlers were followed by later waves of migrants.
Advances in genotyping technology allow variation along the entire genome to be simultaneously interrogated and have revolutionized the study of human genetic diversity, providing new insights into population history and facilitating gene discovery by genome-wide association studies (GWASs).7 However, despite its early and unique place in human global colonization, there has been relatively little investigation of Aboriginal Australian genetic diversity. This is partially due to unease felt by some Aboriginal Australians about genetic research because of the legacy of past research experiences.8 A limited number of previous studies have generally focused on traditional Y chromosome and mtDNA markers and none has surveyed whole genome diversity. GWAS genotyping and analytical approaches are typically geared toward populations of European origin and focused on diseases and conditions that are prevalent in these people. In order to extend gene discovery studies, and their potential longer-term health benefits, to Aboriginal Australians, it will first be necessary to gain a fuller understanding of Australia's population history and the present genetic legacy of that past.9,10
According to genealogical information, the Australians involved in this study have assured maternal Aboriginal ancestry with some Aboriginal, European (Scottish), and other non-Aboriginal paternal connections. They come from the Riverine area of western New South Wales and are a subset of those described in previous investigations of mtDNA variation.11,12 We use the abbreviation AuR for the sample population throughout this report to be consistent with, and for the reasons described in, these publications. Although a single sample population will not necessarily be representative of an entire continent, the Riverine region is especially significant because it includes the Willandra Lakes and Lake Mungo, where some of the earliest Australian human remains have been found.3 Personal contact and ongoing negotiation with the participants has been carried out by one of the authors (S.M.v.H.P.) over the past 18 years. Thirty-eight participants (30 female and eight male) gave informed consent to further genetic study. Community agreement has been granted from Maari Ma Health Aboriginal Corporation, and ethical approval has been given by the Aboriginal Health and Medical Research Council Ethics Committee and the University of New South Wales Human Research Ethics Committee. Local and regional communities have been consulted with regard to this report.
Samples were genotyped for ∼907,000 SNPs on the Affymetrix Genome-Wide Human SNP Array 6.0 platform with standard protocols (Affymetrix). Genotypes were called from the raw intensity (.cel) files with the Birdsuite software13 and a confidence threshold score of 0.1. We subsequently conducted additional quality control on the data by excluding any SNP with a missing genotype rate greater than 5% (66,251 SNPs) and those that were out of Hardy Weinberg equilibrium (HWE) applying a stringent cutoff p < 0.05 (22,300 SNPs). The average missing data per individual across the remaining 824,886 SNPs was 0.2%. Most analysis was restricted to 160,337 SNPs (155,166 autosomal and 5,171 X chromosome) that were genotyped in common between the eleven HapMap3 (n = 988) and 51 Human Genome Diversity Panel (HGDP; n = 940) populations14 (see Table S1 and Table S2, available online, for details).
We first explored the overall relationship of the AuR sample to the HGDP populations. Importantly, the HGDP panel included two samples from Oceania: Papuans (PAP; from New Guinea) and island Melanesians (MEL; from Bougainville), populations thought to be descendents of the region's first inhabitants. The matrix of pairwise interpopulation genetic distances (FST values) was used to construct a neighbor-joining (NJ) phylogenetic tree that summarizes the relationship of the 52 populations to each other (Figure 1A). The tree divides the populations into five broad groups: African, East Asian, West Eurasian (European, Middle Eastern, and Central and South Asian populations), American, and Oceanic. Although the latter branch contains the MEL, PAP, and AuR groups, AuR show a shorter branch length than the others, placing them closer to the trunk of the tree than the other Oceanic populations. This could be the result of greater genetic drift in the MEL and PAP or admixture of the AuR with populations elsewhere on the tree.
Figure 1.
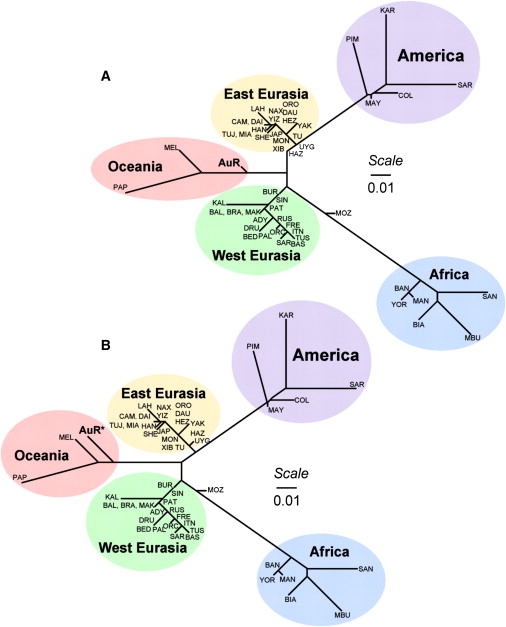
Unrooted Neighbor-Joining Phylogenetic Trees
NJ trees are based on the interpopulation FST matrix calculated from allele frequencies in the 51 HGDP populations and those (A) directly observed in the admixed AuR population sample and (B) reconstructed in STRUCTURE as the unadmixed ancestral AuR∗ population. HGDP population codes can be seen in Table S2. NJ trees were drawn with the PHYLIP package (version 3.68).
Principal component analysis (PCA) of the same data, which examines individual rather than population-level genetic affinities, favors AuR sample admixture. The most important trends (PC1 and PC2) are sufficient to largely differentiate major continental groups (Figure 2A). Some AuR individuals are close to the Oceanic cluster, composed of MEL and PAP individuals, but most occupy a wide range on PC2 between Europeans and East Asians, generally falling in an area occupied by Central and South Asian populations. PC3 separates Amerindians from the other populations. PC4 does the same for the Oceania populations, with MEL and PAP individuals clustered at one pole and all other populations at the opposite extreme (Figure 2B). AuR individuals fall in a broad range between these extremes, supporting substantial admixture from a non-Oceanic source.
Figure 2.
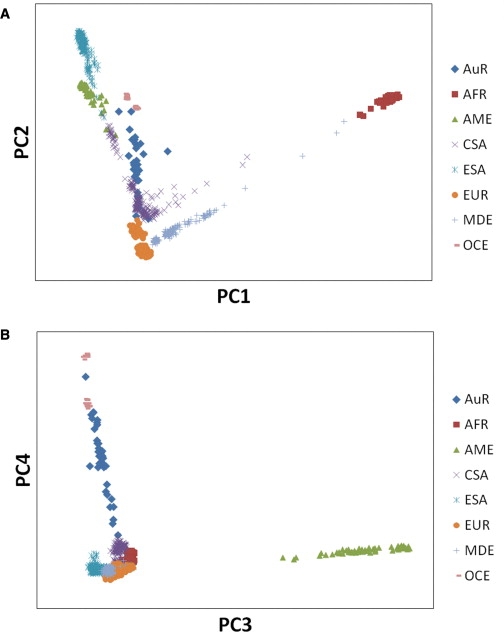
Principal-Component Analysis
(A) PC1 versus PC2 and (B) PC3 versus PC4 derived from the 51 HGDP populations and the AuR sample. HGDP populations have been grouped into seven broader regions: Africa (AFR), America (AME), Central and South Asia (CSA), East Asia (ESA), Europe (EUR), Middle East (MDE), and Oceania (OCE). See Table S2 for further population details. PCA was conducted with EIGENSTRAT33 (version 2).
To investigate the admixture further, we explored the population structure, without initially considering known geographic origin, by using the frappe15 and STRUCTURE16,17 methods, which infer population clusters from the genotypes alone and determine the fractional ancestry of each individual derived from these clusters. Both require the prior specification of the number of populations or clusters (K) into which individuals are to be divided. We carried out a series of runs for K = 2 to K = 8. The frappe analysis used the entire set of 155,166 autosomal SNPs. However, because STRUCTURE requires approximate linkage equilibrium between markers and is computationally slow with large numbers of markers, we divided the 155,166 autosomal SNPs into ten equal sets of ∼15,500 by assigning every tenth SNP, by order along the genome, to a different subset. Given that results from a series of exploratory runs using the different sets were highly correlated, we focused on just one for our main series of K = 2 to K = 8 STRUCTURE analyses.
Results from frappe and STRUCTURE were highly consistent with each other. At K = 5 (Figure 3) there is clear separation of individuals according to the major branches identified in the NJ tree (Figure 1). The Melanesians and Papuans are almost completely assigned to a single Oceanic cluster except for a minority East Asian ancestry component in the Melanesians, probably representing introgression from the Holocene era Austronesian expansion.18 The AuR sample is a clear mixture of two clusters corresponding to Oceanic ancestry and the majority ancestry component of Western Eurasian populations. Recent admixture with Europeans, who began settling the continent in 1788, is an obvious source for this Western Eurasian component and would be consistent with known genealogical information. However, contact with India earlier in the Holocene has also been proposed on the basis of mtDNA19 and Y chromosome20 data. At K > 5, further population distinction emerges in the Western Eurasian cluster, with gradual separation of European from Central and South Asian populations (Figure S1). Although the distinction is never complete, the non-Oceanic component in the AuR sample is most consistent with European ancestry. Such a conclusion is anecdotally supported by the presence of evolutionarily recent alleles (for example, the blue-eye-associated allele of the rs12913832 SNP, near the OCA2 gene21 [MIM 611409] and the red-hair-associated allele of rs1805007 in the MC1R gene22 [MIM 155555]) that are essentially restricted to (primarily northern) Europeans.
Figure 3.

Population Structure Analysis
Individual ancestry proportions in the HGDP and Aboriginal Australian (AuR) samples at K = 5, from (A) frappe analysis, with all 155,166 autosomal markers, and (B) STRUCTURE analysis, with a one-tenth subset (15,516) of all autosomal SNPs. Each horizontal line represents an individual and is divided into K (number of population clusters) colored segments reflecting the estimated ancestry proportion from each cluster. Different geographic samples are divided by black lines with population and region indicated to the right and left of the plot, respectively. See Table S2 for a full explanation of population codes. frappe analysis used 5,000 expectation-maximization (EM) iterations whereas STRUCTURE runs were conducted under the admixture model with a 25,000 replicate burn-in followed by 25,000 Markov chain Monte Carlo (MCMC) iterations.
We attempted to quantify individual ancestry more accurately by conducting further STRUCTURE analysis under a model of K = 2, assuming that the AuR sample is a mixture of an ancient ancestral Aboriginal population and recent European settlers. We therefore only included the AuR sample and European individuals from HapMap3 (TSI and CEU, n = 200, see Table S1) and allowed STRUCTURE to incorporate the origin information of the Europeans in the analysis, effectively rendering them a training set for one parental population. (We used HapMap3 as a discovery data set in this analysis so that we could apply findings downstream in the test HGDP.) STRUCTURE runs were carried out for each of the ten autosomal SNP subsets. Ancestry estimates are highly consistent across all SNP sets, so we averaged across all runs to obtain a single European versus Australian ancestry fraction for each individual. The Australian component ranged from 28% to 100% with an average of 64% (Figure 4). As a result of the strong ascertainment bias of HapMap and Affymetrix SNPs, however, which were largely identified from European ancestry populations, the Aboriginal component might be underestimated.
Figure 4.
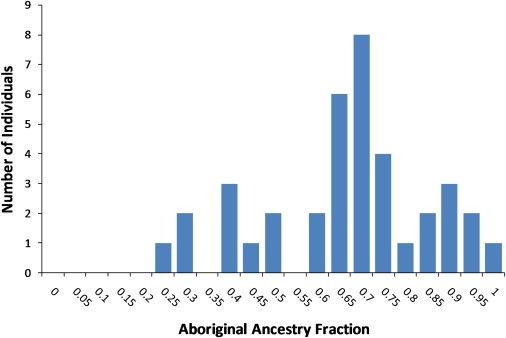
Distribution of Individual Aboriginal Australian Genomic Ancestry Estimates
Values are the fraction of autosomal genomic ancestry assigned to the non-European cluster in STRUCTURE using the 38 AuR individuals, 200 HapMap3 Europeans, and a K = 2. STRUCTURE runs consisted of a 25,000 replicate burn-in followed by 25,000 MCMC iterations.
We also explored the admixture process by examining Y chromosome and mtDNA variation. Previous analysis of AuR mtDNA11,12 showed that 37 individuals are likely to have ancient Australian maternal origin with deep-rooting mtDNA lineages (haplogroups M42a and b, P4b and S1a), with one mtDNA of probable European origin (haplogroup U5). The 257 Y chromosome SNPs successfully genotyped here were not sufficient to infer in full detail the haplogroup of the eight AuR Y chromosomes.23 However, five of these Y chromosomes clearly fall into European haplogroups (R1b1 and I),24 and two are in haplogroup C, one of whose subgroups (C4) is common in Australians.25,26 The final Y chromosome is most likely haplogroup M (but can only be formally assigned KxPxNO), which is also found in Australians.27 From these observations (37.5% male and 97.5% female Aboriginal Australian ancestors), the expected autosomal ancestry fraction is ∼67%, close to the observed value of 64%. Because each X chromosome spends two-thirds of its history in females, the Australian admixture fraction is expected to be higher when X chromosome markers are used. A value of 70% is observed, close to the ∼77% expected from the male/female ancestral bias inferred from Y chromosome and mtDNA markers.
For learning more about the pre-1788 history of the Australian population, a sample with assured Aboriginal paternal and maternal ancestry would obviously be ideal. However, we can use STRUCTURE estimates of parental cluster allele frequencies, which are modeled jointly with ancestry proportions, as an approximation of those in the ancestral population prior to admixture. The STRUCTURE-reconstructed Aboriginal Australian Riverine (AuR∗) allele frequencies, along with those observed in the HGDP samples, were therefore used to recalculate pairwise FST values. We assumed an Australian sample size two-thirds (n = 25) that of the original sample collection in these FST calculations, in line with admixture proportions. (Note: the HGDP were not used in STRUCTURE runs that estimated the AuR∗ allele frequencies.) A NJ phylogenetic tree, derived from the interpopulation FST matrix, again shows five major continental branches (Figure 1B). However, the AuR∗ population now groups more tightly with the Melanesians and Papuans further toward the terminus of the Oceanic branch. The phylogeny supports the common origin of the earliest indigenous inhabitants of Oceania, followed first by the divergence of Australia from Near Oceania and then by the subsequent split of the latter into Papuan and Melanesian populations.
We also used the reconstructed allele frequencies to explore loci that are particularly differentiated between this sample and the rest of the world by calculating the FST between AuR∗ and the entire HapMap3 collection for each autosomal SNP (Figure 5A and 5B). The most highly differentiated SNP (rs12458349) is on chromosome 18q21.33 approximately 30 Kb upstream of the PHLPP gene (MIM 609396). Two further SNPs in the same 0.5 Mb region are also in the top 25 most differentiated SNPs. The derived allele of rs12458349 is at its highest frequency in the reconstructed AuR∗ (61%) and observed AuR (45%) Australian samples (Figure 5C). It occurs at a relatively high frequency in the Papuan sample (30%) but is rare or absent elsewhere in the world. Interestingly, the region has been repeatedly implicated, via linkage and association, in diabetic nephropathy,28–30 which is one of the principal causes of chronic kidney disease (CKD).31 Indigenous Australians have ∼10-fold-higher rates of CKD than nonindigenous Australians.32 It is possible that variants in one or more of the genes in this region also contribute to the increased risk of CKD in Aboriginal Australians. The observed differentiation in the 18q21.33 region may simply be the consequence of stochastic sampling error and/or genetic drift. Genetic drift, or random changes in allele frequencies, is expected to be a major force in a population, like Aboriginal Australia, that has been relatively small and/or isolated for a long period of time. Differentiation could also be explained by natural selection. The presence of several highly differentiated SNPs in the region, spanning nearly 0.5 Mb in length, hints at the presence of a long common haplotype that might be indicative of genetic hitch-hiking and recent positive selection. However, it is difficult to distinguish between possible explanations because the sample is small, with extensive admixture hampering phasing and direct investigation of linkage-disequilibrium-based selection signals.
Figure 5.
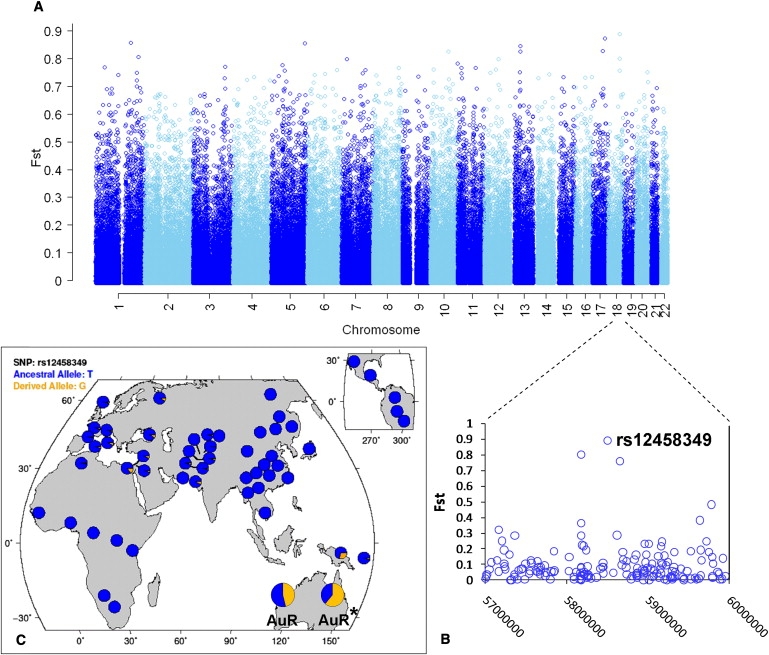
Genomic and Geographic Distribution of Highly Differentiated SNPs
(A) SNP FST values, calculated between the full HapMap3 sample and the reconstructed Aboriginal Australian (AuR∗) allele frequencies, plotted against genomic location.
(B) Detail of the chromosome 18q21.33 region (NCBI-36 coordinates 57,000,000 to 60,000,000) surrounding the highest observed FST at rs12458349.
(C) Geographic distribution of rs12458349 allele frequencies in the HGDP, observed AuR and reconstructed AuR∗ population samples. Adapted from a graphic produced by HGDP Selection browser.49
Whereas the admixture present in the AuR sample presents a potential challenge in conducting traditional association methods for disease gene discovery,33 it opens the possibility of using admixture mapping.34 Admixture mapping is most suitable for traits, like CKD, that differ in frequency between the two parental populations of an admixture group. The approach essentially looks for genomic regions with an excess of higher-risk population ancestry relative to other regions or controls.34 A set of markers, spread across the genome, that are highly informative as to ancestry (ancestry informative markers or AIMs) is an essential requirement for admixture mapping. As an exploratory exercise, we identified a set of 100 Oceanic AIMs, from the top 200 by FST between the AuR∗ and the full HapMap3 population, such that no AIM was within 2 Mb of another (see Table S3 for a full list). With this marker set and STRUCTURE, under K = 2, it was possible to distinguish Oceanic versus non-Oceanic ancestry in the independent HGDP data set with a high degree of accuracy (as judged against earlier frappe and STRUCTURE results obtained with more markers) (Figure 6).
Figure 6.
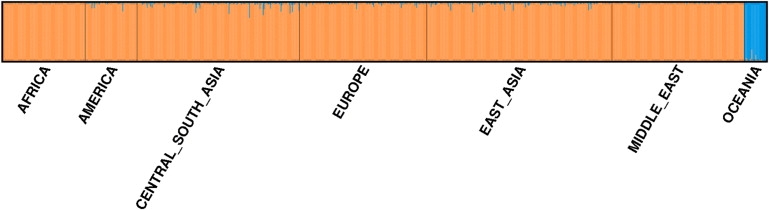
Oceanic Ancestry Informative Markers
Results of a STRUCTURE run (K = 2) using 100 AIMs in the HGDP populations. There is clear ability to distinguish the Oceanic (PAPuans and MELanesians) from non-Oceanic populations. See the Figure 3 legend for a fuller description of the plot. STRUCTURE runs consisted of a 25,000 replicate burn-in followed by 25,000 MCMC iterations.
Our study of whole-genome diversity in a sample of participants with deep Aboriginal ancestry adds to the genetic, archaeological, anthropological, and linguistic evidence that Australia has had a long, rich, and unique population history. Results from the relatively small number of previous genetic diversity studies, typically using single loci, could not definitively resolve between models of initial Sahul settlement. For example, autosomal α-globin locus haplotypes in Australians and New Guineans produced conflicting evidence for both the independent origin hypothesis35 and the common origin hypothesis.36 mtDNA diversity11,19,25,37–41 indicates very deep and diverse maternal ancestry for Australia and New Guinea. The most recent and comprehensive of these studies25 found that the defining ancestral node of some subclades (Q and P) were shared between Australia and New Guinea, but there was little or no sharing of more recently evolved derived lineages within these, indicating a single founding Sahul group with subsequent isolation between regional populations. Other ancient lineages are unique to Australia, suggesting the possibility of different entry points and independent origins. On the paternal side, very few studies are available and are limited in language group representation. However, a single Y chromosome lineage (C4a-DYS390.1 del/M347), which Y microsatellite diversity suggests expanded rapidly beginning in the mid-Holocene about 5000 years ago,20 is apparently unique to the Australian continent.25,26 There is little evidence of Y chromosome sharing between New Guinea and Australia arguing for at least the relatively recent isolation of the two populations.26
Although the Y chromosome and mtDNA are potentially powerful markers of population history, they are each a single locus and potentially prone to high levels of drift that may obscure the window they provide into the very distant past. Our analysis is based on a suite of genome-wide SNPs providing a more robust and broad-based insight on Aboriginal genetic affinities. The clear phylogenetic grouping of the Aboriginal Australians with other Near Oceania samples, from New Guinea and Melanesia, favors the common origin hypothesis for the original settlement of the Pleistocene Sahul continent, if not details of routes and possible entry points. The most parsimonious explanation is a single settlement of the Sahul, which archaeological evidence puts at around 50,000–60,000 years ago,3 followed by differentiation into subregional populations. However, we cannot formally distinguish between this and an initial separation and isolation of the proto-Sahul population in mainland Eurasia followed by multiple ancient migrations to various locations in the Sahul. Our conclusion is consistent with global population genetic affinities gauged from Alu insertion polymorphisms42 and the genetic diversity of the human bacterial parasite, Helicobacter pylori (H. pylori). The haplotypes of H. pylori samples taken from people in New Guinea and Australia form a geographically distinct and phylogenetically ancient group (whose divergence from other global lineages was placed at ∼32,000 years on the basis of accumulated diversity) supporting a single-wave initial Sahul settlement.43
Analysis of finer population structure in the whole-genome SNP data also indicates that these ancient initial settlers were subsequently isolated and relatively undisturbed by later migrations from the Asian mainland. There is little evidence, for instance, of East Asian ancestry that could be attributed to the Austronesian expansion (beginning ∼5,500 years ago), which impacted, to varying degrees, other indigenous Oceanic (Melanesian and Papuan) populations,18,44,45 nor is there any convincing signal of recent contact between Australia and the Indian subcontinent. The presence of phylogenetically ancient and geographically restricted mtDNA lineages (such as haplogroups S and M42) in Australia together with the absence of Austronesian mtDNA (B4a1a1a) and Y chromosome (O-M110, O-M119, O-M324) lineages supports our whole-genome-based conclusions. The results imply that noted archaeological events, such as the mid-Holocene “intensification” (witnessed by increases in both the complexity and density of stone tools at many archaeological sites46) or the arrival of the dingo,47 were not mediated by substantial amounts of migration from mainland Eurasia. It is clear from the data, however, that this period of apparent long-term isolation was ended by the arrival of European settlers beginning in 1788. A significant minority of the biological ancestry of the study participants comes from Europeans and was primarily introduced by males. A marked sex bias or asymmetry has been genetically noted in the history of European miscegenation with indigenous groups—for example, in South Africa.48
Although this study massively increases the number of loci used to investigate Aboriginal Australian genetic diversity, the increased power to uncover population history is mitigated by a relatively small sample population from just one part of Australia, albeit an important Pleistocene habitation region. A single sample will probably not reflect the full later history of an entire continent and its people. The long history of Aboriginal Australians and the large physical distances across a climatically shifting, sometimes arid continent, combined with a hunter-gatherer lifestyle, which does not typically support high population densities, may have allowed considerable isolation and genetic drift between Aboriginal groups in different parts of Australia. Some regions, particularly in northern coastal areas, may have experienced migration events that little impacted other areas. A broader survey of Aboriginal Australian groups is justified to capture the full diversity of the continent. As well as providing new insights into the past, a fuller understanding and appreciation of the continent's genetic diversity will be practically important to extend the latest gene discovery methods, and their potential health benefits, to Aboriginal Australians.
Acknowledgments
The participants in this study are from the main language groups of Barkindji (Paakintji) and Ngiyambaa and from several associated language groups now living in the communities of Wilcannia, Menindee, and Dareton in the Darling River region of western New South Wales, Australia. Their friendship and participation over many years is greatly appreciated. We thank Nick Martin for his role in facilitating and initiating this study and the staff at the Ramaciotti Centre for Gene Function Analysis, University of New South Wales, Sydney, who carried out the genotyping. Experiment costs were provided by the researchers. B.P.M. and P.M.V. are supported by grants from the Australian National Health and Research Council (NHMRC; grants 389892 and 613601) and the Australian Research Council (grant DP0770096). J.M.L. is supported by an NHMRC Peter Doherty Australian Biomedical Fellowship.
Contributor Information
Brian P. McEvoy, Email: brian.mcevoy@qimr.edu.au.
Sheila M. van Holst Pellekaan, Email: s.vanholst@unsw.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Affymetrix, http://www.affymetrix.com
EIGENSTRAT, http://genepath.med.harvard.edu/∼reich/Software.htm
frappe: http://med.stanford.edu/tanglab/software/frappe.html
HGDP Selection Browser: http://hgdp.uchicago.edu/cgi-bin/gbrowse/HGDP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PHYLIP, http://evolution.genetics.washington.edu/phylip.html
References
- 1.Cavalli-Sforza L.L., Feldman M.W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 2003;33(Suppl):266–275. doi: 10.1038/ng1113. [DOI] [PubMed] [Google Scholar]
- 2.Macaulay V., Hill C., Achilli A., Rengo C., Clarke D., Meehan W., Blackburn J., Semino O., Scozzari R., Cruciani F. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science. 2005;308:1034–1036. doi: 10.1126/science.1109792. [DOI] [PubMed] [Google Scholar]
- 3.O'Connell J.F., Allen J. Dating the colonization of Sahul (Pleistocene Australia–New Guinea): A review of recent research. J. Archaeol. Sci. 2004;31:835–853. [Google Scholar]
- 4.Balme J., Davidson I., McDonald J., Stern N., Veth P. Symbolic behaviour and the peopling of the southern arc route to Australia. Quaternary International. 2009;202:59–68. [Google Scholar]
- 5.Oppenheimer S. The great arc of dispersal of modern humans: Africa to Australia. Quaternary International. 2009;202:2–13. [Google Scholar]
- 6.Groube L., Chappell J., Muke J., Price D. A 40,000 year-old human occupation site at Huon Peninsula, Papua New Guinea. Nature. 1986;324:453–455. doi: 10.1038/324453a0. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 8.van Holst Pellekaan S. Genetic research: What does this mean for indigenous Australian communities? Australian Aboriginal Studies. 2000;1/2:65–75. [Google Scholar]
- 9.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Need A.C., Goldstein D.B. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009;25:489–494. doi: 10.1016/j.tig.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 11.van Holst Pellekaan S., Frommer M., Sved J., Boettcher B. Mitochondrial control-region sequence variation in aboriginal Australians. Am. J. Hum. Genet. 1998;62:435–449. doi: 10.1086/301710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Holst Pellekaan S.M., Ingman M., Roberts-Thomson J., Harding R.M. Mitochondrial genomics identifies major haplogroups in Aboriginal Australians. Am. J. Phys. Anthropol. 2006;131:282–294. doi: 10.1002/ajpa.20426. [DOI] [PubMed] [Google Scholar]
- 13.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 15.Tang H., Peng J., Wang P., Risch N.J. Estimation of individual admixture: Analytical and study design considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 16.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayser M., Brauer S., Cordaux R., Casto A., Lao O., Zhivotovsky L.A., Moyse-Faurie C., Rutledge R.B., Schiefenhoevel W., Gil D. Melanesian and Asian origins of Polynesians: mtDNA and Y chromosome gradients across the Pacific. Mol. Biol. Evol. 2006;23:2234–2244. doi: 10.1093/molbev/msl093. [DOI] [PubMed] [Google Scholar]
- 19.Redd A.J., Stoneking M. Peopling of Sahul: mtDNA variation in aboriginal Australian and Papua New Guinean populations. Am. J. Hum. Genet. 1999;65:808–828. doi: 10.1086/302533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redd A.J., Roberts-Thomson J., Karafet T., Bamshad M., Jorde L.B., Naidu J.M., Walsh B., Hammer M.F. Gene flow from the Indian subcontinent to Australia: Evidence from the Y chromosome. Curr. Biol. 2002;12:673–677. doi: 10.1016/s0960-9822(02)00789-3. [DOI] [PubMed] [Google Scholar]
- 21.Sturm R.A., Duffy D.L., Zhao Z.Z., Leite F.P., Stark M.S., Hayward N.K., Martin N.G., Montgomery G.W. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008;82:424–431. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sulem P., Gudbjartsson D.F., Stacey S.N., Helgason A., Rafnar T., Magnusson K.P., Manolescu A., Karason A., Palsson A., Thorleifsson G. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 2007;39:1443–1452. doi: 10.1038/ng.2007.13. [DOI] [PubMed] [Google Scholar]
- 23.Karafet T.M., Mendez F.L., Meilerman M.B., Underhill P.A., Zegura S.L., Hammer M.F. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 2008;18:830–838. doi: 10.1101/gr.7172008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jobling M.A., Tyler-Smith C. The human Y chromosome: An evolutionary marker comes of age. Nat. Rev. Genet. 2003;4:598–612. doi: 10.1038/nrg1124. [DOI] [PubMed] [Google Scholar]
- 25.Hudjashov G., Kivisild T., Underhill P.A., Endicott P., Sanchez J.J., Lin A.A., Shen P., Oefner P., Renfrew C., Villems R., Forster P. Revealing the prehistoric settlement of Australia by Y chromosome and mtDNA analysis. Proc. Natl. Acad. Sci. USA. 2007;104:8726–8730. doi: 10.1073/pnas.0702928104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kayser M., Brauer S., Weiss G., Schiefenhövel W., Underhill P.A., Stoneking M. Independent histories of human Y chromosomes from Melanesia and Australia. Am. J. Hum. Genet. 2001;68:173–190. doi: 10.1086/316949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kayser M. The human genetic history of Oceania: Near and remote views of dispersal. Curr. Biol. 2010;20:R194–R201. doi: 10.1016/j.cub.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 28.McDonough C.W., Bostrom M.A., Lu L., Hicks P.J., Langefeld C.D., Divers J., Mychaleckyj J.C., Freedman B.I., Bowden D.W. Genetic analysis of diabetic nephropathy on chromosome 18 in African Americans: Linkage analysis and dense SNP mapping. Hum. Genet. 2009;126:805–817. doi: 10.1007/s00439-009-0732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewens K.G., George R.A., Sharma K., Ziyadeh F.N., Spielman R.S. Assessment of 115 candidate genes for diabetic nephropathy by transmission/disequilibrium test. Diabetes. 2005;54:3305–3318. doi: 10.2337/diabetes.54.11.3305. [DOI] [PubMed] [Google Scholar]
- 30.Iyengar S.K., Abboud H.E., Goddard K.A., Saad M.F., Adler S.G., Arar N.H., Bowden D.W., Duggirala R., Elston R.C., Hanson R.L., Family Investigation of Nephropathy and Diabetes Research Group Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: The family investigation of nephropathy and diabetes (FIND) Diabetes. 2007;56:1577–1585. doi: 10.2337/db06-1154. [DOI] [PubMed] [Google Scholar]
- 31.Meguid El Nahas A., Bello A.K. Chronic kidney disease: The global challenge. Lancet. 2005;365:331–340. doi: 10.1016/S0140-6736(05)17789-7. [DOI] [PubMed] [Google Scholar]
- 32.Thomas, N., MacRae, A., Burns, J., Catto, M., Debuyst, O., Krom, I., Midford, R., Potter, C., Ride, K., Stumpers, S., and Urquhart, B. (2008). Overview of Australian Indigenous health status, April 2010. http://www.healthinfonet.ecu.edu.au/health-facts/overviews.
- 33.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 34.Seldin M.F. Admixture mapping as a tool in gene discovery. Curr. Opin. Genet. Dev. 2007;17:177–181. doi: 10.1016/j.gde.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsintsof A.S., Hertzberg M.S., Prior J.F., Mickleson K.N., Trent R.J. Alpha-globin gene markers identify genetic differences between Australian aborigines and Melanesians. Am. J. Hum. Genet. 1990;46:138–143. [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts-Thomson J.M., Martinson J.J., Norwich J.T., Harding R.M., Clegg J.B., Boettcher B. An ancient common origin of aboriginal Australians and New Guinea highlanders is supported by alpha-globin haplotype analysis. Am. J. Hum. Genet. 1996;58:1017–1024. [PMC free article] [PubMed] [Google Scholar]
- 37.Friedlaender J., Schurr T., Gentz F., Koki G., Friedlaender F., Horvat G., Babb P., Cerchio S., Kaestle F., Schanfield M. Expanding Southwest Pacific mitochondrial haplogroups P and Q. Mol. Biol. Evol. 2005;22:1506–1517. doi: 10.1093/molbev/msi142. [DOI] [PubMed] [Google Scholar]
- 38.Huoponen K., Schurr T.G., Chen Y., Wallace D.C. Mitochondrial DNA variation in an aboriginal Australian population: Evidence for genetic isolation and regional differentiation. Hum. Immunol. 2001;62:954–969. doi: 10.1016/s0198-8859(01)00294-4. [DOI] [PubMed] [Google Scholar]
- 39.Ingman M., Gyllensten U. Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines. Genome Res. 2003;13:1600–1606. doi: 10.1101/gr.686603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kivisild T., Shen P., Wall D.P., Do B., Sung R., Davis K., Passarino G., Underhill P.A., Scharfe C., Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Holst Pellekaan S.M., Harding R.M. Excavating the mitochondrial genome identifies major haplogroups in Aboriginal Australians. Before Farming. 2006;2006 doi: 10.1002/ajpa.20426. article 3. [DOI] [PubMed] [Google Scholar]
- 42.Stoneking M., Fontius J.J., Clifford S.L., Soodyall H., Arcot S.S., Saha N., Jenkins T., Tahir M.A., Deininger P.L., Batzer M.A. Alu insertion polymorphisms and human evolution: Evidence for a larger population size in Africa. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moodley Y., Linz B., Yamaoka Y., Windsor H.M., Breurec S., Wu J.Y., Maady A., Bernhoft S., Thiberge J.M., Phuanukoonnon S. The peopling of the Pacific from a bacterial perspective. Science. 2009;323:527–530. doi: 10.1126/science.1166083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimura R., Ohashi J., Matsumura Y., Nakazawa M., Inaoka T., Ohtsuka R., Osawa M., Tokunaga K. Gene flow and natural selection in oceanic human populations inferred from genome-wide SNP typing. Mol. Biol. Evol. 2008;25:1750–1761. doi: 10.1093/molbev/msn128. [DOI] [PubMed] [Google Scholar]
- 45.Kayser M., Lao O., Saar K., Brauer S., Wang X., Nürnberg P., Trent R.J., Stoneking M. Genome-wide analysis indicates more Asian than Melanesian ancestry of Polynesians. Am. J. Hum. Genet. 2008;82:194–198. doi: 10.1016/j.ajhg.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulvaney D.J., Kamminga J. Allen & Unwin; St. Leonards, NSW: 1999. Prehistory of Australia. [Google Scholar]
- 47.Savolainen P., Leitner T., Wilton A.N., Matisoo-Smith E., Lundeberg J. A detailed picture of the origin of the Australian dingo, obtained from the study of mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 2004;101:12387–12390. doi: 10.1073/pnas.0401814101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quintana-Murci L., Harmant C., Quach H., Balanovsky O., Zaporozhchenko V., Bormans C., van Helden P.D., Hoal E.G., Behar D.M. Strong maternal Khoisan contribution to the South African coloured population: A case of gender-biased admixture. Am. J. Hum. Genet. 2010;86:611–620. doi: 10.1016/j.ajhg.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pickrell J.K., Coop G., Novembre J., Kudaravalli S., Li J.Z., Absher D., Srinivasan B.S., Barsh G.S., Myers R.M., Feldman M.W., Pritchard J.K. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.