

Abstract
Cholesterol is an essential component of both the peripheral and central nervous systems of mammals. Over the last decade, evidence has accumulated that disturbances in cholesterol metabolism are associated with the development of various neurological conditions. In addition to genetically defined defects in cholesterol synthesis, which will be covered in another review in this Thematic Series, defects in cholesterol metabolism (cerebrotendinous xanthomatosis) and intracellular transport (Niemann Pick Syndrome) lead to neurological disease. A subform of hereditary spastic paresis (type SPG5) and Huntington's disease are neurological diseases with mutations in genes that are of importance for cholesterol metabolism. Neurodegeneration is generally associated with disturbances in cholesterol metabolism, and presence of the E4 isoform of the cholesterol transporter apolipoprotein E as well as hypercholesterolemia are important risk factors for development of Alzheimer's disease. In the present review, we discuss the links between genetic disturbances in cholesterol metabolism and the above neurological disorders.
Keywords: Alzheimer's disease, cholesterol/metabolism, cholesterol/trafficking, lipoproteins/assembly, nuclear receptors, omega oxidation
THE ROLE OF CHOLESTEROL AND ITS TURNOVER IN THE NERVOUS SYSTEM
The mammalian nervous system contains a disproportionate amount of cholesterol. In the brain, the cholesterol content is about 10-fold greater than in any other organ (1). The development of the capacity to synthesize and store such a large amount of cholesterol indicates that there is a close link between the evolution of the nervous system and a specific role for cholesterol. Within the brain, some 70% of cholesterol is present in myelin, where it fulfills a critical insulating role. It is likely that the requirement for efficient signaling despite a small transverse diameter of axons was a key selective pressure driving the accretion of cholesterol in the mammalian brain (1, 2). The requirement for large amounts of cholesterol is further underscored by the long half-life of brain cholesterol; overall brain cholesterol turns over some 250–300 times slower than that in the circulation (3).
At a cellular level, the myelin sheath consists of sections of plasma membrane repeatedly wrapped around an axon, with the extrusion of virtually all of the cytoplasm. Myelin is formed by 2 very specialized cells: the oligodendrocyte in the central nervous system (CNS) and the Schwann cell in the peripheral nervous system (PNS). Because an individual axon may be ensheathed by myelin from several oligodendrocytes, periodic gaps are present in the sheath. These are called the “nodes of Ranvier” and are the site of propagation of the action potential. Myelin can thus be regarded as a discontinuous insulation that enables the saltatory conduction of the action potential (1, 2). In addition to a large lipid component, myelin also contains many specific proteins such as proteolipid protein and myelin basic protein. Recently, evidence has been presented that cholesterol can regulate the correct targeting of one of the major membrane proteins of the PNS and thereby myelin compaction. These data extend the role of cholesterol in myelin from an essential structural component to a regulator of overall myelin structure (4).
The remaining 30% of brain cholesterol is divided between glial cells (20%) and neurons (10%). According to various in vitro studies with cultured cells, astrocytes synthesize at least 2- to 3-fold more cholesterol than neurons, and oligodendrocytes have an even higher capacity for cholesterol synthesis, at least during periods of active myelination [for a more detailed review, see ref (3)]. It has also been suggested that in the mature state, the neurons synthesize only a relatively small amount of cholesterol, and instead they rely almost totally on the supply of cholesterol from glial cells, especially astrocytes in the CNS (5). Studies using conditional ablation of a key gene in cholesterol synthesis have provided evidence that at least some adult neurons can survive without any de novo cholesterol synthesis (6, 7).
It is noteworthy that even minor changes in the structure of the constituent sterols of the nervous system, e.g., the presence of one additional double bond, leads to a change in the biophysical properties of the cell membrane with profound clinical effects. This is illustrated by the severity of the neurological phenotypes associated with defects in cholesterol synthesis, which lead to the incorporation of a cholesterol precursor such as desmosterol or 7-dehydrocholesterol into cell membranes. For a detailed review, see the review from Porter in this Thematic Series.
In the cells outside the CNS, the overall cholesterol homeostasis is achieved by de novo synthesis, uptake of lipoprotein-bound cholesterol, and efflux of cholesterol. In addition to this, liver can eliminate cholesterol by conversion into polar bile acids. When challenged with high levels of exogenous cholesterol, the excess can be stored in an esterified form.
The strategy used by nature for cholesterol homeostasis in the brain and spinal cord is different from that in other parts of the body. Due to the efficient blood-brain barrier, there is no passage of lipoprotein-bound cholesterol from the circulation to the brain (1, 3). The blood-brain barrier thus prevents diffusion of large molecules at the level of tight junctional attachments between adjacent capillary endothelial cells. In addition to this, there is also no transvesicular movement of solution across the capillaries. It is possible that one or more members of the ATP binding cassette transporter superfamily may be involved in the exclusion of circulating cholesterol from the brain, but conclusive evidence is lacking for this.
Thus, all the cholesterol present in the brain is formed by de novo synthesis, and the same is true also for the cholesterol in the PNS. Except for the active phase of specific pathological conditions, almost all (at least 99%) cholesterol in the nervous system is unesterified. In the adult brain, most of its synthesis is balanced by formation of a hydroxylated metabolite, 24S-hydroxycholesterol (24S-OHC), which is able to pass across the blood-brain barrier and enter the circulation (3, 8, 9). In addition, there is a small efflux of cholesterol from the brain in the form of apolipoprotein (APO) E containing lipoproteins via the cerebrospinal fluid (10). The mechanism by which cholesterol is eliminated from the PNS is not known with certainty, and the spinal cord does not appear to be able to synthesize 24S-OHC.
The stability of the bulk of cholesterol in the CNS and in the spinal cord is in marked contrast to the rapidly changing levels of plasma lipoproteins under different conditions. The turnover of the cholesterol present in membranes of the neuronal cells and in astrocytes may, however, change in response to different factors. Under normal conditions, cholesterol 24-hydroxylase (CYP46A1), the enzyme system responsible for formation of 24S-OHC, is present only in neuronal cells, with particularly high levels in the pyramidal neurons in various layers of the cortex, in the hippocampus, and in Purkinje cells in the cerebellum (9). The uptake of cholesterol by these cells may thus be balanced by the secretion of 24S-OHC. The 24S-OHC secreted from the neuronal cells may be of importance for regulation of cholesterol synthesis and secretion of this cholesterol in APOE-bound form from astroglia. The latter effects may be mediated by the liver X receptor (LXR) (5, 11).
In the liver, the conversion of cholesterol into bile acids is regulated by highly sophisticated mechanisms (12). In the brain, however, the expression of CYP46A1 appears to be resistant to regulatory axes, which are known to regulate cholesterol homeostasis and bile acid synthesis (13). Indeed, the putative promoter region of CYP46A1 has the characteristics of a housekeeping gene (13), although recent data indicate that epigenetic mechanisms may regulate CYP46A1 expression (14, 15). In contrast, cholesterol synthesis appears to be regulated by similar mechanisms both outside and inside the brain, with hydroxy-methyl-glutaryl CoA reductase (HMGCR) being the most important regulatory enzyme (2). However, in the brain, cholesterol synthesis via the 7-dehydrodesmosterol pathway seems to be preferred over the 7-dehydrocholesterol pathway, and disruption of the gene coding for the Δ24-reductase results in the accumulation of desmosterol without any accumulation of 7-dehydrodesmosterol (16).
In the Cyp46a1 null mouse, loss of the main pathway for cholesterol elimination results in a compensatory 40% decrease in cholesterol synthesis, while the overall content of brain cholesterol is normal. There are, however, significant behavioral changes and reduced memory function, indicating that a certain rate of flux through the mevalonate pathway is necessary to support memory consolidation (17). In subsequent studies, the isoprenoid geranylgeraniol was identified as the important intermediate, and it was suggested the Cyp46a1-mediated removal of cholesterol from neurons is critical to prevent cholesterol accumulation, feedback inhibition of cholesterol synthesis, and a decreased production of essential isoprenoid intermediates [reviewed in (9)].
Although formation of 24S-OHC is responsible for the removal of about two-thirds of cholesterol from the brain, under normal conditions, APOE seems to be the most important carrier of cholesterol in the brain extracellular fluid. In view of this, it is surprising that deletion of APOE does not appear to lead to any major functional abnormalities in the brain, although there is a compensatory increase in the expression of APOD (18). In aged APOE knockout mice, however, there is a lipid deposition in astrocytes in hippocampus and thalamus (19). In addition to APOE and APOD, there are other apolipoproteins in the brain, such as APOJ (also known as clusterin) and APOA1, that may be of importance (3).
THE ROLE OF O XYSTEROLS
Side-chain oxidized oxysterols (SCOx) such as 24S-OHC have the capacity to traverse biological membranes, and there is a concentration-driven flux of this oxysterol from the brain into the circulation (20). This flux corresponds to an elimination of 6–7 mg cholesterol/24 h (3). The oxysterol is taken up by the liver and further oxidized to bile acids (8). In addition to the role of 24S-OHC in the elimination of cholesterol, it may be of importance in connection with neurodegeneration. Under in vitro conditions, this oxysterol stimulates α-secretase activity and inhibits β-secretase activity in neuronal cell lines (21–23). This is consistent with a preventive effect of 24S-OHC on generation of amyloid. In accordance with this, it was recently reported that injection of adeno-associated vector encoding CYP46A1 in the cortex and hippocampus of amyloid precursor protein (APP)-mutated mice before the onset of amyloid deposits markedly reduced these deposits (24). In addition, the treatment caused improvement in spatial memory. Further support for the contention that increased formation of 24S-OHC is beneficial was presented in a very recent report demonstrating that disruption of the gene coding for the esterification enzyme ACAT1 in a mouse model of Alzheimer's disease (AD) had a beneficial effect on amyloid generation and memory function (25). Evidence was given that at least part of this effect was mediated by increased levels of 24S-OHC.
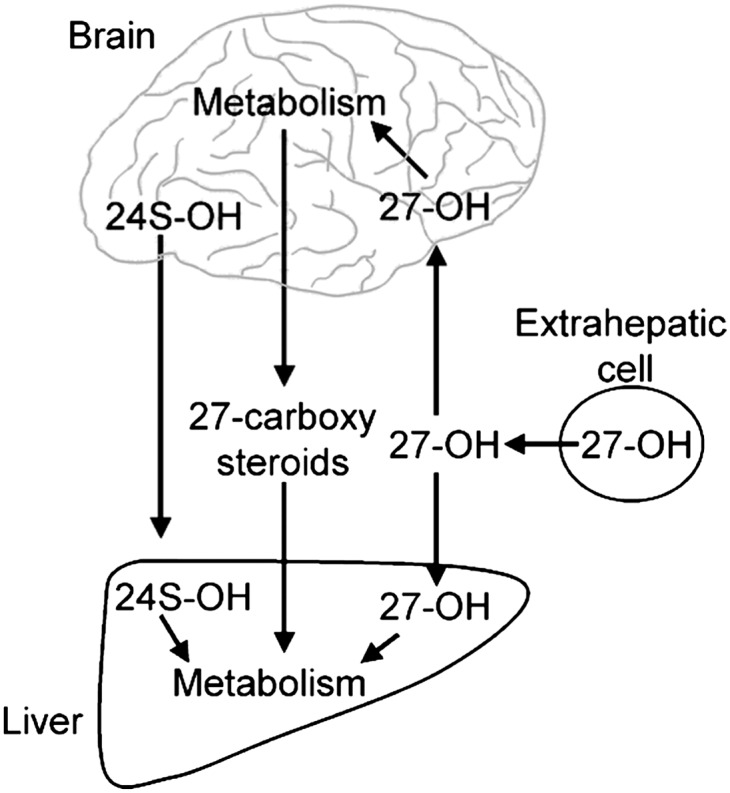
A related SCOx, 27-hydroxycholesterol (27-OHC), has similar physiochemical properties to 24S-OHC and is also able to pass the blood-brain barrier. There is only a low production of this oxysterol in CNS, and most of the 27-OHC present in the brain and cerebrospinal fluid originates from extracerebral sources. Although there is a substantial uptake of 27-OHC by the brain, about 5 mg/24 h (26), the steady-state levels within the brain are very low due to a rapid metabolism into a steroidal acid (27). This acid fluxes from the brain into the circulation and is rapidly metabolized in the liver (27). Oxysterol 7α-hydroxylase (CYP7B1) seems to be the most critical enzyme in this conversion. Similarly to CYP46A1, this enzyme seems to be exclusively located to the neuronal cells. Figure 1 summarizes the flux of oxysterols across the blood-brain barrier.
Fig. 1.
Cross-talk of oxysterols across the blood-brain barrier (3, 27).
27-OHC is a potent inhibitor of cholesterol synthesis and has been shown to antagonize the beneficial effects of 24S-OHC to reducing amyloid accumulation in cultured neuronal cells (22). Based on this, we have suggested that the balance between 24S-OHC and 27-OHC in the brain is of importance for neurodegeneration (28, 29). In light of the importance of an adequate flux of intermediates through the mevalonate pathway, the entry of an inhibitory compound into the brain may have deleterious downstream consequences for cognition.
DEFECTS IN CHOLESTEROL SYNTHESIS
In view of the importance of cholesterol, it is not surprising that relatively few disorders in the synthesis of this molecule are compatible with life. The disorders are associated with accumulation of a cholesterol precursor (lathosterol, desmosterol, or 7-dehydrocholesterol) and severely debilitating and frequently fatal birth defects. For a detailed review, see the review from Porter in this Thematic Series.
Cerebrotendinous xanthomatosis
The patients are characterized by dementia, ataxia, cataracts, and xanthomas in the tendons and in the nervous system. Typical onsets of the disease consists of early bilateral cataracts and diarrhea, followed by cerebellar and pyramidal signs, mental retardation, and xanthomas.
In contrast to the situation in familial hypercholesterolemia, the xanthomas contain high levels of cholestanol, the 5α-saturated analog of cholesterol.
This rare autosomal recessive genetic disorder is caused by a mutation in CYP27A1 that encodes the cytochrome P-450 enzyme sterol 27-hydroxylase [for reviews, see (30, 31)]. The gene contains nine exons and spans 18.6 kb of DNA. The enzyme comprises 498 amino acids and contains putative binding sites for heme ligands and adrenodoxin. CYP27A1 is located at the inner mitochondrial membranes.
CYP27A1 is important for the normal oxidation of the steroid side-chain in connection with biosynthesis of bile acids (12) and is the rate limiting step in the conversion of the C27-steroid into a C24-steroid. The preferred substrates for the enzyme are thus 7α-hydroxylated intermediates in bile acid synthesis. The enzyme is, however, also active on cholesterol. Because the enzyme is likely to be present and active in most cells in the body, and all cells contain cholesterol, there is some production of 27-OHC in most cells. Because 27-OHC is able to pass cell membranes, there is a continuous concentration-dependent flux of 27-OHC to the two organs containing efficient metabolizing systems, i.e., the liver and the brain. In the liver, 27-OHC and its peripheral metabolites are further oxidized into bile acids. The flux of 27-OHC and its metabolites to the liver can be regarded as an alternative mechanism to the classical HDL-dependent reversed cholesterol transport and may be regarded as antiatherogenic (20, 32). In accordance with this, the levels of CYP27A1 activity are particularly high in macrophages and endothelial cells (20, 31).
The activity of CYP27A1 is dependent upon NADPH and the availability of two electron transporters, adrenodoxin and adrenodoxin reductase (12). Additionally, it seems likely that specific transporters are required to transport cholesterol to CYP27A1 on the inner mitochondrial membranes. At present, no mutation causing cerebrotendinous xanthomatosis (CTX) has been defined in any other protein than CYP27A1. There is, however, a description of a CTX patient with a heterozygous mutation in CYP27A1 who is likely to have a mutation in some additional protein, possibly a protein responsible for transport of cholesterol into the mitochondria (33). The possibility of an exon deletion was excluded. This subject has very low levels of 27-OHC in the circulation. Normally, subjects with a heterozygote mutation in the CYP27A1 gene have levels of 27-OHC about one-half those of normal and have no symptoms.
Currently, more than 300 CTX cases have been described in the literature, with distinctive clusters in Japan, The Netherlands, and Israel. About 30 different mutations in the CYP27A1 gene have been described (missense, nonsense, frameshift, and splice junction).
The activity of the enzyme is dependent upon a binding of the electron carrier adrenodoxin as well as binding of heme, and the mutations causing CTX are all located in the adrenodoxin binding or heme ligand binding sites (31). Diagnosis of CTX is obtained by assay of 27-OHC, cholestanol, and bile alcohols in the circulation and MRI. Cholesterol levels are increased in the tissues but not in the circulation. The most serious consequence of the disease is the development of xanthomas in the brain and the neurological symptoms caused by this. The preferential site of the brain xanthomas is in the white matter of the cerebellum (30).
Generation of cholestanol is likely to be the driving force in this disease, and accumulation of cholesterol is likely to be secondary to this. Most of the excess cholestanol appears to be derived from 7α-hydroxylated intermediates during the synthesis of bile acids. Because CYP27A1 is required for oxidation of the steroid side chain in the normal biosynthesis of bile acids, there is an accumulation of 7α-hydroxylated precursors to bile acids containing an intact side chain, in particular 7α-hydroxy-4-cholesten-3-one (Fig. 2). This accumulation is further increased as a consequence of the markedly reduced formation of the bile acid chenodeoxycholic acid. The latter bile acid is the most effective suppressor of the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1) in humans (34). Thus, there is a marked upregulation of CYP7A1 in CTX, and the levels of 7α-hydroxy-4-cholesten-3-one in the circulation may be increased up to 100-fold (35). The latter steroid is able to pass the blood-brain barrier and may be converted into cholestanol in the brain (36). It is noteworthy that treatment with chenodeoxycholic acid reverses the biochemical changes, and even the xanthomas in the brain may be reduced or disappear as a consequence of such treatment (37). In some of the latter patients, the treatment with chenodeoxycholic acid was shown to improve cognition (36).
Fig. 2.

Metabolic relation between bile acid synthesis and formation of cholestanol in patients with CTX (30, 36).
The reason for the accumulation of cholesterol in parallel with cholestanol in the xanthomas is not clear. Cholestanol is, however, less efficient than cholesterol in suppressing cholesterol synthesis (38), and a dilution of a cholesterol pool with cholestanol may cause a compensatory increased synthesis of cholesterol. In some experiments with experimental animals, dietary treatment with cholestanol has been shown to increase cholesterol synthesis (39, 40).
In addition to formation of cholestanol, the accumulation of 7α-hydroxylated intermediates in bile acid synthesis leads to formation of 7α- and 25-hydroxylated alcohols, e.g., 5β-cholestane-3α,7α,12α,25-tetrol. These alcohols may be secreted in bile and urine in gram amounts, and detection of these compounds is generally used in the diagnosis of the disease.
It is noteworthy that a disruption of the gene coding for the Cyp27a1 in mice does not lead to formation of xanthomas in tendons or brain (41). There is some accumulation of cholestanol in these structures, in particular in female mice, but this accumulation is less marked and is not accompanied by accumulation of cholesterol (Björkhem, unpublished observation). The reason for the difference between human CTX and the corresponding mouse model is not known with certainty. Part of the explanation may be that the degree of upregulation of the CYP7A1 as a consequence of the lack of the CYP27A1 is less marked in the mouse model than in patients with CTX.
Hereditary spastic paresis type SPG5
Hereditary spastic paresis (HSP) is a genetically and clinically very heterogenous group of upper motor neuron degenerative diseases (42–46). The disease is characterized by selective axonal loss in the corticospinal tracts and dorsal columns. The cardinal features consist of progressive spasticity of the lower limbs and muscle weakness, frequently associated with deep sensory loss and urinary urgency. These features define “pure” HSP, while “complex” or “complicated” HSP are accompanied by other neurological signs such as ataxia, mental retardation, dementia, visual dysfunction, and epilepsy. The different subgroups of HSP are associated with all modes of inheritance and about 40 loci have been described. The first pure autosomal-recessive HSP locus defined was called SPG5, located on chromosome 8q12.3.
Surprisingly, this locus was very recently identified as the gene coding for the CYP7B1 (46). To date, more than 20 unrelated, HSP-affected families with 17 different mutations in the CYP7B1 gene have been described (42–46). Most patients with the SPG5 are “pure” with progressive lower limb spasticity as the only symptom, but a few of the patients can be regarded as “complicated” and also present ataxia, mental retardation, and neurological symptoms. At this time, it is not known whether it will be possible to relate the site of the mutation and the degree of inhibition of the CYP7B1 activity to the clinical picture.
CYP7B1 is a 506 amino acid enzyme that catalyses the 7α-hydroxylation of several steroids and SCOx [for a recent review, see (47)]. The enzyme is located in the endoplasmic reticulum and is dependent upon NADPH and cytochrome P-450 oxidoreductase for activity. The enzyme has a broad tissue distribution and is present in liver, kidney, brain, and endocrine tissues. The substrate specificity is broad and includes the two oxysterols 25-hydroxycholesterol (25-OHC) and 27-OHC (48). In addition, a number of C19 and C21-steroids, e.g., dehydroepiandrosterone and pregnenolone, are substrates for the enzyme (49).
CYP7B1 is evolutionarily conserved and is of importance both for bile acid synthesis and for metabolism of neurosteroids. Its role in bile acid synthesis is well defined, because the alternative pathway to bile acids starting with a 27-hydroxylation of cholesterol as the first step in the liver or in extrahepatic tissues is dependent on CYP7B1 (12). The metabolism of 27-OHC in the brain also requires CYP7B1 (27). The importance of CYP7B1 in connection with the turnover of the neurosteroids dehydroepiandrosterone and pregnenolone is less well defined. As anticipated, SPG5 patients have increased plasma levels of 27- OHC (6- to 9-fold) and 25-OHC (100-fold) (50). A 30- to 50- fold increase in 27-OHC was found in cerebrospinal fluid.
A disruption of the gene coding for Cyp7b1 in mice leads to accumulation of 27-OHC and 25-OHC in the circulation, but otherwise the mice have no obvious phenotype (51). This is in contrast to the situation in humans; mice with a knockout of Cyp7b1 do not develop spastic paresis in spite of the elevated levels of 25-OHC and 27-OHC. A transgenic mouse model overexpressing human CYP27A1 has also been developed and characterized (52). As with the Cyp7b1 knockout mice, the CYP27A1 transgenic mice have increased levels of 27-OHC, levels similar to those in Cyp7b1-deficient mice. As with the Cyp27a1 knockout mice, the CYP27A1 transgenic mice do not present any obvious phenotype.
It may be concluded from the experiments with Cyp7b1-deficient and Cyp27a1-overxpressing mice that high levels of 25-OHC and 27-OHC are not sufficient to cause obvious neurological deficits. The situation is different in humans, however. The absolute plasma levels of the two SCOx are about 5-fold higher in patients with the SPG5 disease than in the above mouse models. Another factor that may be of importance is the relatively small corticospinal tract and its redundancy in rodents compared with humans (49).
Thus, we do not know if it is the high levels of the oxysterols or the defective metabolism of neurosteroids that is the most important pathogenetic factor in patients with the SPG5 disease. SCOx such as 25-OHC and 27-OHC are known to be cytotoxic to cultured cells (20, 53). It has been reported that 25-OHC induces production of interleukin-1β and interleukin-8 from human macrophages (54, 55) as well as altering the production of IgA (56). We have discussed the possibility that a local interleukin release as a consequence of the high levels of 25-OHC may be a pathogenetic factor in the SPG5 patients (50).
The brains of patients with AD have reduced levels of CYP7B1 (57), probably as a consequence of the neuronal location of this enzyme and the neuronal loss in this disease. In accordance with this, the brains of these patients contain elevated levels of 27-OHC (58). The possibility has been discussed that this accumulation may be of some pathogenetic importance (28). If the accumulation of 25-OHC and 27-OHC is the most important pathogenetic factor in the SPG5 disease, it may be beneficial to reduce the levels of these oxysterols. Because it is known that substrate availability is a limiting factor for CYP27A1 activity, we have suggested that a possible therapeutic strategy could be to treat the patients with a cholesterol synthesis inhibitor (50). Such treatment is thus known to reduce the levels of 27-OHC in parallel with cholesterol.
HSP is, however, not the only pathological consequence of a mutation in the CYP7B1 gene. A decade before the link between SPG5 and CYP7B1 was reported, a fatal case with mutations in the CYP7B1 gene was reported, an infant who died in the neonatal stage with liver failure (59). Whether or not the mutation was causative or a contributing factor to the death of the infant is not known with certainty. The 10-week-old boy presented with severe cholestasis, cirrhosis, and liver failure. The plasma levels of 27-OHC were increased more than 1,000 times those of normal controls, considerably higher than in adult patients with SPG5. Another oxysterol, 24S-OHC, was increased by a similar magnitude in spite of the fact that this oxysterol is not a substrate for CYP7B1 (60). The latter finding suggests that other factors than the loss of the CYP7B1 activity must have contributed. More recently, another neonatal case was described with severe cholestatic liver disease, a mutation in the CYP7B1 gene and a fatal outcome (61). The levels of oxysterols were never measured in this patient. To our knowledge, neonatal cholestasis has not been reported in any adult patient with the SPG5 disease. Thus, it seems likely that other factors than a CYP7B1 mutation must have contributed to the severe cholestasis in the two neonatal patients described thus far. The situation may be similar to that in patients with CTX. A few neonatal cases of this disease have been described with severe cholestasis (62, 63).
Huntington's disease
Huntington's disease (HD) is an inherited dominant neurodegenerative disorder characterized by a glutamine expansion within the N terminus of the huntingtin protein (HTT) (64). It has a prevalence of 3–10 affected subjects/100,000 individuals in Western Europe and North America. The CAG trinucleotide repeats is located within the coding region of exon 1 of the huntingtin gene, which is located on the short arm of chromosome 4 (4p63).
HTT is a protein composed of more than 3,100 amino acids with a molecular weight of about 349 kDa, depending on the exact number of glutamine residues. A polymorphic proline-rich segment follows the polyglutamine tail. Within the cell, wild-type HTT is mainly localized in the cytoplasm but is also associated with mitochondria, Golgi apparatus, endoplasmic reticulum, synaptic vesicles, and several components of the cytoskeleton. A small amount of HTT can also be found in the nucleus. The wild-type HTT is essential for normal embryonic development, because knockout mutations to exons 4 or 5 or to the promoter results in embryonic lethality. HTT is widely expressed throughout the body, and it has been ascribed numerous roles in various intracellular functions, including protein trafficking, vesicle transport and anchoring, clathrin-mediated endocytosis, postsynaptic signaling, and transcriptional regulation, as well as antiapoptotic function (65). In the HD brain, particularly in striatum and cerebral cortex, an altered intracellular localization and perinuclear accumulation of mutant HTT is observed, with the formation of neuronal intranuclear inclusions (66) and aggregates in dystrophic neurites (67). Such aggregates are considered the pathological hallmarks of HD and can be present in gene-positive premanifest subjects who are close to the estimated age-at-onset (68).
The expanded polyglutamine is thought to confer a new function to HTT that is toxic to the cell and impairs the correct function of the protein. The mutant protein tends to auto-aggregate and to promote aberrant protein-protein interactions in several subcellular locations (69), leading to the disruption of several intracellular pathways. The loss of function contributes to the disruption of intracellular homeostasis, leading to neuronal dysfunction and death. The contribution of both toxic gain of function and loss of function of wild-type HTT culminates in neuronal dysfunction as an overall consequence of activation of protease, protein misfolding, transcriptional deregulation, proteasome impairment, oxidative injury, mitochondrial dysfunction, and disruption of axonal transport and synaptic function (64).
A classic degenerative feature of HD is the gradual atrophy of the striatum (caudate nucleus and putamen) together with astrogliosis (70). The most afflicted neuronal populations are the medium-sized projection spiny neurons, which correspond to 95% of the striatal neuronal population. Because these γ-amino butyric acid-utilizing neurons are inhibitory, it is thought that the loss of their inhibition is the underlying cause of the uncontrolled movement characteristic of HD (71). According to MRI investigations, there is also a severe cortical atrophy combined with striatal degeneration. The loss in brain mass is quite substantial in HD: during the 15–20 years of the disease course from the onset of symptoms to death, the brain weight declines in patients from an average of 1.350 g to <1.100 g (71). The clinical associates of such dramatic neurodegeneration, mainly occurring in the striatum and cortex (72), are progressive motor deterioration including chorea and dystonia. In addition there, are psychiatric and behavioral abnormalities, which may precede or accompany the motor onset and cognitive decline (73). Neuropathological findings in HD are mirrored by evidence of progressive atrophy of the striatum on MRI scans (68).
The expression of some important genes involved in the cholesterol biosynthetic pathway, HMGCR, sterol 14-α demethylase, and 7-dehydrocholesterol 7-reductase, is reduced in inducible mutant HTT cell lines as well as in striatum and cortex from transgenic R6/2 HTT-fragment mice (74). These changes are also present in brain and fibroblasts from HD patients (75). Additional studies showed that cholesterol synthesis and accumulation (as indicated by content of the cholesterol precursors lanosterol and lathosterol and by decreased enzymatic activity of HMGCR) are significantly reduced in the brain of several rodent models such as the R6/2 mice, the yeast artificial chromosome mice, the (HdhQ111/111) Hdh knockin mice, and others (76, 77) (Valenza and Leoni, unpublished observations). The degree of reduction of cholesterol synthesis and accumulation was found to increase with the length of the CAG repeats, the amount of mutated HTT, and age. Thus, the levels of cholesterol and precursors are only slightly reduced in young animals and much more reduced in older animals (Valenza and Leoni, unpublished observations). The molecular mechanism underlying this dysfunction appears to be a mutant HTT-dependent decrease in the amount of active gene coding for sterol regulatory element binding protein (SREBP), resulting in less activation of SREBP-controlled genes. The molecular mechanism behind the mutation in the huntingtin gene and the reduced level of SREBP is not known with certainty, however.
Interestingly, wild-type HTT is able to bind to nuclear receptors involved in lipid metabolism like LXR, PPARγ, and vitamin D receptor (78). Overexpression of HTT thus activates LXR, whereas in cells with a lack of HTT, there is an inhibition of LXR-mediated transcription. The possibility must be considered that in the case of HD, the mutated HTT is less able to upregulate LXR and LXR-targeted genes, including SREBP. Such a mechanism is a possible link between the HTT mutation and the disturbances in cholesterol metabolism. Further work is needed, however, to establish this.
Neurodegeneration, with loss of neurons, would be expected to lead to reduced levels of CYP46A1 with subsequent reduction in the formation of 24S-OHC and a lower efflux from the brain to the circulation. In accordance with this, the 24S-OHC content was reduced in both brain and circulation of yeast artificial chromosome 128 mice (76). In a large cohort of controls, HD patients and gene-positive premanifesting patients, a significant reduction of plasma levels in 24-OHC was observed in clinically manifesting patients. Notably, this reduction was correlated to the shrinking of striatum as estimated by MRI. In the case of preHD, it was found that patients closer to the onset of symptoms had levels similar to the HD stage 1 patients, and those far from onset had levels similar to those of controls (79). It is likely that the observed reduction of cholesterol turnover is a consequence of a loss of metabolically active neurons in brain. In a population of gene-positive, pre-HD individuals, we observed a reverse relationship between length of the CAG repeats and plasma levels of 24S-OHC (Leoni, unpublished observations).
Niemann-Pick disease
Niemann Pick disease Type C (NPC) is a rare autosomal recessive neurovisceral lipid storage disease with no known cure (80). Progressive neurological disease is a hallmark and is responsible for disability and premature death beyond early childhood. The neurological symptoms include ataxia, dysathria, dysphagia, tremor, and epilepsy. In the terminal stages, the patients have a loss volitional movements and are severely demented. Mutations in the NPC type C1 (NPC1) and NPC type C2 (NPC2) specific genes have been identified as the cause of the disease, with mutations in NPC1 responsible for the vast majority (95%) of clinical cases (81). Unusually, despite very similar clinical manifestations, the NPC1 and NPC2 proteins are unrelated. NPC1 is a large membrane-anchored protein with homology to HMGCR, SREBP cleavage activating protein, and patched 1, a gene involved in Hedgehog signaling (82, 83). In contrast, NPC2 is a small soluble glycoprotein (81). The common clinical picture is thought to be a consequence of the fact that both NPC1 and NPC2 participate in the movement of lipids, in particular cholesterol, out of the endolysosomal system. This has led to the consensus that NPC disease is a cholesterol storage disease. NPC patients have a markedly impaired capacity for cholesterol esterification and accumulate free cholesterol, which may be directly observed by staining cultured fibroblasts from the patients with fluorescent probes such as filipin (84). The latter procedure is regarded to be the best diagnostic test.
As mentioned above, almost all cases of NPC disease arise from mutations in NPC1. NPC1 is a large (1,278 amino acid) transmembrane protein with 13 putative membrane-spanning regions interspersed by various loops projecting into the lumen of endosomes or lysosomes (82). There is significant homology between helices 3-7 and the so-called sterol sensing domain of other proteins involved in cholesterol turnover (SREBP cleavage activating protein and HMGCR) and hedgehog signaling (patched 1), indicating that there may be a common mechanism for sterol sensing. Using a photoactivable cholesterol analog, Ohgami et al. (85) showed that sterol binding to NPC1 required a functional sterol sensing domain. More recent studies by Infante et al. (86) have confirmed and extended these observations. In an unbiased screen to identify a membrane-bound oxysterol binding protein, Infante et al. (86) reported, somewhat unexpectedly, that intact recombinant NPC1 was capable of binding SCOx as well as cholesterol. The highest affinity binding occurred with 24S-, 25-, and 27-OHC. Notably, the authors reported that while SCOx were able to impair the binding of cholesterol, the reverse was not true, implying that there was more than one sterol binding site in the molecule and indeed suggesting that separate cholesterol and oxysterol binding sites are present (86). In a subsequent study, the same group defined a well-conserved, 240 amino acid loop region of NPC1 as the sterol binding site. Using a recombinant protein, these authors were able to demonstrate that sterol binding was saturable in this region, with a 13-fold higher affinity for 25-OHC compared with cholesterol (Kd 10 nM vs. 130 nM, respectively), indicating that this is the oxysterol binding site on the protein (87). Because the brain contains about 0.1 mM levels of 24S-OH, it can be speculated that this site is largely saturated with this oxysterol under in vivo conditions. The potential functional role of such binding is impossible to evaluate with the present knowledge, although it is possible that it may influence cholesterol supply to the ER, where 24S-OHC is synthesized. Emerging studies have indicated that the expression of NPC1mRNA and protein are regulated by LXR both in vitro and in vivo. Oxysterols such as 24S-OHC (86–88) are thought to be responsible for the activation in vivo. In this connection, it is of interest that treatment with LXR agonists increased the lifespan of NPC null mice (89). In the brain in particular, the convergence of different pathways on the 24S-OHC underscores the importance of this metabolite for brain cholesterol homeostasis.
In contrast to NPC1, NPC2 is a small (132 amino acid) soluble glycoprotein present in both the lysosomes and in various secreted fluids [to date, NPC2 has been identified in milk, epididymal fluid, plasma, and bile (88–90)]. The structure of NPC2 has been determined, and functional studies have confirmed that the loosely packed hydrophobic core is an authentic cholesterol binding pocket and that NPC2 binds cholesterol with low micromolar binding affinities (91). Available data indicate that cholesterol is oriented such that the 3β-hydroxy group is at the entrance to the binding site, and the remainder of the hydrophobic cholesterol molecule is buried within the NPC2. Notably, it has also been shown using competition assays that SCOx are unable to bind to NPC2 (92). Despite the clear data on cholesterol binding to NPC2, studies have shown that although some point mutations do not significantly affect the capacity for cholesterol binding, they do impair the ability of exogenous NPC2 to correct NPC2 deficiency in cell systems. It has been proposed that NPC2 facilitates the rapid movement of cholesterol between lipid bilayers to NPC1, providing a plausible explanation for how mutations in either of these unrelated proteins may lead to clinical manifestations of the disease (93).
Accumulation of free cholesterol in tissues is a consistent finding in NPC patients, including the brain (94), where cholesterol may also be redistributed within individual neurons (95). It should be noted that the extremely long half-life of the majority of brain cholesterol has obscured the direct identification of cholesterol accumulation using analytical biochemical methods. However, using filipin microfluorodensitometry, Treiber-Held et al. (94) demonstrated a significant accumulation of cholesterol in the brains of NPC1 null mice, with the cholesterol accumulating before the onset of a disease phenotype. A consequence of cholesterol accumulation of this is that the neurons become distorted; NPC patients often have ectopic dendrite formation and neurofibrillary tangles similar to those see in AD (96). The Purkinje cells of the cerebellum seem particularly vulnerable to the effects of cholesterol accumulation, perhaps related to the capacity of the cerebellum to synthesize cholesterol.
In addition to cholesterol, several other lipids have been found to accumulate in NPC disease, most notably sphingomyelin in the periphery and glycosphingolipids in the brain (for details, see the review from Xu et al. in this Thematic Series). Very recently, it was proposed that the earliest defect in NPC disease is accumulation of sphingosine (97). Using the class II amphiphile U18666A to induce NPC phenotype in cultured cells, Lloyd-Evans et al. (97) were able to show a very rapid (within 10 min) accumulation of sphingosine followed by the accumulation of cholesterol and other lipids. It should be noted, however, that U18666A may interact with many other proteins, complicating the interpretation of this model. This compound is an inhibitor of oxidosqualene cyclase, a key enzyme in the synthesis of cholesterol (98). The authors speculated that the accumulation of sphingosine disturbs lysosomal calcium homeostasis and leads to impaired vesicle trafficking and fusion (97). Indeed, these authors report the accumulation of sphingosine (3-fold) and sphingosine-1-phosphate (7-fold) in the brain of NPC1 null mice. Moreover, by treating the mouse model with a large dose of curcumin (150 mg/kg/day), a compound able to elevate cytosolic calcium levels, the authors were able to increase the life expectancy of the mice. It should be noted that S1P has recently been recognized as an endogenous histone deacetylase inhibitor and that by inhibiting CBP/p300 histone acetyl transferase activity, curcumin is able to modulate gene expression (99). Given that there is accumulating evidence that sterol homeostasis is under epigenetic control, the proposed mechanistic explanation for the action of curcumin may not be the entire story (14, 15, 100).
A recent report indicates that the sphingosine analog FTY720 can stimulate formation of 27-OHC and activate LXR (101). This implies that enhanced formation of SCOx may be a general mechanism engaged to reduce cholesterol in the endolysosomal system. The recent recognition that the STARt domain containing protein MLN64 is able to promote movement of cholesterol to the mitochondria (the site of 27-OHC formation) in the absence of functional NPC1 adds weight to the concept that formation of 27-OHC is a compensatory mechanism (102). Very recent data on the levels of 27-OHC in the plasma of NPC1 null mice has provided some support for this; these levels are markedly elevated (103). However, given the role of CYP7B1 in the metabolism of 27-OHC, it cannot be excluded that metabolism is impaired rather than synthesis increased. In contrast, cultured fibroblasts from NPC patients have a decreased level of 27-OHC (104, 105).
The very recent report on the effectiveness of cyclodextrin therapy further supports the role of cholesterol rather than sphingosine as the pathogenic factor. Treatment of NPC1 knockout mice with cyclodextrin effectively overcame the transport defect in NPC disease and led to excretion of cholesterol (as bile acids) from the affected mice, with the exception of the lungs (106). Early treatment (7 days postnatally) resulted in a significant increase in the lifespan of the affected animals (107). A human trial of this therapy is currently underway.
Very recent data has been presented indicating that the plasma concentration of certain oxysterols, in particular the auto-oxidation products 7-oxocholesterol and cholestane 3,5,6-triol, is elevated in NPC disease, indicating a possible role for measurement of these sterols in the diagnosis and/or monitoring of NPC disease (103, 108). These data are consistent with the previous demonstration by Alvelius et al. (109) that the urine of an NPC patient contains unusual 7-oxygenated bile acid sulfates, possibly representing the elimination product of 7-oxocholesterol. It is difficult to understand the origin of these sterols, which are likely to be formed by autooxidation rather than by enzymes. It has been reported that ferritin is deficient in NPC disease, suggesting a block in iron homeostasis and an inability to correctly store iron, which is consistent with the known pathway for the internalization of transferrin (110, 111). Because iron is known to be able to catalyze the formation of oxysterols from cholesterol via the Fenton reaction, it is feasible that the accumulation of cholesterol leads to the expansion of a pool of substrate for this reaction in vivo in conjunction with iron accumulation. Chodhury et al. (112) have demonstrated that increased membrane cholesterol disturbs the endolysosomal pathway in a general sense, which would be anticipated to impair the transfer of iron from transferring to ferritin. Moreover, transcriptomic studies have indicated that there is a modest increase in the expression of genes involved in oxidative stress in NPC cells, proving some support for this idea (113).
CHOLESTEROL AND AD
AD is a devastating neurodegenerative disorder. In the Western population, it has been estimated that about 10% of the population over the age of 65 years and one-half those over age 85 years have AD (114, 115). The most important characteristic of AD pathology is the presence of extracellular β-amyloid containing senile plaques and τ-protein containing neurofibrillary tangles in the neuronal cells. In rare cases of familiar forms of AD, three distinct genes have been identified that are of importance for generation of amyloid: the APP gene and the presenilin 1 and presenilin 2 genes. Mutations in these genes result in overproduction of amyloid β (Aβ)-peptides or the proportion of the longer Aβ42 form. The longer form is regarded to be more amyloidogenic and toxic than the shorter form due to its highly aggregative nature (116). The clinical consequences of the mutations in the above genes have led to the “amyloid hypothesis” according to which increased synthesis or decreased degradation of Aβ is the most important factor behind the neuroinflammation, neurodegeneration, memory loss, and senile dementia in AD (116). In addition to the amyloid-containing plaques, oligomers of Aβ in the cytoplasm of neurons are thought to be important for the pathogenesis.
Except for the rare cases with a defined mutation in one of the above critical genes (<2% of the patients), AD is a multifactoral disease. It is possible that the amyloid cascade is not always the driving force and there is an imperfect histopathological correlation of plaque deposition on one hand and neuronal loss and dementia on the other hand. At present, the possibility cannot be excluded that AD often is a consequence of a disruption of more fundamental cellular functions that, when impaired, kill neuronal cells and promote plaque formation in the process (117).
Hypercholesterolemia in mid-life is one of several factors associated with a moderately increased risk to develop AD (118, 119). The most important cholesterol-associated risk factor is, however, presence of the ϵ4 isoform of the cholesterol transporter APOE [for reviews, see (120, 121)]. In addition, recent genome wide association studies have provided compelling evidence that genetic variations in APOJ are associated with the risk of developing AD (122–124). However, the precise mechanistic connection(s) between these genes and AD remain poorly described. This is made all the more difficult by the fact that current data indicates a role for APOE in both brain sterol balance and clearance of β-amyloid (125). Regardless of the specific role of APOE (and possibly APOJ) in β-amyloid metabolism, it is evident that there must be significant overlap with pathways governing cholesterol balance, and expression of APOE is known to be regulated by cholesterol.
The concentration of cholesterol in the critical membranes is of importance for the balance between α- and β-secretase activity, with consequences for the rate of generation of amyloid (126). Both APP and Aβ are at least in part associated with cholesterol-rich domains and increased concentrations of cholesterol leads to increased production of Aβ. Conversely, however, β-amyloid may affect cholesterol synthesis or efflux and the concentration of cholesterol in the membranes (127, 128). Thus, it is possible that there is a negative feedback cycle in which increased cholesterol increases production of Aβ that in turn inhibits cholesterol synthesis. Under some conditions, this balance may, however, be disrupted with a switching toward the amyloidogenic pathway. Also, the state of τ phosphorylation is affected by cholesterol levels in the lipid rafts, with reduced cholesterol stimulating phosphorylation (129).
A detailed description of the great number of different factors involved in neurodegeneration possible to modulate by cholesterol is, however, beyond the scope of this article. Here, we focus on two established genetic risk factors for AD: hypercholesterolemia and presence of the ϵ4 isoform of APOE.
Hypercholesterolemia as a risk factor for AD
Hypercholesterolemia is a risk factor not only for atherosclerosis but also for AD. Increased cholesterol levels in midlife have been linked to AD-type pathology in autopsy studies (130, 131). Treatment of rabbits and mice with cholesterol-enriched diets has been reported to result in cerebral accumulation of amyloid (132, 133). The highly unphysiological conditions used for the animal experiments makes it difficult, however, to draw firm conclusions. In a recent study, it was found that the induction of AD-like neuropathology by dietary cholesterol depends on the quality of the water that the animal was drinking (134).
In view of the fact that lipoprotein-bound cholesterol in the circulation does not pass the blood-brain barrier under normal conditions, it is difficult to understand the mechanism by which hypercholesterolemia increases the risk for neurodegeneration. We have suggested that the oxysterol 27-OHC could be a link between hypercholesterolemia and neurodegeneration (28, 29). In contrast to cholesterol, 27-OHC passes the blood-brain barrier and there is a positive correlation between levels of cholesterol and 27-OHC in the circulation (135). Thus, it is likely that hypercholesterolemia leads to a higher uptake of 27-OHC. The latter steroid has been shown to antagonize the preventive effect of 24S-OHC on generation of amyloid under in vitro conditions (22, 23). In addition, 27-OHC has a downregulating effect on the “memory protein” activity-regulated cytoskeleton-associated protein in mouse brain (136). The same effect is obtained by treatment of the mice with a cholesterol-enriched diet, consistent with the possibility that 27-OHC is the link between the hypercholesterolemia and the effect. Further studies are needed, however, to clarify this.
If high cholesterol in the circulation is a pathogenetic factor in AD, a lowering of these levels may be beneficial. In view of the fact that hypercholesterolemia increases the risk for AD by a factor of about 2 (119, 121), only relatively small effects can be expected after treatment with lipid-lowering drugs. A great number of clinical studies of different design have been performed with statins [for a review see (137)]. Several of these studies have shown a positive effect ,whereas others have been negative. Two large, very well-designed, double-blind, randomized, placebo-controlled trials of statins in people at risk for dementia were, however, negative (138) and at present it is not possible to conclude that statins have a protective effect on AD.
Role of APOE and its different isoforms in AD and other neurodegenerative diseases
The strongest risk factor known for nonfamiliar AD is the genotype for the cholesterol transporter APOE [for general and more detailed reviews, see (120, 121)]. The fact that the ApoE genotype might account for at least 50% of AD in a Caucasian population is one of the most obvious indications of the importance of cholesterol homeostasis for development of a neurodegenerative disease. It should be emphasized, however, that given the pleiotrophic physiological functions of APOE, part of the genetic association with neuropathological disease may be unrelated to the known role of the protein in cholesterol metabolism.
APOE, a 34 kDa protein composed of 299 amino acids, is the most important lipoprotein for uptake of lipoproteins by the LDL receptor, the LDL receptor related protein, the APOE receptor 2, the VLDL receptor, and megalin (119, 120). In humans, the APOE gene shows polymorphism with three different alleles (E2, E3, and E4) that gives rise to six different phenotypes (E2/2, E2/3, E2/4, E3/3, E3/4, E4/4). APOE3 is the most common isoform (77–78%), while APOE2 is found in 7–8% and APOE4 in 14–16% in most Western populations. Subjects with one or two copies of APOE4 have a higher risk of developing AD compared with carriers of other isoforms. APOE also reduces the median age for AD onset from 84 in noncarriers to 68 in E4 homozygotes. The E4 allele occurs in about 60% of AD patients (about 15% with E4/4, 40% with E3/4, and <5% with APOE2/4). The 2% of the population with the E4/4 genotype carries about 15 times the risk of the 60% of the population that has the E3/3 genotype. It has been reported that no APOE4/4 carrier has been shown to reach the age 90 without having AD (139).
From a structural point of view, the difference among the three isoforms is minor, involving only two amino acid positions [E2 (cys112,cys158), E3 (cys112,arg158), and E4 (arg112,arg158)].
These changes affect the three-dimensional structure as well as the lipid binding properties of the isoforms. Thus, APOE3 and APOE2 preferentially bind HDL, whereas APOE4 preferentially binds VLDL. It is likely that this change will affect the capacity of APOE to participate in the distribution of lipids necessary for proliferation, synaptogenesis, and myelinization of axons. It important to note that humans are the only species that has arg61 in APOE, which leads to domain interaction as a second layer of structural complexity.
The evolutionary history of APOE genotypes in humans is unique and has been proposed to be a result of adaptive changes (120, 121). According to current concepts, the APOE4 allele is the ancestral gene, existing alone until 300,000 years ago, at which time the APOE3 allele appeared. The APOE2 allele mutated from the E3 allele about 200,000 years ago. Introduction of the new APOE genotypes provided better cognitive and cardiovascular function to those rare subjects who survived beyond 60 years of age at that time. The retention of more elderly subjects with intact cognitive function in the population may have been important for accumulation of knowledge and expansion of human populations (139).
A great number of studies have appeared demonstrating that the association of APOE4 to AD affects the majority of the pathological events in AD (Aβ-generation and deposition, formation of neurofibrillary tangles, neuronal survival, lipid homeostasis, intracellular signaling) (121). With respect to Aβ-generation, APOE4 has been shown to stimulate endocytic recycling of APP, resulting in increased net production (140). In contrast to APOE4, APOE3, and APOE2 seem to have a protective role by inhibiting Aβ aggregation and/or favoring Aβ-clearance (141).
Under in vitro conditions, APOE3 but not APOE4 forms a stable complex with τ. Phosphorylation of τ inhibits its interaction with APOE3, suggesting that APOE3 only binds to nonphosphorylated τ. Based on this, it has been suggested that APOE3 might be able to prevent the abnormal hyperphosphorylation occurring in AD and the destabilization of the neuronal cytoskeleton (142). The carboxy terminal-truncated form of APOE4 is neurotoxic and stimulates τ phosphorylation (143). The enzymatic cleavage of APOE to give the carboxy-terminal truncated form may thus be important, and it has been shown that the serin protease involved in this hydrolysis cleaves APOE4 more efficiently than APOE3. The APOE fragments are generated inside cultured neuronal cells and in AD brains and may interact with phospho-τ and phosphorylated neurofilaments of high molecular weight, resulting in large filamentous intracellular inclusions in the neuronal cells (144). Studies with transgenic mice have shown increased phosphorylation of τ in mice expressing human APOE4 in neurons, but not in mice expressing this protein in astrocytes (145). The effect of APOE4 thus seems to be specific for neuronal cells.
Induction of APOE synthesis by neuronal cells has been suggested to protect them from injury or to promote intraneuronal repair and maintenance of synapto-dendritic connections (121). When synthesized by neurons, however, the above-mentioned serine protease is also active and cleaves APOE, resulting in fragments that are detrimental to the repair/maintenance process. Because of its conformation and reactivity, APOE4 is more sensitive to cleavage than APOE3.
Cholesterol uptake by cultured neuronal cells is lower when the lipid is bound to APOE4 compared with APOE2 and APOE3 (146). APOE4 is less efficient than the other forms to stimulate cholesterol efflux from astrocytes and neuronal cells. Experiments with transgenic mouse models in which the murine APOE has been replaced with human APOE4 or APOE3 have shown that APOE4 but not APOE3 transgenic mice have an age-dependent disruption of synaptic organization (147). It has been shown in humans that APOE4 carriers show a poor compensation of neuronal loss in different brain regions, whereas non-E4 carriers exhibit marked regenerative changes in the same areas (120, 148).
In all the above cases, the presence of APOE4 is either negative or does not have a positive effect in relation to that of other isoforms. The negative effect is, however, not restricted to AD, as the presence of APOE4 has also negative effects on other neurological diseases, including stroke, severity of head trauma, Parkinsons's disease, amyotrophic lateral sclerosis, multiple sclerosis, Lewy body disease, and CNS ischemia [for a detailed review, see (121)]. Thus, it is tempting to suggest that APOE might participate in molecular mechanisms that are common to several disorders (120, 121).
Footnotes
Abbreviations:
- Aβ
- amyloid β
- AD
- Alzheimer's disease
- APO
- apolipoprotein
- APP
- amyloid precursor protein
- CNS
- central nervous system
- CTX
- cerebrotendinous xanthomatosis
- CYP7A1
- gene coding for cytochrome P-450 7A1, cholesterol 7α-hydroxylase
- CYP7B1
- gene coding for cytochrome P-450 7B1, oxysterol 7α hydroxylase
- CYP27
- gene coding for cytochrome P-450 CYP27A1, sterol 27-hydroxylase
- HD
- Huntington's disease
- HMGCR
- gene coding for 3-hydroxy-3-methylglutaryl CoA reductase
- HSP
- hereditary spastic paresis
- HTT
- huntingtin protein
- LXR
- liver X receptor
- NPC
- Niemann-Pick disease Type C
- 24S-OHC
- 24S-hydroxycholesterol
- 25-OHC
- 25-hydroxycholesterol
- 27-OHC
- 27-hydroxycholesterol
- PNS
- peripheral nervous system
- SCOx
- side-chain oxidized oxysterol
- SREBP
- gene coding for sterol regulatory element binding protein
The investigations performed in the authors' laboratory have been supported by grants from the Swedish Research Council, Swedish Brain Power, and the Blanceflor Foundation nee' Bildt.
REFERENCES
- 1.Dietschy J. M., Turley S. D. 2004. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45: 1375–1397. [DOI] [PubMed] [Google Scholar]
- 2.Snipe G., Suter U. 1997. Cholesterol and myelin. Subcellular Biochemistry, Vol. 28 Bittman R., editor Plenum Press, New York: pp 173–204. [DOI] [PubMed] [Google Scholar]
- 3.Bjorkhem I., Meaney S. 2004. Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24: 806–815. [DOI] [PubMed] [Google Scholar]
- 4.Saher G., Quintes S., Mobius W., Wehr M. C., Kramer-Albers E. M., Brugger B., Nave K. A. 2009. Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J. Neurosci. 29: 6094–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfrieger F. W. 2003. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays. 25: 72–78. [DOI] [PubMed] [Google Scholar]
- 6.Funfschilling U., Saher G., Xiao L., Mobius W., Nave K. A. 2007. Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saher G., Brugger B., Lappe-Siefke C., Mobius W., Tozawa R., Wehr M. C., Wieland F., Ishibashi S., Nave K. A. 2005. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8: 468–475. [DOI] [PubMed] [Google Scholar]
- 8.Bjorkhem I., Andersson U., Ellis E., Alvelius G., Ellegard L., Diczfalusy U., Sjovall J., Einarsson C. 2001. From brain to bile. Evidence that conjugation and omega-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J. Biol. Chem. 276: 37004–37010. [DOI] [PubMed] [Google Scholar]
- 9.Russell D. W., Halford R. W., Ramirez D. M., Shah R., Kotti T. 2009. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78: 1017–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pitas R. E., Boyles J. K., Lee S. H., Foss D., Mahley R. W. 1987. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta. 917: 148–161. [DOI] [PubMed] [Google Scholar]
- 11.Abildayeva K., Jansen P. J., Hirsch-Reinshagen V., Bloks V. W., Bakker A. H., Ramaekers F. C., de Vente J., Groen A. K., Wellington C. L., Kuipers F., et al. 2006. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 281: 12799–12808. [DOI] [PubMed] [Google Scholar]
- 12.Russell D. W. 2003. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72: 137–174. [DOI] [PubMed] [Google Scholar]
- 13.Ohyama Y., Meaney S., Heverin M., Ekstrom L., Brafman A., Shafir M., Andersson U., Olin M., Eggertsen G., Diczfalusy U., et al. 2006. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J. Biol. Chem. 281: 3810–3820. [DOI] [PubMed] [Google Scholar]
- 14.Shafaati M., O'Driscoll R., Bjorkhem I., Meaney S. 2009. Transcriptional regulation of cholesterol 24-hydroxylase by histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 378: 689–694. [DOI] [PubMed] [Google Scholar]
- 15.Nunes M. J., Milagre I., Schnekenburger M., Gama M. J., Diederich M., Rodrigues E. Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of Cyp46a1 gene transcription. J. Neurochem. 113: 418–431. [DOI] [PubMed] [Google Scholar]
- 16.Wechsler A., Brafman A., Shafir M., Heverin M., Gottlieb H., Damari G., Gozlan-Kelner S., Spivak I., Moshkin O., Fridman E., et al. 2003. Generation of viable cholesterol-free mice. Science. 302: 2087. [DOI] [PubMed] [Google Scholar]
- 17.Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., Russell D. W. 2006. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. USA. 103: 3869–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansen P. J., Lutjohann D., Thelen K. M., von Bergmann K., van Leuven F., Ramaekers F. C., Monique M. 2009. Absence of ApoE upregulates murine brain ApoD and ABCA1 levels, but does not affect brain sterol levels, while human ApoE3 and human ApoE4 upregulate brain cholesterol precursor levels. J. Alzheimers Dis. 18: 319–329. [DOI] [PubMed] [Google Scholar]
- 19.Mato M., Ookawara S., Mashiko T., Sakamoto A., Mato T. K., Maeda N., Kodama T. 1999. Regional difference of lipid distribution in brain of apolipoprotein E deficient mice. Anat. Rec. 256: 165–176. [DOI] [PubMed] [Google Scholar]
- 20.Bjorkhem I., Diczfalusy U. 2002. Oxysterols: friends, foes, or just fellow passengers? Arterioscler. Thromb. Vasc. Biol. 22: 734–742. [DOI] [PubMed] [Google Scholar]
- 21.Brown J., 3rd, Theisler C., Silberman S., Magnuson D., Gottardi-Littell N., Lee J. M., Yager D., Crowley J., Sambamurti K., Rahman M. M., et al. 2004. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J. Biol. Chem. 279: 34674–34681. [DOI] [PubMed] [Google Scholar]
- 22.Famer D., Meaney S., Mousavi M., Nordberg A., Bjorkhem I., Crisby M. 2007. Regulation of alpha- and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem. Biophys. Res. Commun. 359: 46–50. [DOI] [PubMed] [Google Scholar]
- 23.Prasanthi J. R., Huls A., Thomasson S., Thompson A., Schommer E., Ghribi O. 2009. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 4: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudry E., Van Dam D., Kulik W., De Deyn P. P., Stet F. S., Ahouansou O., Benraiss A., Delacourte A., Bougneres P., Aubourg P., et al. 2010. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol. Ther. 18: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryleva E. Y., Rogers M. A., Chang C. C., Buen F., Harris B. T., Rousselet E., Seidah N. G., Oddo S., LaFerla F. M., Spencer T. A., et al. 2010. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA. 107: 3081–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heverin M., Meaney S., Lutjohann D., Diczfalusy U., Wahren J., Bjorkhem I. 2005. Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J. Lipid Res. 46: 1047–1052. [DOI] [PubMed] [Google Scholar]
- 27.Meaney S., Heverin M., Panzenboeck U., Ekstrom L., Axelsson M., Andersson U., Diczfalusy U., Pikuleva I., Wahren J., Sattler W., et al. 2007. Novel route for elimination of brain oxysterols across the blood-brain barrier: conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J. Lipid Res. 48: 944–951. [DOI] [PubMed] [Google Scholar]
- 28.Bjorkhem I., Cedazo-Minguez A., Leoni V., Meaney S. 2009. Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 30: 171–179. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkhem I., Heverin M., Leoni V., Meaney S., Diczfalusy U. 2006. Oxysterols and Alzheimer's disease. Acta Neurol. Scand. Suppl. 185: 43–49. [DOI] [PubMed] [Google Scholar]
- 30.Bjorkhem I., Muri-Boberg K., Lietersdorf E. 2001. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. The Metabolic Basis of Inherited Diseases. Scrivener C., Beaudet A., Sly W., Valle D., McGraw-Hill, New York: 2961–2988. [Google Scholar]
- 31.Gallus G. N., Dotti M. T., Federico A. 2006. Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol. Sci. 27: 143–149. [DOI] [PubMed] [Google Scholar]
- 32.Babiker A., Andersson O., Lund E., Xiu R. J., Deeb S., Reshef A., Leitersdorf E., Diczfalusy U., Bjorkhem I. 1997. Elimination of cholesterol in macrophages and endothelial cells by the sterol 27-hydroxylase mechanism. Comparison with high density lipoprotein-mediated reverse cholesterol transport. J. Biol. Chem. 272: 26253–26261. [DOI] [PubMed] [Google Scholar]
- 33.Hansson M., Olin M., Floren C. H., von Bahr S., van't Hooft F., Meaney S., Eggertsen G., Bjorkhem I. 2007. Unique patient with cerebrotendinous xanthomatosis. Evidence for presence of a defect in a gene that is not identical to sterol 27-hydroxylase. J. Intern. Med. 261: 504–510. [DOI] [PubMed] [Google Scholar]
- 34.Ellis E., Axelson M., Abrahamsson A., Eggertsen G., Thorne A., Nowak G., Ericzon B. G., Bjorkhem I., Einarsson C. 2003. Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology. 38: 930–938. [DOI] [PubMed] [Google Scholar]
- 35.Bjorkhem I., Skrede S., Buchmann M. S., East C., Grundy S. 1987. Accumulation of 7 alpha-hydroxy-4-cholesten-3-one and cholesta-4,6-dien-3-one in patients with cerebrotendinous xanthomatosis: effect of treatment with chenodeoxycholic acid. Hepatology. 7: 266–271. [DOI] [PubMed] [Google Scholar]
- 36.Panzenboeck U., Andersson U., Hansson M., Sattler W., Meaney S., Bjorkhem I. 2007. On the mechanism of cerebral accumulation of cholestanol in patients with cerebrotendinous xanthomatosis. J. Lipid Res. 48: 1167–1174. [DOI] [PubMed] [Google Scholar]
- 37.Berginer V. M., Salen G., Shefer S. 1984. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N. Engl. J. Med. 311: 1649–1652. [DOI] [PubMed] [Google Scholar]
- 38.Lund E., Bjorkhem I. 1994. Down-regulation of hepatic HMG-CoA reductase in mice by dietary cholesterol: importance of the delta 5 double bond and evidence that oxidation at C-3, C-5, C-6, or C-7 is not involved. Biochemistry. 33: 291–297. [DOI] [PubMed] [Google Scholar]
- 39.Bjorkhem I., Buchmann M. S., Skrede S. 1985. On the structural specificity in the regulation of the hydroxymethylglutaryl-CoA reductase and the cholesterol-7 alpha-hydroxylase in rats. Effects of cholestanol feeding. Biochim. Biophys. Acta. 835: 18–22. [DOI] [PubMed] [Google Scholar]
- 40.Shefer S., Hauser S., Salen G., Zaki F. G., Bullock J., Salgado E., Shevitz J. 1984. Comparative effects of cholestanol and cholesterol on hepatic sterol and bile acid metabolism in the rat. J. Clin. Invest. 74: 1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosen H., Reshef A., Maeda N., Lippoldt A., Shpizen S., Triger L., Eggertsen G., Bjorkhem I., Leitersdorf E. 1998. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J. Biol. Chem. 273: 14805–14812. [DOI] [PubMed] [Google Scholar]
- 42.Biancheri R., Ciccolella M., Rossi A., Tessa A., Cassandrini D., Minetti C., Santorelli F. M. 2009. White matter lesions in spastic paraplegia with mutations in SPG5/CYP7B1. Neuromuscul. Disord. 19: 62–65. [DOI] [PubMed] [Google Scholar]
- 43.Criscuolo C., Filla A., Coppola G., Rinaldi C., Carbone R., Pinto S., Wang Q., de Leva M. F., Salvatore E., Banfi S., et al. 2009. Two novel CYP7B1 mutations in Italian families with SPG5: a clinical and genetic study. J. Neurol. 256: 1252–1257. [DOI] [PubMed] [Google Scholar]
- 44.Goizet C., Boukhris A., Durr A., Beetz C., Truchetto J., Tesson C., Tsaousidou M., Forlani S., Guyant-Marechal L., Fontaine B., et al. 2009. CYP7B1 mutations in pure and complex forms of hereditary spastic paraplegia type 5. Brain. 132: 1589–1600. [DOI] [PubMed] [Google Scholar]
- 45.Schule R., Brandt E., Karle K. N., Tsaousidou M., Klebe S., Klimpe S., Auer-Grumbach M., Crosby A. H., Hubner C. A., Schols L., et al. 2009. Analysis of CYP7B1 in non-consanguineous cases of hereditary spastic paraplegia. Neurogenetics. 10: 97–104. [DOI] [PubMed] [Google Scholar]
- 46.Tsaousidou M. K., Ouahchi K., Warner T. T., Yang Y., Simpson M. A., Laing N. G., Wilkinson P. A., Madrid R. E., Patel H., Hentati F., et al. 2008. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am. J. Hum. Genet. 82: 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stiles A. R., McDonald J. G., Bauman D. R., Russell D. W. 2009. CYP7B1: one cytochrome P450, two human genetic diseases, and multiple physiological functions. J. Biol. Chem. 284: 28485–28489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwarz M., Lund E. G., Lathe R., Bjorkhem I., Russell D. W. 1997. Identification and characterization of a mouse oxysterol 7alpha-hydroxylase cDNA. J. Biol. Chem. 272: 23995–24001. [DOI] [PubMed] [Google Scholar]
- 49.Rose K. A., Stapleton G., Dott K., Kieny M. P., Best R., Schwarz M., Russell D. W., Bjorkhem I., Seckl J., Lathe R. 1997. Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7alpha-hydroxy dehydroepiandrosterone and 7alpha-hydroxy pregnenolone. Proc. Natl. Acad. Sci. USA. 94: 4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schule R., Siddique T., Deng H. X., Yang Y., Donkervoort S., Hansson M., Madrid R. E., Siddique N., Schols L., Bjorkhem I. 2010. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J Lipid Res. 51: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li-Hawkins J., Lund E. G., Turley S. D., Russell D. W. 2000. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J. Biol. Chem. 275: 16536–16542. [DOI] [PubMed] [Google Scholar]
- 52.Meir K., Kitsberg D., Alkalay I., Szafer F., Rosen H., Shpitzen S., Avi L. B., Staels B., Fievet C., Meiner V., et al. 2002. Human sterol 27-hydroxylase (CYP27) overexpressor transgenic mouse model. Evidence against 27-hydroxycholesterol as a critical regulator of cholesterol homeostasis. J. Biol. Chem. 277: 34036–34041. [DOI] [PubMed] [Google Scholar]
- 53.Clare K., Hardwick S. J., Carpenter K. L., Weeratunge N., Mitchinson M. J. 1995. Toxicity of oxysterols to human monocyte-macrophages. Atherosclerosis. 118: 67–75. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y., Hulten L. M., Wiklund O. 1997. Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arterioscler. Thromb. Vasc. Biol. 17: 317–323. [DOI] [PubMed] [Google Scholar]
- 55.Rosklint T., Ohlsson B. G., Wiklund O., Noren K., Hulten L. M. 2002. Oxysterols induce interleukin-1beta production in human macrophages. Eur. J. Clin. Invest. 32: 35–42. [DOI] [PubMed] [Google Scholar]
- 56.Bauman D. R., Bitmansour A. D., McDonald J. G., Thompson B. M., Liang G., Russell D. W. 2009. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA. 106: 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yau J. L., Rasmuson S., Andrew R., Graham M., Noble J., Olsson T., Fuchs E., Lathe R., Seckl J. R. 2003. Dehydroepiandrosterone 7-hydroxylase CYP7B: predominant expression in primate hippocampus and reduced expression in Alzheimer's disease. Neuroscience. 121: 307–314. [DOI] [PubMed] [Google Scholar]
- 58.Heverin M., Bogdanovic N., Lutjohann D., Bayer T., Pikuleva I., Bretillon L., Diczfalusy U., Winblad B., Bjorkhem I. 2004. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J. Lipid Res. 45: 186–193. [DOI] [PubMed] [Google Scholar]
- 59.Setchell K. D., Schwarz M., O'Connell N. C., Lund E. G., Davis D. L., Lathe R., Thompson H. R., Weslie Tyson R., Sokol R. J., Russell D. W. 1998. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Invest. 102: 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Norlin M., Toll A., Bjorkhem I., Wikvall K. 2000. 24-Hydroxycholesterol is a substrate for hepatic cholesterol 7alpha-hydroxylase (CYP7A). J. Lipid Res. 41: 1629–1639. [PubMed] [Google Scholar]
- 61.Ueki I., Kimura A., Nishiyori A., Chen H. L., Takei H., Nittono H., Kurosawa T. 2008. Neonatal cholestatic liver disease in an Asian patient with a homozygous mutation in the oxysterol 7alpha-hydroxylase gene. J. Pediatr. Gastroenterol. Nutr. 46: 465–469. [DOI] [PubMed] [Google Scholar]
- 62.Clayton P. T., Verrips A., Sistermans E., Mann A., Mieli-Vergani G., Wevers R. 2002. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J. Inherit. Metab. Dis. 25: 501–513. [DOI] [PubMed] [Google Scholar]
- 63.von Bahr S., Bjorkhem I., Van't Hooft F., Alvelius G., Nemeth A., Sjovall J., Fischler B. 2005. Mutation in the sterol 27-hydroxylase gene associated with fatal cholestasis in infancy. J. Pediatr. Gastroenterol. Nutr. 40: 481–486. [DOI] [PubMed] [Google Scholar]
- 64.Agorogiannis E. I., Agorogiannis G. I., Papadimitriou A., Hadjigeorgiou G. M. 2004. Protein misfolding in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 30: 215–224. [DOI] [PubMed] [Google Scholar]
- 65.Gil J. M., Rego A. C. 2008. Mechanisms of neurodegeneration in Huntington's disease. Eur. J. Neurosci. 27: 2803–2820. [DOI] [PubMed] [Google Scholar]
- 66.DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 277: 1990–1993. [DOI] [PubMed] [Google Scholar]
- 67.Sapp E., Penney J., Young A., Aronin N., Vonsattel J. P., DiFiglia M. 1999. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J. Neuropathol. Exp. Neurol. 58: 165–173. [DOI] [PubMed] [Google Scholar]
- 68.Aylward E. H. 2007. Change in MRI striatal volumes as a biomarker in preclinical Huntington's disease. Brain Res. Bull. 72: 152–158. [DOI] [PubMed] [Google Scholar]
- 69.Graham R. K., Deng Y., Slow E. J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., et al. 2006. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 125: 1179–1191. [DOI] [PubMed] [Google Scholar]
- 70.Vonsattel J. P., Myers R. H., Stevens T. J., Ferrante R. J., Bird E. D., Richardson E. P., Jr 1985. Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44: 559–577. [DOI] [PubMed] [Google Scholar]
- 71.Vonsattel J. P., DiFiglia M. 1998. Huntington disease. J. Neuropathol. Exp. Neurol. 57: 369–384. [DOI] [PubMed] [Google Scholar]
- 72.Rosas H. D., Liu A. K., Hersch S., Glessner M., Ferrante R. J., Salat D. H., van der Kouwe A., Jenkins B. G., Dale A. M., Fischl B. 2002. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 58: 695–701. [DOI] [PubMed] [Google Scholar]
- 73.Marder K., Zhao H., Myers R. H., Cudkowicz M., Kayson E., Kieburtz K., Orme C., Paulsen J., Penney J. B., Jr., Siemers E., et al. 2000. Rate of functional decline in Huntington's disease. Huntington Study Group. Neurology. 54: 452–458. [DOI] [PubMed] [Google Scholar]
- 74.Sipione S., Rigamonti D., Valenza M., Zuccato C., Conti L., Pritchard J., Kooperberg C., Olson J. M., Cattaneo E. 2002. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 11: 1953–1965. [DOI] [PubMed] [Google Scholar]
- 75.Valenza M., Rigamonti D., Goffredo D., Zuccato C., Fenu S., Jamot L., Strand A., Tarditi A., Woodman B., Racchi M., et al. 2005. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J. Neurosci. 25: 9932–9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valenza M., Carroll J. B., Leoni V., Bertram L. N., Bjorkhem I., Singaraja R. R., Di Donato S., Lutjohann D., Hayden M. R., Cattaneo E. 2007. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum. Mol. Genet. 16: 2187–2198. [DOI] [PubMed] [Google Scholar]
- 77.Valenza M., Leoni V., Tarditi A., Mariotti C., Bjorkhem I., Di Donato S., Cattaneo E. 2007. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 28: 133–142. [DOI] [PubMed] [Google Scholar]
- 78.Futter M., Diekmann H., Schoenmakers E., Sadiq O., Chatterjee K., Rubinsztein D. C. 2009. Wild-type but not mutant huntingtin modulates the transcriptional activity of liver X receptors. J. Med. Genet. 46: 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leoni V., Mariotti C., Tabrizi S. J., Valenza M., Wild E. J., Henley S. M., Hobbs N. Z., Mandelli M. L., Grisoli M., Bjorkhem I., et al. 2008. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington's disease. Brain. 131: 2851–2859. [DOI] [PubMed] [Google Scholar]
- 80.Vanier M. T., Millat G. 2003. Niemann-Pick disease type C. Clin. Genet. 64: 269–281. [DOI] [PubMed] [Google Scholar]
- 81.Storch J., Xu Z. 2009. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta. 1791: 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davies J. P., Ioannou Y. A. 2000. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J. Biol. Chem. 275: 24367–24374. [DOI] [PubMed] [Google Scholar]
- 83.Kuwabara P. E., Labouesse M. 2002. The sterol-sensing domain: multiple families, a unique role? Trends Genet. 18: 193–201. [DOI] [PubMed] [Google Scholar]
- 84.Pentchev P. G., Comly M. E., Kruth H. S., Vanier M. T., Wenger D. A., Patel S., Brady R. O. 1985. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. USA. 82: 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohgami N., Ko D. C., Thomas M., Scott M. P., Chang C. C., Chang T. Y. 2004. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. USA. 101: 12473–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Infante R. E., Abi-Mosleh L., Radhakrishnan A., Dale J. D., Brown M. S., Goldstein J. L. 2008. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283: 1052–1063. [DOI] [PubMed] [Google Scholar]
- 87.Infante R. E., Radhakrishnan A., Abi-Mosleh L., Kinch L. N., Wang M. L., Grishin N. V., Goldstein J. L., Brown M. S. 2008. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283: 1064–1075. [DOI] [PubMed] [Google Scholar]
- 88.Klein A., Amigo L., Retamal M. J., Morales M. G., Miquel J. F., Rigotti A., Zanlungo S. 2006. NPC2 is expressed in human and murine liver and secreted into bile: potential implications for body cholesterol homeostasis. Hepatology. 43: 126–133. [DOI] [PubMed] [Google Scholar]
- 89.Larsen L. B., Ravn P., Boisen A., Berglund L., Petersen T. E. 1997. Primary structure of EPV20, a secretory glycoprotein containing a previously uncharacterized type of domain. Eur. J. Biochem. 243: 437–441. [DOI] [PubMed] [Google Scholar]
- 90.Legare C., Thabet M., Gatti J. L., Sullivan R. 2006. HE1/NPC2 status in human reproductive tract and ejaculated spermatozoa: consequence of vasectomy. Mol. Hum. Reprod. 12: 461–468. [DOI] [PubMed] [Google Scholar]
- 91.Friedland N., Liou H. L., Lobel P., Stock A. M. 2003. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. USA. 100: 2512–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu S., Benoff B., Liou H. L., Lobel P., Stock A. M. 2007. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 282: 23525–23531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Infante R. E., Wang M. L., Radhakrishnan A., Kwon H. J., Brown M. S., Goldstein J. L. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA. 105: 15287–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Treiber-Held S., Distl R., Meske V., Albert F., Ohm T. G. 2003. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J. Pathol. 200: 95–103. [DOI] [PubMed] [Google Scholar]
- 95.Karten B., Vance D. E., Campenot R. B., Vance J. E. 2002. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 83: 1154–1163. [DOI] [PubMed] [Google Scholar]
- 96.Walkley S. U., Suzuki K. 2004. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta. 1685: 48–62. [DOI] [PubMed] [Google Scholar]
- 97.Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14: 1247–1255. [DOI] [PubMed] [Google Scholar]
- 98.Sexton R. C., Panini S. R., Azran F., Rudney H. 1983. Effects of 3 beta-[2-(diethylamino)ethoxy]androst-5-en-17-one on the synthesis of cholesterol and ubiquinone in rat intestinal epithelial cell cultures. Biochemistry. 22: 5687–5692. [DOI] [PubMed] [Google Scholar]
- 99.Giandomenico V., Simonsson M., Gronroos E., Ericsson J. 2003. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol. Cell. Biol. 23: 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chittur S. V., Sangster-Guity N., McCormick P. J. 2008. Histone deacetylase inhibitors: a new mode for inhibition of cholesterol metabolism. BMC Genomics. 9: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Blom T., Back N., Mutka A. L., Bittman R., Li Z., de Lera A., Kovanen P. T., Diczfalusy U., Ikonen E. 2010. FTY720 stimulates 27-Hydroxycholesterol production and confers atheroprotective effects in human primary macrophages. Circ Res. 106: 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Charman M., Kennedy B. E., Osborne N., Karten B. 2010. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J Lipid Res. 51: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ory D. S., Porter F. D. 2009. Biomarkers for Niemann-Pick C disease and related disorders. United States patent US20090286272. 2009 Apr 1. [Google Scholar]
- 104.De Windt A., Rai M., Kytomaki L., Thelen K. M., Lutjohann D., Bernier L., Davignon J., Soini J., Pandolfo M., Laaksonen R. 2007. Gene set enrichment analyses revealed several affected pathways in Niemann-pick disease type C fibroblasts. DNA Cell Biol. 26: 665–671. [DOI] [PubMed] [Google Scholar]
- 105.Frolov A., Zielinski S. E., Crowley J. R., Dudley-Rucker N., Schaffer J. E., Ory D. S. 2003. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 278: 25517–25525. [DOI] [PubMed] [Google Scholar]
- 106.Liu B., Ramirez C. M., Miller A. M., Repa J. J., Turley S. D., Dietschy J. M. 2009. Cyclodextrin overcomes the transport defect in nearly every organ of the newborn or mature NPC1 mouse leading to excretion of the sequestered cholesterol as bile acid. J. Lipid Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. 2009. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE. 4: e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ory D., Porter F., Scherrer D., Lanier M., Langmade S., Molugu V., Gale S., Olzeski D., Sidhu R., Wassif C., et al. 2010. Cholesterol oxidation productsnext term are sensitive and specific blood-based biomarkers for Niemann–Pick C1 disease. Mol. Genet. Metab. 99: S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alvelius G., Hjalmarson O., Griffiths W. J., Bjorkhem I., Sjovall J. 2001. Identification of unusual 7-oxygenated bile acid sulfates in a patient with Niemann-Pick disease, type C. J. Lipid Res. 42: 1571–1577. [PubMed] [Google Scholar]
- 110.Christomanou H., Harzer K. 1996. Ouchterlony double immunodiffusion method demonstrates absence of ferritin immunoreactivity in visceral organs from nine patients with Niemann-Pick disease type C. Biochem. Mol. Med. 58: 176–183. [DOI] [PubMed] [Google Scholar]
- 111.Christomanou H., Kellermann J., Linke R. P., Harzer K. 1995. Deficient ferritin immunoreactivity in visceral organs from four patients with Niemann-Pick disease type C. Biochem. Mol. Med. 55: 105–115. [DOI] [PubMed] [Google Scholar]
- 112.Choudhury A., Sharma D. K., Marks D. L., Pagano R. E. 2004. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol. Biol. Cell. 15: 4500–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reddy J. V., Ganley I. G., Pfeffer S. R. 2006. Clues to neuro-degeneration in Niemann-Pick Type C disease from global gene expression profiling. PLoS ONE. 1: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rocca W. A., Hofman A., Brayne C., Breteler M. M., Clarke M., Copeland J. R., Dartigues J. F., Engedal K., Hagnell O., Heeren T. J., et al. 1991. Frequency and distribution of Alzheimer's disease in Europe: a collaborative study of 1980–1990 prevalence findings. The EURODEM-Prevalence Research Group. Ann. Neurol. 30: 381–390. [DOI] [PubMed] [Google Scholar]
- 115.European Collaboration on Dementia. 2009. Prevalence of dementia. Accessed at http://alzheimer.vo.lu/site/Our-Research/European-Collaboration-on-Dementia.
- 116.Hardy J., Selkoe D. J. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 297: 353–356. [DOI] [PubMed] [Google Scholar]
- 117.Herz J. 2007. Overview: the long and winding road to understanding Alzheimer's disease. Neuron. 53: 477–479. [DOI] [PubMed] [Google Scholar]
- 118.Kivipelto M., Helkala E. L., Hanninen T., Laakso M. P., Hallikainen M., Alhainen K., Soininen H., Tuomilehto J., Nissinen A. 2001. Midlife vascular risk factors and late-life mild cognitive impairment: a population-based study. Neurology. 56: 1683–1689. [DOI] [PubMed] [Google Scholar]
- 119.Solomon A., Kivipelto M., Wolozin B., Zhou J., Whitmer R. A. 2009. Midlife serum cholesterol and increased risk of Alzheimer's and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 28: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cedazo-Minguez A. 2007. Apolipoprotein E and Alzheimer's disease: molecular mechanisms and therapeutic opportunities. J. Cell. Mol. Med. 11: 1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mahley R. W., Weisgraber K. H., Huang Y. 2006. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 103: 5644–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Williams A., et al. 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41: 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lambert J. C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M. J., Tavernier B., et al. 2009. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41: 1094–1099. [DOI] [PubMed] [Google Scholar]
- 124.van Es M. A., van den Berg L. H. 2009. Alzheimer's disease beyond APOE. Nat. Genet. 41: 1047–1048. [DOI] [PubMed] [Google Scholar]
- 125.Deane R., Sagare A., Hamm K., Parisi M., Lane S., Finn M. B., Holtzman D. M., Zlokovic B. V. 2008. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118: 4002–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Puglielli L., Tanzi R. E., Kovacs D. M. 2003. Alzheimer's disease: the cholesterol connection. Nat. Neurosci. 6: 345–351. [DOI] [PubMed] [Google Scholar]
- 127.Grimm M. O., Grimm H. S., Hartmann T. 2007. Amyloid beta as a regulator of lipid homeostasis. Trends Mol. Med. 13: 337–344. [DOI] [PubMed] [Google Scholar]
- 128.Umeda T., Mori H., Zheng H., Tomiyama T. 2010. Regulation of cholesterol efflux by amyloid beta secretion. J Neurosci Res. 88: 1985–1994. [DOI] [PubMed] [Google Scholar]
- 129.Michikawa M. 2006. Role of cholesterol in amyloid cascade: cholesterol-dependent modulation of tau phosphorylation and mitochondrial function. Acta Neurol. Scand. Suppl. 185: 21–26. [DOI] [PubMed] [Google Scholar]
- 130.Launer L. J., White L. R., Petrovitch H., Ross G. W., Curb J. D. 2001. Cholesterol and neuropathologic markers of AD: a population-based autopsy study. Neurology. 57: 1447–1452. [DOI] [PubMed] [Google Scholar]
- 131.Pappolla M. A., Bryant-Thomas T. K., Herbert D., Pacheco J., Fabra Garcia M., Manjon M., Girones X., Henry T. L., Matsubara E., Zambon D., et al. 2003. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 61: 199–205. [DOI] [PubMed] [Google Scholar]
- 132.Refolo L. M., Malester B., LaFrancois J., Bryant-Thomas T., Wang R., Tint G. S., Sambamurti K., Duff K., Pappolla M. A. 2000. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 7: 321–331. [DOI] [PubMed] [Google Scholar]
- 133.Sparks D. L., Scheff S. W., Hunsaker J. C., 3rd, Liu H., Landers T., Gross D. R. 1994. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126: 88–94. [DOI] [PubMed] [Google Scholar]
- 134.Sparks D. L. 2008. The early and ongoing experience with the cholesterol-fed rabbit as a model of Alzheimer's disease: the old, the new and the pilot. J. Alzheimers Dis. 15: 641–656. [DOI] [PubMed] [Google Scholar]
- 135.Babiker A., Dzeletovic S., Wiklund B., Pettersson N., Salonen J., Nyyssonen K., Eriksson M., Diczfalusy U., Bjorkhem I. 2005. Patients with atherosclerosis may have increased circulating levels of 27-hydroxycholesterol and cholestenoic acid. Scand. J. Clin. Lab. Invest. 65: 365–375. [DOI] [PubMed] [Google Scholar]
- 136.Mateos L., Akterin S., Gil-Bea F. J., Spulber S., Rahman A., Bjorkhem I., Schultzberg M., Flores-Morales A., Cedazo-Minguez A. 2009. Activity-regulated cytoskeleton-associated protein in rodent brain is down-regulated by high fat diet in vivo and by 27-hydroxycholesterol in vitro. Brain Pathol. 19: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Solomon A. 2009. Cholesterol and late-life cognition: an epidemiological and clinical approach. University of Kuopio, Kuopio, Finland. [Google Scholar]
- 138.McGuinness B., Craig D., Bullock R., Passmore P. 2009. Statins for the prevention of dementia. Cochrane Database Syst. Rev. CD003160. [DOI] [PubMed] [Google Scholar]
- 139.Ashford J. W. 2004. APOE genotype effects on Alzheimer's disease onset and epidemiology. J. Mol. Neurosci. 23: 157–165. [DOI] [PubMed] [Google Scholar]
- 140.Ye S., Huang Y., Mullendorff K., Dong L., Giedt G., Meng E. C., Cohen F. E., Kuntz I. D., Weisgraber K. H., Mahley R. W. 2005. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. USA. 102: 18700–18705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Holtzman D. M., Bales K. R., Wu S., Bhat P., Parsadanian M., Fagan A. M., Chang L. K., Sun Y., Paul S. M. 1999. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J. Clin. Invest. 103: R15–R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Strittmatter W. J., Weisgraber K. H., Goedert M., Saunders A. M., Huang D., Corder E. H., Dong L. M., Jakes R., Alberts M. J., Gilbert J. R., et al. 1994. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol 125: 163–171; discussion 172–164. [DOI] [PubMed] [Google Scholar]
- 143.Harris F. M., Brecht W. J., Xu Q., Tesseur I., Kekonius L., Wyss-Coray T., Fish J. D., Masliah E., Hopkins P. C., Scearce-Levie K., et al. 2003. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA. 100: 10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang Y., Liu X. Q., Wyss-Coray T., Brecht W. J., Sanan D. A., Mahley R. W. 2001. Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. USA. 98: 8838–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tesseur I., Van Dorpe J., Spittaels K., Van den Haute C., Moechars D., Van Leuven F. 2000. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol. 156: 951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rapp A., Gmeiner B., Huttinger M. 2006. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie. 88: 473–483. [DOI] [PubMed] [Google Scholar]
- 147.Buttini M., Orth M., Bellosta S., Akeefe H., Pitas R. E., Wyss-Coray T., Mucke L., Mahley R. W. 1999. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J. Neurosci. 19: 4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Arendt T. 2001. Disturbance of neuronal plasticity is a critical pathogenetic event in Alzheimer's disease. Int. J. Dev. Neurosci. 19: 231–245. [DOI] [PubMed] [Google Scholar]