

Abstract
Protein prenylation is a post-translational modification that is present in a large number of proteins; it has been proposed to be responsible for membrane association and protein-protein interactions which contribute to its role in signal transduction pathways. Research has been aimed at inhibiting prenylation with farnesyltransferase inhibitors (FTIs) based on the finding that the farnesylated protein Ras is implicated in 30% of human cancers. Despite numerous studies on the enzymology of prenylation in vitro, many questions remain about the process of prenylation as it occurs in living cells. Here we describe the preparation of a series of farnesylated peptides that contain sequences recognized by protein farnesyltransferase. Using a combination of flow cytometry and confocal microscopy, we show that these peptides enter a variety of different cell types. A related peptide where the farnesyl group has been replaced by a disulfide-linked decyl group is also shown to be able to efficiently enter cells. These results highlight the applicability of these peptides as a platform for further study of protein prenylation and subsequent processing in live cells.
INTRODUCTION
Oncogenically mutated forms of the prenylated protein Ras are found in over 30% of human cancers (1); it has also been demonstrated that the levels of farnesyl diphosphate and geranylgeranyl diphosphate, but not cholesterol, are elevated in the brains of patients with Alzheimer’s Disease, indicating a potentially disease-specific targeting of isoprenoid regulation independent of HMG-CoA-reductase and cholesterol (2, 3). These findings together with the estimation that approximately 2% of all proteins are prenylated (4) underscore the importance of improving our understanding of protein prenylation and isoprenoid diphosphate utilization in vivo to facilitate disease intervention. Protein prenylation is a post translational modification that serves to direct membrane association of many proteins (5). It involves the addition of a farnesyl (15 carbon) or geranylgeranyl (20 carbon) isoprenoid moiety to a C-terminal cysteine residue of a protein containing a CAAX box motif, where C represents cysteine, A represents an aliphatic amino acid and X represents a specific amino acid that controls the length of isoprenoid addition (6–8). Specifically, the amino acids serine, methionine, and glutamine signal farnesylation by the enzyme farnesyltransferase while leucine signals geranylgeranylation by the enzyme geranylgeranyltransferase (9). Upon prenylation, the protein is further processed by an endoprotease (RCE1 protease) that cleaves the “AAX” residues followed by methylation of the new C-terminus by isoprenylcysteine carboxyl methyltransferase (ICMT) (10).
Substantial effort has been focused on the development of inhibitors of the prenyltransferase enzymes and some success has been realized, including the creation of several farnesyltransferase inhibitors (FTIs) that are in stage 2 and 3 clinical trials (11–13) for conditions such as chronic myeloid leukemia and advanced non small cell lung cancer (14, 15). Unfortunately, FTIs have proved to be less useful than expected due to fact that K-Ras, the most frequently mutated form of Ras in human cancers, is able to be alternatively prenylated by the enzyme geranylgeranyltransferase I, thus rendering it able to bypass the effects of an FTI (16, 17). In addition to the development of FTIs, studies detailing the role of prenylation in membrane association (5), protein-protein interactions (18), as well as its effects on signal transduction (19, 20) have been undertaken, with most studies having been conducted in vitro. Even with an abundant knowledge of prenylation in vitro, much remains unclear about the process as it occurs in living cells. For example, prenylation takes place in the cytosol but the resulting proteins are translocated to the plasma membrane via the secratory pathway (21); additionally, prenylated proteins appear to cycle in a dynamic fashion between the plasma membrane and the Golgi. Peptides can provide useful information concerning protein prenylation, but to date, most peptides have been microinjected into cells (22). To gain insight into the processing and localization of prenylated proteins, we have sought to create cell permeable prenylated peptides that can be used to study prenylation in living cells. Recently, we reported on the preparation and cellular distribution of a class of prenylated peptides that freely enter HeLa cells (23). Those molecules, based on the C-terminus of CDC42 include a geranylgeranylated cysteine residue embedded within the CAAX-box sequence CVLL, thus mimicking the C-terminus of a naturally geranylgeranylated protein and rendering them substrates for RCE1 and ICMT. Here, we extend that work to peptides that incorporate the CAAX-box sequences CVLS and CVIM that are present in farnesylated proteins. Flow cytometry and confocal laser scanning microscopy (CLSM) were used to quantify the levels and evaluate the localization patterns of farnesylated peptides in HeLa cells. Several additional cell lines including COS-1 (African green monkey kidney cells), NIH/3T3 Fibroblast (Swiss mouse embryo fibroblasts), D1TNC1 astrocytes (rat astrocyte cell line), and PC-12 cells (derived from rat adrenal pheochromocytoma (24)) were also evaluated for their ability to take up these peptides. These specific cell lines were chosen both for their common laboratory use as well as their utility for future experiments with these peptides. Finally, a peptide incorporating a lipid linked to the CAAX-box cysteine residue thiol via a disulfide was prepared and evaluated for its cell penetrating ability. Entry of such a probe into cells should result in reductive cleavage of the lipid to reveal a CAAX-box containing peptide that can serve as a substrate for protein farnesyltransferase and the subsequent modifying enzymes.
MATERIALS AND METHODS
General
HeLa cells were the generous gift of Dr. Audrey Minden (Department of Chemical Biology, Rutgers University). Petri dishes (35 mm) fitted with microwells (14 mm) and a No. 1.5 coverglass were from MatTek Corporation. Wheat germ agglutinin Alexa Fluor 594 conjugate, Hoechst 34850, and DMEM (Dulbecco’s Modified Eagle Medium) were from Invitrogen. Fetal bovine serum (FBS) was from Intergen Company. Horse Serum (HS) was from Fischer Scientific. F12K media was from ATCC. Mouse β-Nerve Growth Factor was purchased from Raybiotech. Vydac 218TP54 and 218TP1010 columns were used for analytical and preparative RP-HPLC, respectively. All analytical and preparative RP-HPLC solvents, water and CH3CN, contained 0.10% TFA and were of HPLC grade. PAL-PEG-PS was from Applied Biosystems. Preloaded CLEAR-Acid resins were from Peptides International. 5-Carboxyfluorescein was from Berry & Associates. All other reagents were from Sigma Aldrich.
Synthesis
Peptide Synthesis
Peptides were synthesized on an Applied Biosystems 433A automated synthesizer according to manufacturer protocols. 5-Carboxyfluorescein was coupled to the peptide while still on resin in the following manner: In a small test tube, 2.0 equivalents of 1-Hydroxybenzotriazole (HOBt, 0.2 mmol), 2.0 equivalents of N,N′-Diisopropylcarbodiimide (DIC, 0.2 mmol), and 2.1 equivalents of 5-carboxyfluorescein (5-Fam, 0.2 mmol) were pre-incubated for 5 min in 1.2 mL N,N-Dimethylformamide (DMF). This solution was added to the peptide on resin in a syringe with a fritted filter and reacted overnight on a rotisserie wrapped in aluminum foil. The resin was then rinsed with DMF (5x, 3.0 mL each) followed by CH2Cl2 (5x, 3.0 mL each). After drying the crude peptide in vacuo for 2 h, it was cleaved from the resin using freshly prepared Reagent K (25) (TFA/phenol/thioanisole/water/ethanedithiol, 82.5:5:5:5:2.5) for 2h. Following resin cleavage, the peptide was precipitated by the addition of 50 mL diethyl ether (Et2O), centrifuged to form a pellet which was rinsed twice with Et2O, and frozen at −20°C until later purification.
5-Fam-KKSRRC(Acm)VIM (1a)
Synthesized on Fmoc protected methionine CLEAR Acid Resin. The peptide was purified by RP-HPLC on a C18 column using a gradient of 1% B per minute (solvent A: 0.1% aqueous TFA, solvent B: 0.1% TFA in CH3CN) affording a light yellow solid. Product eluted at 31% B, verified with mass spectrometry (deconvoluted ESI-MS calculated for C70H104N18O18S2 1548.7, found 1547.7).
5-Fam-KKSRRC(Scm)VIM (2a)
A 0.27 M stock solution of methoxycarbonylsulfenyl chloride was prepared by adding 5.0 μL of methoxycarbonylsulfenyl chloride to 200 μL acetonitrile; the stock solution was cooled on ice. The concentration of the peptide used was determined by UV spectroscopy of the 5-Fam chromophore (ε492=79,000 M−1cm−1, 492 nm, pH=9.0); this method was used throughout the synthesis to calculate peptide concentration. After the peptide concentration was determined, 1 equivalent of solid peptide (15.1 mg, 9.7 μmol) was dissolved in a 1:1 mixture of DMF and CH3CN (7.0 mL total, approximately 1.0 mL solvent per 1.0 mg peptide). The peptide solution was cooled on ice and 3 equivalents of the 0.27 M methoxycarbonylsulfenyl chloride stock solution (108 μL, 29.2 μmol) was added. The reaction was stirred at rt for 3h in the dark and purified by RP-HPLC on a C18 column using a gradient of 1% B per minute (solvent A: 0.1% aqueous TFA, solvent B: 0.1% TFA in CH3CN) yielding a green solid (9.3 mg, 58%). Product eluted at 30.5% B and was verified with mass spectrometry (deconvoluted ESI-MS calculated for C69H101N17O19S3 1567.7, found 1567.3).
5-Fam-KKSRRCVIM (3a)
1.0 mg of Scm-protected peptide 2a was dissolved in 10 mL of 20 mM Tris (pH=8.0). To this peptide solution, approximately 0.5 mg of solid dithiothreitol (DTT) was added. The reaction was stirred at rt in the dark for approximately 30 min. The product was purified by RP-HPLC on a C18 column using a gradient of 1% B per minute (solvent A: 0.1% aqueous TFA, solvent B: 0.1% TFA in CH3CN) yielding a green solid (0.9 mg, 83%). Product eluted at 28% B and was verified with MS (deconvoluted ESI-MS calculated for C67H99N17O17S2 1477.7, found 1477.5).
5-Fam-KKSRRC(farnesyl)VIM (4a)
100 mM Zn(OAc)2 stock solution was prepared by dissolving 22.0 mg Zn(OAc)2 in 1.0 mL of 0.1% aq. TFA. 1.0 equivalent of 3a (0.6 mg, 0.4 μmol) was dissolved in 600 μL of solvent (DMF/1-butanol/0.1% aqueous TFA, 2:1:1), to which 20.1 μL of the 100 mM stock Zn(OAc)2 solution (5.0 eq., 2.0 μmol) was added. To this solution, 0.4 μL (4.0 eq., 1.6 μmol) of farnesyl bromide was added. The solution was stirred at rt, in the dark, overnight and purified by RP-HPLC on a C18 column using a gradient of 1% B per minute (solvent A: 0.1% aqueous TFA, solvent B: 0.1% TFA in CH3CN) which afforded a light green solid (0.07 mg, 11.7%). Product eluted at 55%B and was verified with high-resolution ESI-MS (deconvoluted ESI-MS for C82H123N17O17S2 1681.8724, found 1681.8888).
5-Fam-KKSRRC(S-decyl)VIM (5a)
A 7.0 mM decanethiol solution was prepared by adding 1.4 μL of decanethiol to 1.0 mL DMF. A 17 mM Zn(OAc)2 stock solution was also prepared by dissolving 3.7 mg of Zn(OAc)2 in 1.0 mL water. Peptide 2a (1.0 eq., 0.5 μmol, 0.7 mg) was dissolved in 100 μL DMF. To the peptide solution, 64.3 μL of the 7.0 mM stock decanethiol solution was added (1.0 eq., 0.5 μmol). After cooling this solution on ice, 132.4 μL of the 17 mM Zn(OAc)2 stock solution was slowly added (5.0 eq., 2.3 μmol). The reaction was stirred at rt in the dark for 4 h and was purified by RP-HPLC on a C18 column using a gradient of 1% B per minute (solvent A: 0.1% aqueous TFA, solvent B: 0.1% TFA in CH3CN) yielding a light green solid (0.08 mg, 11.4%). Product eluted at 52%B and was verified with high-resolution ESI-MS (deconvoluted ESI-MS for C77H119N17O17S3 1649.8132, found 1649.8288).
5-Fam-KKSRRC(Acm)VLS (1b)
Prepared similar to 1a using Fmoc-L-Ser(tBu)-PEG-PS resin. Product eluted at approximately 28% B, verified with mass spectrometry (deconvoluted ESI-MS calculated for C68H100N18O19S 1504.7, found 1504.8).
5-Fam-KKSRRC(Scm)VLS (2b)
Prepared similar to 2a. Reaction scale = 5.7 mg, 3.8 μmol peptide. Yield 3.4 mg (58.9%). Product eluted at 30% B and was verified with ESI-MS (deconvoluted ESI-MS calculated for C67H97N17O20S2 1523.7, found 1523.2).
5-Fam-KKSRRCVLS (3b)
Prepared similar to 3a. Reaction scale = 1.5 mg, 1.0 μmol peptide. Yield 1.4 mg (95.7%). Product eluted at 28% B and was verified with MS (deconvoluted ESI-MS calculated for C65H95N17O18S 1433.7, found 1433.8).
5-Fam-KKSRRC(farnesyl)VLS (4b)
Prepared similar to 4a. Reaction scale = 0.3 mg, 0.2 μmol. Yield 0.04 mg (11.1%). Product eluted at 50%B and was verified with MS (deconvoluted ESI-MS calculated for C80H119N17O18S 1637.9, found 1637.8).
5-Fam-KKSRRC(S-decyl)VLS (5b)
Prepared similar to 5a. Reaction scale = 1.3 mg, 0.9 μmol. Yield 0.4 mg (29.2%). Product eluted at 48%B and was verified with MS (deconvoluted ESI-MS calculated for C75H115N17O18S2 1605.8, found 1605.4).
Cell Experiments
For cell experiments involving HeLa, COS-1, NIH/3T3, and D1TNC1 astrocyte cells, 7.8 × 103 cells/cm2 were seeded in culture dishes and grown to approximately 50% confluency (generally 24 h); PC-12 cells were seeded at 3.9 × 103 cells/cm2. PC-12 cells were differentiated into neurons before analysis (see below). HeLa cells were maintained in DMEM supplemented with 10% FBS; PC-12 were maintained in F12K supplemented with 10% HS and 5% FBS.
Microscopy
The cells were washed twice with PBS (1 mL), followed by the addition of serum-free DMEM (or F12K + 1% HS for PC-12) and incubated for either 1h or 2h at 37°C with 5.0% CO2 with the desired peptide at the desired concentration (typically 1 or 3 μM). Hoechst 34580 nucleus stain was added to a final concentration of 1 μg/mL during the final 20 min of incubation. Additionally, wheat germ agglutinin Alexa Fluor 594 conjugate was added to a final concentration of 5 μg/mL during the final 10 min of incubation. After the incubation period all cells were washed twice with PBS (1mL) and placed back in serum-free DMEM (F12K + 1% HS for PC-12). The cells were then imaged using an Olympus FluoView 1000 confocal microscope with a 60X objective. The fluorophores were monitored at their maximum emission wavelengths of 520 nm, 450 nm, and 620 nm for 5-Fam, Hoechst 34580 nuclear stain, and wheat germ agglutinin Alexa Fluor 594 conjugate, respectively.
Flow cytometry
The cells were rinsed twice with PBS (1 mL) and incubated for 1 h at 37°C with 5.0% CO2 with the desired peptide in serum-free DMEM (or F12K + 1% HS for PC-12) at varying concentrations. Following incubation, the media was aspirated and the cells rinsed with PBS (2X, 1 mL each). The cells were removed from the plate by trypsinization for 15 min (to remove membrane bound peptide) at 37°C with 5.0% CO2 (0.2 mL of a 0.0625% trypsin/versene solution). Cells were re-suspended in 1.8 mL complete DMEM (or F12K for PC-12) and transferred to a 15 mL falcon tube. The cells were pelleted by centrifugation at 100 g for 5 min. The media was aspirated from the cells followed by the addition of 2 mL PBS to resuspend the pellet. Cells were added to a 12 × 75 mm test tube for flow cytometry analysis. A total of 10,000 events for each sample were analyzed using a BD FACSCalibur (BD Biosciences). For ATP depletion, cells were washed twice with glucose-free, serum-free DMEM and then incubated for 2 h at 37°C and 5.0% CO2 in glucose-free, serum-free DMEM supplemented with 12 μM rotenone and 15 mM 2-deoxyglucose (26); then the peptides were added and incubation proceeded as above.
Neuron Differentiation for PC-12
PC-12 cells were differentiated similarly to previously described methods (24). Briefly, PC-12 cells were seeded at 3.9 × 103 cells/cm2 on poly lysine coated plates (27) and grown 24 h at 37°C with 5.0% CO2. Cells were serum starved for 24h with F12K + 1% HS. Addition of F12K media containing 1% HS and 100 ng/mL of β-NGF initiated neuron differentiation. Incubation with β-NGF proceeded for 6–8 days; fresh media was added every 2 days.
Octanol/water partition coefficient (LogP) measurement
LogP values were measured following a previously established procedure (28). Briefly, 100 μL of a peptide solution (50 or 100μM) in 10mM Tris (pH=7.4) was added to 100 μL octanol in a microtube (1.5 mL). A buffered Tris solution was used to ensure the peptide was kept in a state similar to physiological pH. The solution was vortexed for 2 min and centrifuged for 2 min. 25 μL of each layer was removed and diluted either in 100 μL 3:1 methanol/Tris or methanol/octanol for a final composition of 3:1:1 methanol/octanol/Tris. The aqueous layer was further diluted 2–4 fold so the absorbance (at 492 nM for 5-Fam) was in the linear range of the instrument. For measurements at lower concentration, the octanol absorbance was measured directly. The log (A492 of the organic layer/A492 of the aqueous layer) yielded logP. This was repeated twice to yield a mean logP ± standard deviation.
RESULTS AND DISCUSSION
Peptide Synthesis
Synthesis of peptides 1a and 1b was accomplished via standard SPPS with an automated synthesizer utilizing HBTU coupling. After SPPS, on-resin addition of 5-Fam was accomplished in one step using peptide coupling conditions (HOBt, DIC) followed by removal of the acid labile protecting groups and cleavage from the resin with Reagent K (25). It is important to note that the acetamidomethyl (Acm) protecting group on cysteine is not removed during this treatment, which allowed its later conversion to an S-carbomethoxysulfenyl (Scm) group. Conversion of Acm protected peptides 1a and 1b to the Scm protected peptides 2a and 2b was accomplished using a previously described method (23).
In route to peptides 4a and 4b, the free thiol precursors 3a and 3b were first generated by reducing the Scm disulfide protecting groups of 2a and 2b with DTT. After reduction, farnesylation of 3a and 3b was conducted using an acidic, aqueous medium and zinc acetate catalyzed coupling conditions as described by Xue et al. (29). The disulfide linked lipidated peptides 5a and 5b were also generated starting with the Scm protected peptides 2a and 2b. In those cases, disulfide exchange with decanethiol was used to complete the synthesis of 5a and 5b.
Peptide design and uptake in HeLa cells
In earlier work, we investigated the cell penetrating ability of a number of different peptides based on the naturally geranylgeranylated protein CDC42 containing the C-terminal sequence KKSRRCVLL. Those experiments unequivocally demonstrated that such peptides could be introduced into live HeLa cells (23). Here we sought to expand the range of cell penetrating prenylated peptides that can be studied to include sequences that mimic those present in farnesylated proteins. Accordingly, the CAAX box sequences CVIM and CVLS were chosen as they are present in the naturally occurring oncogenic proteins K or N-Ras and H-Ras, respectively (30). The same sequence upstream of the CAAX box in CDC42 was used in the design of these new peptides to facilitate comparison of their uptake characteristics with those previously reported for peptides bearing geranylgeranylation sequences (Figure 1).
Figure 1.

Fluorescently labeled prenylated peptides for in vivo analysis. In all cases, the ‘a’ series of peptides contain the CVIM sequence while the ‘b’ series of peptides contain the CVLS sequence.
Initially, confocal laser scanning microscopy (CLSM) was used to monitor the uptake of the peptides in HeLa cells (Figure 2). Inspection of the CLSM images in Figure 2 clearly indicates significant uptake into HeLa cells for 4a and 4b as well as for the disulfide linked peptides 5a and 5b (all other synthesized peptides showed no cellular uptake). The punctate fluorescence in the cytoplasm is similar to what has been observed previously in images of live cells treated with related geranylgeranylated peptides (23). Next, flow cytometry was used to quantify the uptake of the peptides in live HeLa cells. Cells were incubated with peptides at various concentrations for 1 h, followed by trypsinization for 15 minutes to remove any surface bound peptide and thereby allow the measurement of only the internalized peptide (31). As the concentration of peptide was increased, the uptake increased, although not in a linear fashion (Figure 3). It was previously noted for related geranylgeranylated peptides that longer incubation times result in increased uptake (23), and this trend was observed here for the farnesylated peptides as well (data not shown). Uptake was also compared to a peptide that has a similar sequence but is geranylgeranylated rather than farnesylated (Figure 3, peptide GG, sequence: Ac-K(5-Fam)AKKSRRC(gg)VLL where ‘gg’ represents a geranylgeranyl moiety). While there is a small difference in the sequence, the uptake of the geranylgeranylated peptide at a similar concentration (1μM) is approximately 4 times greater than that of the farnesylated peptide. Increased uptake is presumed to be because of the more hydrophobic geranylgeranyl group. To explore this idea, octanol/water partition coefficient (LogP) values were measured for peptides 4b and GG. With a LogP value of −0.81 ± 0.26, the geranygeranylated peptide GG is noticeably more hydrophobic than the farnesylated peptide 4a, with a LogP of −1.37 ± 0.37. While this difference is small compared to the uptake difference, it is important to note that previous studies by our group have shown that the hydrophobic moiety on cysteine is crucial for uptake, regardless of peptide sequence (23, 32). Furthermore, the enantiomer of peptide 4b was synthesized with D amino acids (D-4b)and uptake was measured by flow cytometry (data not shown). The resulting uptake was similar to that of the standard L amino acid peptide. This data, coupled with the aforementioned observations, indicates that uptake is likely not dependent on a specific receptor transport system. Lastly, both peptide 4a and 4b exhibit similar uptake; however, because methionine is prone to oxidation to methionine sulfoxide (33), peptide 4b was used in subsequent experiments to avoid possible ambiguity due to the presence of multiple related but different species.
Figure 2.
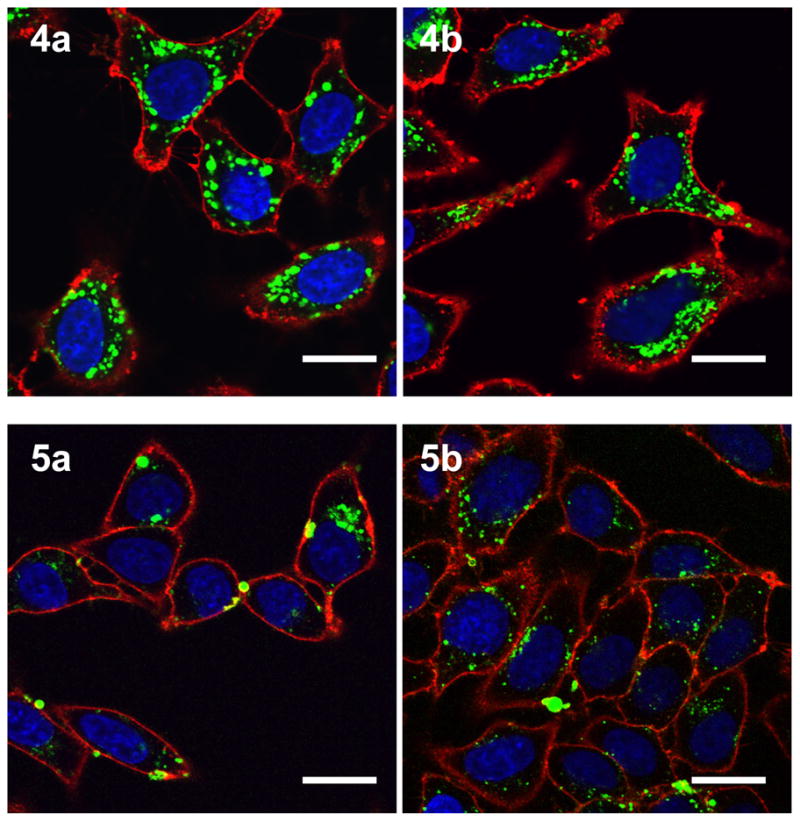
Confocal laser scanning microscopy (CLSM) images of live HeLa cells after incubation with various peptides. Peptides 4a and 4b were incubated for 2h at 3μM while peptides 5a and 5b were incubated for 1h at 1μM. Hoechst 34850 was used to stain the nucleus blue and wheat germ agglutinin Alexa Fluor 594 conjugate was used to stain the plasma membrane red. The peptide is visualized as green fluorescence. Size bar represents 20 μm.
Figure 3.

Flow cytometry analysis of fluorescently labeled prenylated peptides incubated with HeLa cells at 37°C for 1h at varying concentrations. After incubation, cells were trypsinized for 15 min to remove membrane-bound peptide. Each bar represents the geometric mean fluorescence of 10,000 cells. Peptide GG represents a similar sequence that is geranylgeranylated for comparison (sequence Ac-K(Fam)-AKKSRRC(gg)VLL; because peptide characterization and synthesis has been previously described (23), only the 1 μM concentration was used for comparison).
Utility of the peptides in diverse cell types
Previous studies investigating cell penetrating prenylated peptides have focused solely on HeLa cells, concluding that the uptake of the prenylated peptides is ATP-independent, possibly relying on a diffusional process across the cell membrane (23). It is likely that various cells types exhibit different membrane compositions; even within cell cultures themselves the composition of membranes is highly affected by cell–cell contact and culture density (34). To explore the utility of these cell penetrating peptides beyond HeLa cells (a cervical cancer cell line), their ability to enter a diverse range of cell types was studied by both CLSM and flow cytometry. Initial incubation of 4b with COS-1 and NIH/3T3 fibroblast cells showed moderate uptake (Figure 4, panels A and B). Subsequent experiments used D1TNC1 astrocytes and PC-12 cells, which are used as a model of neurons. Both cell lines showed significant uptake (Figure 4, panels C and D). Live cells were used for the above study as it has been shown that cell fixation can cause an artificial redistribution of cell penetrating peptides in the cytoplasm (31).
Figure 4.

CSLM images of various live cells after incubation with peptide 4b (VS) at 1 μM for 2 h. A) COS-1 cells. B) NIH/3T3 fibroblasts. C) D1 astrocytes. D) PC-12 differentiated neurons. The top panel is an overlay of all fluorescence channels, while the bottom panel represents only the green channel for clarity. Hoechst 34850 was used to stain the nucleus blue and wheat germ agglutinin Alexa Fluor 594 conjugate was used to stain the plasma membrane red. The peptide is visualized as green fluorescence. Size bar represents 20 μm.
The uptake pattern of the prenylated peptides reported here can be characterized as punctate (see Figures 2 and 4), which is often indicative of an endocytotic process (35). To explore this issue, flow cytometry was performed under ATP depletion conditions in all cell lines and compared with uptake data obtained under normal conditions (no ATP depletion); a comparison for each cell type is given in Figure 5. No significant difference was observed with uptake under ATP depletion conditions in HeLa and COS-1 cells, suggesting that farnesylated peptide 4b enters in an ATP-independent, non-endocytotic manner similar to what was observed with the aforementioned geranylgeranlyated peptides. In contrast, reduced uptake under ATP depleting conditions was observed in NIH/3T3 and PC-12 cells. The significance of these latter results are unclear since many of the cells did not survive the depletion conditions; additionally, ATP depletion in astrocytes resulted in essentially complete cell death, possibly because this rapidly growing cell is not able to survive extended periods without ATP. Thus, it is possible that entry of 4b into NIH/3T3, PC-12 and astrocytes involves an ATP-dependent process although this appears unlikely in view of the results obtained in the other cell lines. Finally, it is also noteworthy that the amount of peptide that entered both the astrocytes and the PC-12 cells appears to be significantly higher than was observed in the other cell lines tested. This may be significant, suggesting that these cells are more permeable or it may simply reflect the larger size and internal volume of the cells themselves.
Figure 5.

Flow cytometry analysis of peptide 4b incubated with various cells at 1 μM for 2h. Cells were incubated at either 37°C or under conditions of ATP depletion. After incubation, cells were trypsinized for 15 min to remove membrane-bound peptide. Each bar represents the geometric mean fluorescence of 10,000 cells. Experiments performed in triplicate with the results expressed as the mean fluorescence intensity ± standard deviation.
Evaluation of a probe to study in vivo prenylation
From the experiments described above, we demonstrated that peptides containing farnesylation sequences were introduced into living cells. We next sought to develop a peptide that could serve as a substrate for protein farnesyltransferase. If this could be accomplished, it would then be possible to monitor the complete processing of prenylated peptides that includes prenylation, proteolysis and methylation. Accordingly, the disulfide linked peptides 5a and 5b (see Figure 1) were constructed to explore the cell penetrating ability of probes containing a lipid group that would be reductively cleaved upon entry into the reducing environment of the cytoplasm (36). Such a process would reveal a complete CAAX sequence suitable for subsequent modification by the endogenous prenylation machinery (37).
The uptake of the disulfide-linked peptides 5a and 5b into HeLa cells was studied by flow cytometry analysis. As can be seen in Figure 3, the disulfide linked peptides 5a and 5b have greater uptake than the corresponding farnesylated peptides even though they contain only a 10 carbon alkyl chain compared with the farnesyl group (15 carbons) present in 4a and 4b. This may be a result of the straight alkyl chain having more favorable interactions with similarly structured saturated lipids in the plasma membrane, while the isoprenoid chain contains branches and elements of unsaturation that may render it less likely to integrate with and pass through the membrane. In support of this hypothesis, the calculated octanol/water partition coefficient (ClogP) value of N-acetylcysteine methyl ester with a farnesyl group is lower (3.82) than that of the same amino acid conjugated to a disulfide linked decyl group (5.31), suggesting that the decyl group is more hydrophobic and may have more favorable membrane interactions.
Finally, the significant cell penetrating capability of these disulfide-linked peptides led us to investigate their ability to enter other cell types. Flow cytometry analysis (see Figure 6) was used to quantify their entry into astrocytes and PC12 cells. Interestingly, 5b was able to enter these cells with efficiency comparable to that observed with HeLa cells. In light of this finding, both astrocytes and PC-12 cells were used in these experiments. The finding that the disulfide linked peptides 5a and 5b are able to enter these cells (Figure 6) opens the door for studies of prenylation in the central nervous system in order to better understand the molecular mechanisms underlying the prenylation state in Alzheimer’s Disease. Experiments using this probe to study prenylation in live cells are underway.
Figure 6.
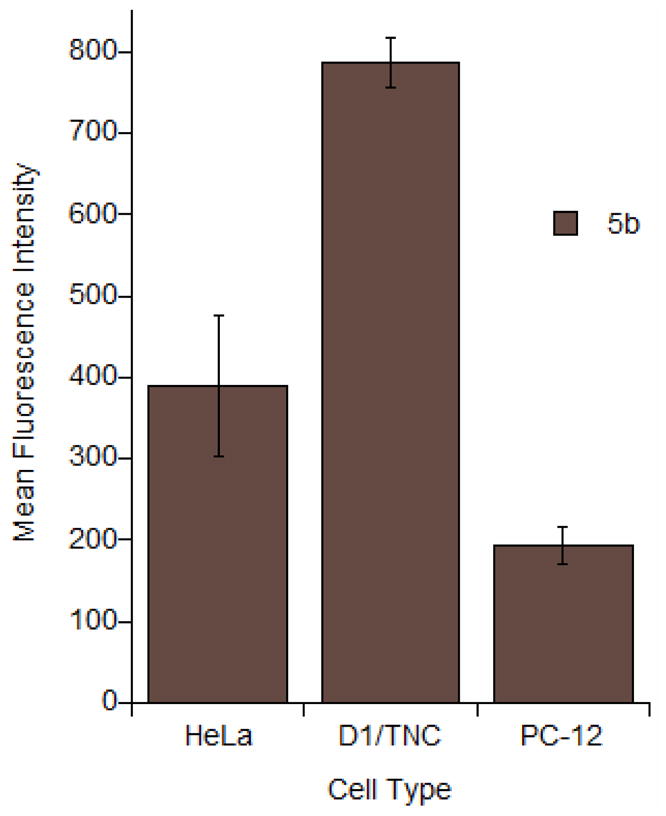
Flow cytometry analysis of peptide 5b incubated with various cells at 1 μM for 2h at 37°C. After incubation, cells were trypsinized for 15 min to remove membrane-bound peptide. Each bar represents the geometric mean fluorescence of 10,000 cells. Experiments performed in triplicate with the results expressed as the mean fluorescence intensity ± standard deviation.
CONCLUSIONS AND FUTURE DIRECTIONS
We have demonstrated that fluorescently labeled farnesylated peptides based on the C-terminus of CDC42 are able to translocate the membrane of live cells in an ATP-independent manner. The peptides in this study are also able to enter diverse cell types, suggesting that they will be useful for a variety of different types of studies. Of particular interest, peptides 5a and 5b, which contain a 10 carbon alkyl chain linked through a disulfide bridge in place of the isoprenoid, are able to efficiently enter live cells. In previous work by Krylov and coworkers, fluorescently labeled peptides were used to study the kinetics of farnesyltransferase as well as to detect the processing steps of prenylation in single cells (38–40). However, using single cell methods, the authors were not able to detect the processing of the substrate peptide, probably because the uptake of the peptide was insufficient. Using peptides 5a and 5b should allow this limitation to be overcome and will expand the available tools for examining prenyl processing in vivo. In addition to applications in cancer biology, recent studies have indicated that the levels of farnesyl diphosphate and geranygeranyl diphosphate are elevated in the brains of patients with Alzheimers Disease, suggesting that prenylation may play some role in the neuropathophysiology of this disease (2). Thus, we are currently employing the peptides reported here to probe the process of prenylation in both cancer and neuronal cells.
Acknowledgments
PC-12 cells were the generous gift of Bob Sorenson. We would like to thank Prof. George Barany for the use of an automated peptide synthesizer. We would like to acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute, supported in part by P30 CA77598. This work was supported by the National Institutes of Health (Grants GM58442 MDD, AG23524 and AG18357 WGW).
Abbreviations
- Acm
acetamidomethyl
- CLEAR
cross-linked ethoxylate acrylate resin
- CLSM
confocal laser scanning microscopy
- DIC
N,N′-diisopropylcarbodiimide
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-dimethylformamide
- DTT
dithiothreitol
- ESI-MS
electrospray ionization mass spectrometry
- 5-Fam
5-carboxyfluorescein
- FBS
fetal bovine serum
- Fmoc
9-fluorenylmethyloxycarbonyl
- FPP
farnesyl diphosphate
- FTI
farnesyltransferase inhibitor
- GGPP
geranylgeranyl diphosphate
- HBTU
O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate
- HMG-CoA reductase
3-hydroxy-3-methyl-glutaryl-CoA reductase
- HOBt
1-hydroxybenzotriazole
- HS
horse serum
- ICMT
isoprenylcysteine carboxyl methyltransferase
- PBS
phosphate buffered saline
- pFTase
protein farnesyltransferase
- RCE1
Ras-converting enzyme 1
- Scm
S-carbomethoxysulfenyl
- SPPS
solid-phase peptide synthesis
- TFA
trifluoroacetic acid
References
- 1.Ohkanda J, Knowles DB, Blaskovich MA, Sebti SM, Hamilton AD. Inhibitors of protein farnesyltransferase as novel anticancer agents. Curr Top Med Chem. 2002;2(3):303–323. doi: 10.2174/1568026023394281. [DOI] [PubMed] [Google Scholar]
- 2.Eckert GP, Hooff GP, Strandjord DM, Igbavboa U, Volmer DA, Muller WE, et al. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol Dis. 2009;35(2):251–257. doi: 10.1016/j.nbd.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooff GP, Volmer DA, Wood WG, Mueller WE, Eckert GP. Isoprenoid quantitation in human brain tissue: a validated HPLC-fluorescence detection method for endogenous farnesyl- (FPP) and geranylgeranylpyrophosphate (GGPP) Anal Bioanal Chem. 2008;392(4):673–680. doi: 10.1007/s00216-008-2306-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epstein WW, Lever D, Leining LM, Bruenger E, Rilling HC. Quantitation of prenylcysteines by a selective cleavage reaction. Proc Natl Acad Sci U S A. 1991;88(21):9668–9670. doi: 10.1073/pnas.88.21.9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seabra MC. Membrane association and targeting of prenylated Ras-like GTPases. Cell Signal. 1998;10(3):167–172. doi: 10.1016/s0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- 6.Benetka W, Koranda M, Eisenhaber F. Protein Prenylation: An (Almost) Comprehensive Overview on Discovery History, Enzymology, and Significance in Physiology and Disease. Monatsh Chem. 2006;137(10):1241–1281. [Google Scholar]
- 7.Casey PJ. Biochemistry of protein prenylation. J Lipid Res. 1992;33(12):1731–1740. [PubMed] [Google Scholar]
- 8.Clarke S. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu Rev Biochem. 1992;61:355–386. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- 9.Zhang FL, Casey PJ. Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annual Review of Biochemistry. 1996;65(1):241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 10.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5(5):405–412. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 11.Blum R, Kloog Y. Tailoring Ras-pathway-Inhibitor combinations for cancer therapy. Drug Resist Updates. 2005;8(6):369–380. doi: 10.1016/j.drup.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Karp JE, Lancet JE. Development of Farnesyltransferase Inhibitors for Clinical Cancer Therapy: Focus on Hematologic Malignancies. Cancer Invest. 2007;25(6):484–494. doi: 10.1080/07357900701359437. [DOI] [PubMed] [Google Scholar]
- 13.Zhu K, Hamilton AD, Sebti SM. Farnesyltransferase inhibitors as anticancer agents: current status. Curr Opin Invest Drugs. 2003;4(12):1428–1435. [PubMed] [Google Scholar]
- 14.Adjei AA, Mauer A, Bruzek L, Marks RS, Hillman S, Geyer S, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21(9):1760–1766. doi: 10.1200/JCO.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 15.Cortes J, Albitar M, Thomas D, Giles F, Kurzrock R, Thibault A, et al. Efficacy of the farnesyl transferase inhibitor R115777 in chronic myeloid leukemia and other hematologic malignancies. Blood. 2003;101(5):1692–1697. doi: 10.1182/blood-2002-07-1973. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee J, Guha A. Ras signaling pathways and farnesyltransferase inhibitors. Handb Brain Tumor Chemother. 2006:173–184. [Google Scholar]
- 17.Sousa SF, Fernandes PA, Ramos MJ. Farnesyltransferase Inhibitors: A Detailed Chemical View on an Elusive Biological Problem. Current Medicinal Chemistry. 2008;15:1478–1492. doi: 10.2174/092986708784638825. [DOI] [PubMed] [Google Scholar]
- 18.Lindorfer MA, Sherman NE, Woodfork KA, Fletcher JE, Hunt DF, Garrison JC. G protein gamma subunits with altered prenylation sequences are properly modified when expressed in Sf9 cells. J Biol Chem. 1996;271(31):18582–18587. doi: 10.1074/jbc.271.31.18582. [DOI] [PubMed] [Google Scholar]
- 19.Fenton JW, II, Jeske WP, Catalfamo JL, Brezniak DV, Moon DG, Shen GX. Statin drugs and dietary isoprenoids downregulate protein prenylation in signal transduction and are antithrombotic and prothrombolytic agents. Biochemistry (Moscow, Russ Fed) 2002;67(1):85–91. doi: 10.1023/a:1013956215394. [DOI] [PubMed] [Google Scholar]
- 20.Workman P. Signal transduction pathways: a gold mine for therapeutic targets. Farnesyltransferase Inhib Cancer Ther. 2001:1–20. [Google Scholar]
- 21.Roy M-O, Leventis R, Silvius JR. Mutational and Biochemical Analysis of Plasma Membrane Targeting Mediated by the Farnesylated, Polybasic Carboxy Terminus of K-ras4B. Biochemistry. 2000;39(28):8298–8307. doi: 10.1021/bi000512q. [DOI] [PubMed] [Google Scholar]
- 22.Waldmann H, Schelhaas M, Nagele E, Kuhlmann J, Wittinghofer A, Schroeder H, et al. Chemoenzymic synthesis of fluorescent N-Ras lipopeptides and their use in membrane localization studies in vivo. Angew Chem, Int Ed Engl. 1997;36(20):2238–2241. [Google Scholar]
- 23.Wollack JW, Zeliadt NA, Mullen DG, Amundson G, Geier S, Falkum S, et al. Multifunctional Prenylated Peptides for Live Cell Analysis. J Am Chem Soc. 2009;131(21):7293–7303. doi: 10.1021/ja805174z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci. 1976;73(7):2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King DS, Fields CG, Fields GB. A cleavage method which minimizes side reactions following Fmoc solid phase peptide synthesis. Int J Pept Protein Res. 1990;36(3):255–266. doi: 10.1111/j.1399-3011.1990.tb00976.x. [DOI] [PubMed] [Google Scholar]
- 26.Meriin AB, Yaglom JA, Gabai VL, Zon L, Ganiatsas S, Mosser DD, et al. Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol Cell Biol. 1999;19(4):2547–2555. doi: 10.1128/mcb.19.4.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazia D, Schatten G, Sale W. Adhesion of cells to surfaces coated with polylysine. Applications to electron microscopy. J Cell Biol. 1975;66(1):198–200. doi: 10.1083/jcb.66.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yousif LF, Stewart KM, Horton KL, Kelley SO. Mitochondria-penetrating peptides: sequence effects and model cargo transport. Chembiochem. 2009;10(12):2081–2088. doi: 10.1002/cbic.200900017. [DOI] [PubMed] [Google Scholar]
- 29.Xue CB, Becker JM, Naider F. Efficient regioselective isoprenylation of peptides in acidic aqueous solution using zinc acetate as catalyst. Tetrahedron Lett. 1992;33(11):1435–1438. [Google Scholar]
- 30.Moores SL, Schaber MD, Mosser SD, Rands E, O’Hara MB, Garsky VM, et al. Sequence dependence of protein isoprenylation. J Biol Chem. 1991;266(22):14603–14610. [PubMed] [Google Scholar]
- 31.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 32.Wollack JW, Zeliadt NA, Ochocki JD, Mullen DG, Barany G, Wattenberg EV, et al. Investigation of the sequence and length dependence for cell-penetrating prenylated peptides. Bioorg Med Chem Lett. 2010;20(1):161–163. doi: 10.1016/j.bmcl.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50(4–5):301–307. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 34.Du W, Cui Z, Tsui ZC, Chen Q, Willingham MC. In situ immunocytochemical detection of altered membrane composition induced by cell-cell contact in cultured mammalian cells. Microsc Res Tech. 2008;71(10):749–759. doi: 10.1002/jemt.20616. [DOI] [PubMed] [Google Scholar]
- 35.Massodi I, Bidwell GL, 3rd, Raucher D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J Control Release. 2005;108(2–3):396–408. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Stewart EJ, Aslund F, Beckwith J. Disulfide bond formation in the Escherichia coli cytoplasm: an in vivo role reversal for the thioredoxins. Embo J. 1998;17(19):5543–5550. doi: 10.1093/emboj/17.19.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47(5):883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Arkhipov SN, Berezovski M, Jitkova J, Krylov SN. Chemical cytometry for monitoring metabolism of a Ras-mimicking substrate in single cells. Cytometry, Part A. 2005;63A(1):41–47. doi: 10.1002/cyto.a.20100. [DOI] [PubMed] [Google Scholar]
- 39.Berezovski M, Li W-P, Poulter CD, Krylov SN. Measuring the activity of farnesyltransferase by capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis. 2002;23(19):3398–3403. doi: 10.1002/1522-2683(200210)23:19<3398::AID-ELPS3398>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Jitkova J, Carrigan CN, Poulter CD, Krylov SN. Monitoring the three enzymatic activities involved in posttranslational modifications of Ras proteins. Anal Chim Acta. 2004;521(1):1–7. [Google Scholar]