

Abstract
Amyloids are thought to be involved in various types of neurodegenerative disorders. Several kinds of intermediates, differing in morphology, size, and toxicity, have been identified in the multistep amyloidogenesis process. However, the mechanisms explaining amyloid toxicity remain unclear. We previously generated a toxic mutant of the nontoxic HET-s(218-289) amyloid in yeast. Here we report that toxic and nontoxic amyloids differ not only in their structures but also in their assembling process. We used multiple and complementary methods to investigate the intermediates formed by these two amyloids. With the methods used, no intermediates were observed for the nontoxic amyloid; however, under the same experimental conditions, the toxic mutant displayed visible oligomeric and fibrillar intermediates.
Introduction
Amyloid deposits are associated with >40 human pathologies, including type II diabetes, Alzheimer's disease and prion disease (for review see (1,2)). Despite extensive research into amyloid toxicity, the mechanisms that cause this cytotoxicity are far from clear. Several studies have suggested the involvement of intermediate species such as oligomers (3–5), annular or short/quiescent fibrils (6–8), and membrane pores/channels (9,10); others have shown toxicity to be associated with mature fibrils (11,12). Amyloid toxicity may be independent of the polypeptide length, primary sequence, or chirality (13). It has also been suggested that the toxic species could interact inappropriately with a wide range of cellular components, particularly biological membranes (14–16).
Amyloids may be very diverse in their primary sequences. However, they all share typical self-assembly into fibrils, fibers, or aggregates with a cross-β architecture (4.7 Å x-ray diffraction pattern), which is characteristically proteinase-resistant. When stained by Congo Red, amyloid fibers also exhibit birefringence by observation under cross-polarized light microscopy (17).
HET-s is a 289-amino-acid protein of the filamentous fungus Podospora anserina. It has the characteristic features of prion proteins: infectivity and amyloid folding. The prion-forming domain extending from amino acid 218 to 289 (HET-s(218–289)) has been well characterized as amyloid fibers by solid-state NMR (18), and is not toxic when expressed in yeast Saccharomyces cerevisiae (1). In previous studies, we used error-prone polymerase chain reaction mutagenesis to generate several mutants that were toxic to the yeast (1) and characterized a new toxic mutant called M8, an amyloid protein showing different types of structured aggregates (19). It formed unusual short fibrils with β-sheets in an antiparallel orientation. In this study, we investigated the features that may explain the toxicity of this M8 mutant. We thus used various approaches to investigate and compare the dynamics of the polymerization of this mutant and of the wild-type (WT) protein. We neatly showed that, indeed, the M8 toxic mutant assembly occurs through a totally different amyloidogenic pathway, involving structured on-pathway intermediates that are not observed during the nontoxic amyloid fibrilization.
Materials and Methods
Prion-forming domain expression and purification
The C-terminal histidine-tagged HET-s(218–289) constructs (WT and M8 ; Fig. 1 A) were introduced in Escherichia coli BL21(DE3) pLysS Gold cells. Bacteria were grown to 1OD in 2xYT medium (16 g /L Tryptone, 10 g/L Yeast Extract, and 5.0 g/L NaCl), and expression was induced by addition of 1 mM isopropyl-D-thiogalactoside (Euromedex, Souffelweyersheim, France). After 4 h induction, cells were harvested by centrifugation and frozen at −20°C. Cells were sonicated 4 × 1 min in buffer A (150 mM NaCl and 100 mM Tris-HCl, pH 8.0). The lysate was centrifuged for 30 min at 20,000 g. The pellet was washed in the buffer A and resuspended in denaturing buffer (6 M guanidinium/HCl in buffer A). The lysate was incubated with 2 mL TALON resin (Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France) for 3 h at room temperature. The resin was then washed twice with 35 mL of 8 M urea/buffer A, by centrifuging 10 min at 900 g. The peptides were eluted from the resin in the same buffer containing 250 mM imidazole (Sigma-Aldrich, Saint Louis, MO) and kept aliquoted at −80°C. This yields ∼2–4 mg of peptide per liter of culture. The peptide was pure as judged by analysis on 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by Coomassie Blue staining. Protein concentrations were determined by quantitative amino-acid analysis.
Figure 1.
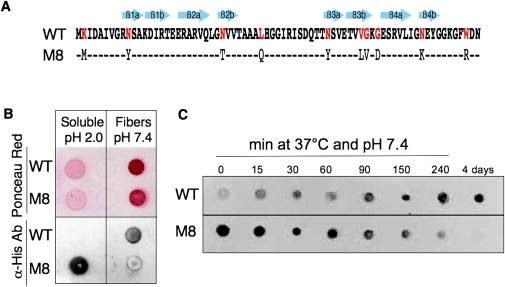
Soluble and aggregated forms of the two proteins display different antibody-binding characteristics during amyloidogenesis. (A) Sequence of HET-s(218-289) (WT) and M8 toxic mutant. (B) Detection of soluble proteins (pH 2.0 just after desalting) and amyloid fibers formed after several days at pH 7.4 with Ponceau Red and antihistidine antibody (α-His Ab). (C) Kinetics of polymerization followed with anti-His-tag antibody demonstrate that this antibody shows almost no binding affinity for soluble WT (T0 at pH 7.4) or M8 amyloid fibers. Dots represent 2-μL drops of 20 μM proteins grown without agitation at pH 7.4 and 37°C.
Fiber polymerization
For renaturation, proteins were subjected to gel filtration on a Hi-Trap Sephadex G-25 column (GE Healthcare Europe, Orsay, France) at 4°C, using 10 mM HCl pH 2.0 as the eluent. pH was then increased to 7.4 by adding a final concentration of 1× phosphate-buffered saline (PBS; MP Biomedicals Europe, Illkirch, France). Fiber polymerization was usually considered to be complete after three days at 37°C without agitation. Anti-His-tag antibodies (Amersham, Piscataway, NJ) and Ponceau Red (Sigma-Aldrich) were used to detect aggregates on nitrocellulose membranes (Optitran BA-S83; Schleicher & Schuell, Dassel, Germany). Antibody binding was visualized using a Dura chemoluminescence kit (Pierce, Thermo Fisher Scientific, San Jose, CA) with the Quantity One software/VersaDoc Imaging system (Bio-Rad, Hercules, CA).
Congo Red binding properties
All kinetics experiments were performed at 37°C and with 20 μM proteins in PBS. A 20-μM solution of Congo Red (CR; cat. No. C.I. 22120; Sigma-Aldrich) in PBS was prepared (extinction coefficient at 498 nm ∼3.7 104 M−1 cm−1) and filtered through a 0.22-μm polyether sulfone filter. Absorbance spectra with Congo Red (5 μL CR for 100 μL samples) were collected with a POLARstar Omega microplate reader (BMG Labtech, Champigny-sur-Marne, France). Red shifts were observed and respective displacement peaks were recorded. For microscopic analysis of CR staining, aggregates were centrifuged and spread on microscope slides. CR binding and birefringence were observed using an ECLIPSE E600FN microscope (Nikon, Tokyo, Japan) with a PlanFluor 40× objective and a DXM1200 digital camera (Nikon) equipped with optimally aligned cross-polarizers. Three independent kinetics studies were performed with different freshly desalted samples.
Transmission electronic microscopy
A 10-μL aliquot of aggregates was adsorbed 10 min onto Formvar-coated, carbon-stabilized copper grids (200 mesh), washed four times with water, and air-dried. Grids were then negatively stained with 15–20 drops of freshly prepared 2% uranyl acetate in double-distilled water and filtered with a 0.22-μm membrane (Millipore, Bedford, MA), dried with filter paper, and examined with a TECNAI Biotwin transmission electron microscope (Philips, Amsterdam, The Netherlands) at an accelerating voltage of 120 kV. Transmission electron microscopy (TEM) was performed at the Sercomi platform of the Bordeaux Imaging Center, and image processing and analysis were done with ImageJ Ver. 10.2 (National Institutes of Health, Bethesda, MD).
Dynamic light scattering
For dynamic light scattering (DLS) data, mean particle size and intensity size distribution were measured with a Malvern Instrument ALV/CGS-3 multiangle compact goniometer system (Malvern Instrument, Orsay, France) containing a HeNe laser source (λ = 632.8 nm and 22 mW output power) under a scattering angle of 90°. The temperature of the sample holder was controlled via a recirculating water bath. The freshly prepared sample (2 mL; 10 μM at pH 7.4) was filtered through a 0.22-μm filter, gently mixed, and placed in a borosilicate 10 mm-diameter cylindrical cuvette. The estimated hydrodynamic radii were calculated using an ALV-Correlator 3.0 software (ALV, Langen, Germany); the experiments were carried out using an autopilot function accumulating 30 measurements per sample. DLS measurements were performed three times on three different desalted samples.
Attenuated total reflectance-Fourier transform infrared spectroscopy
To resolve the structure at pH 2.0, soluble proteins were desalted in 10 mM DCl/D2O, under nitrogen environment to avoid H2O exchange (Sigma-Aldrich; and Euriso-Top, Saint Aubain, France), and Fourier-transform infrared (FTIR) spectra were recorded in solution using a CaF2 cell (50 μm). Proteins in DCl were at 200 μM (WT) and 165 μM (M8). FTIR and attenuated total reflectance (ATR)-FTIR spectra were recorded on a Nexus 870 FTIR spectrometer equipped with a deuterated triglycine sulfate detector (Nicolet, Thermo Fisher Scientific), with a spectral resolution of 4 cm−1 and a one-level zero filling. One-hundred interferograms, representing an acquisition time of 3.5 min, were co-added. The methods used for analysis and deconvolutions of ATR-FTIR spectra are presented in Table S1 in the Supporting Material. For kinetics studies, the samples were desalted in 10 mM HCl, and then were buffered with 1× PBS at pH 7.4 to a final concentration of 50 μM. Time 0 (T0) of kinetics corresponded to the addition of PBS. Fibers were grown at 37°C without agitation. The first measurement was done after 5 min drying of a 5-μL drop onto the germanium ATR crystal (Specac, Orpington, UK). All the aliquots generated during the kinetics were analyzed after 5 min drying. For CR binding experiments on soluble proteins, CR was added at T0 and the spectra were recorded on ATR-FTIR after three days. The fiber samples (final) obtained previously were also analyzed in presence of CR in the same way.
Results
Soluble forms of WT and M8 are structurally distinct
The M8 mutant primary sequence presents 10 mutations in comparison to WT (86% identical; Fig. 1 A), but proteins remain quite similar in terms of MW, pI, net charge, and hydrophobicity (see Table S1 and Fig. S1). Both purified proteins bear a His-tag sequence that may be recognized by specific antibodies. This tag in the WT amyloid protrudes outside of the axis fiber (18). However, this antibody did not recognize the soluble form of WT proteins, but instead was recognizing only WT fibers that appear during the incubation (Fig. 1 B). At the opposite, it was only binding to soluble M8 but not to the amyloid M8 fibers formed during the aggregation process (Fig. 1 C). Our results are consistent with the progressive burial of the His-tag in the M8 fiber structure during its formation. One possible explanation for the nondetection of the native WT is that the C-terminal end of the protein is contained within a random coil or loop conformation, impeding access of antibodies to the His-tag. We observed another unusual pattern of behavior when we analyzed the kinetics of Congo Red (CR) staining (Fig. 2 A). As expected, CR binding to WT and M8 increased over time with the formation of aggregates. However, at T0, soluble M8 binds efficiently to CR with a strong red shift and high turbidity (Fig. 2 A). It was therefore astounding to discover that soluble M8 was indeed immediately precipitated in presence of CR. These CR/M8 aggregates analyzed by ATR-FTIR have the same antiparallel β-sheet organization as the M8 fibers stained a posteriori by CR (Fig. 2 B). This observation led us to conclude that CR seemed to catalyze M8 polymerization into amyloids. We observed the same kind of precipitation of M8 with Thioflavine T and 8-Anilino-1-naphthalenesulfonic acid (data not shown). In contrast, CR inhibited the fibrilization of WT by keeping the soluble proteins in a random coil conformation as observed by ATR-FTIR (Fig. 2B). This role of CR is consistent with previous findings that characterized this dye as an inhibitor of amyloid polymerization (20). However, in 2010, two distinct publications (21,22) reported that Aβ(1–40) interacted with CR as M8, promoting β-sheet formation and peptide aggregation. Thus, this is the third report of CR-amyloid binding behavior of toxic amyloids differing from all other previous reports. Our results clearly suggest the existence of different amyloids with different behaviors, which may explain the differences in toxicity between the two proteins in yeast.
Figure 2.
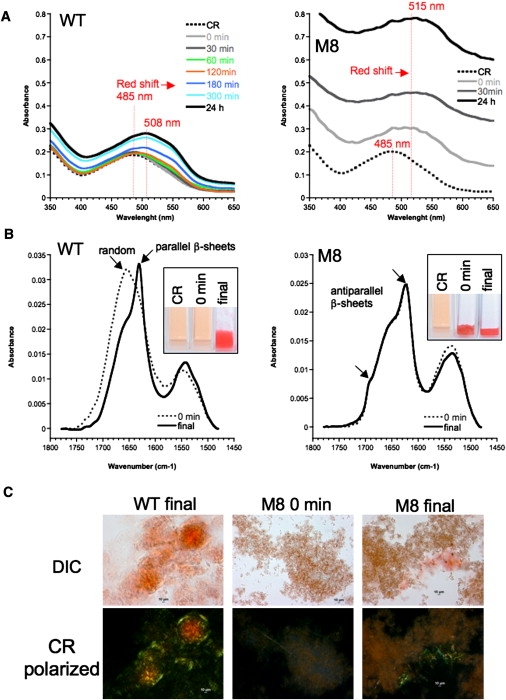
Aggregation of WT and M8 in presence of Congo Red (CR). (A) Kinetics of amyloid formation followed by CR binding. CR has a classical absorbance peak at 485 nm. CR was added after polymerization and absorbance spectra recorded. For WT, the binding increases over time during the formation of amyloid fibers and a red shift is seen at 508 nm. For M8, an immediate precipitation occurs in the presence of Congo Red, concomitant with a high turbidity and a red shift at 515 nm. (B) ATR-FTIR analysis of species formed in presence of CR. After three days, soluble WT species in the presence of CR remain in random coils (dotted spectra), whereas fiber aggregates exhibit specific parallel β-sheets. In such conditions, CR is an inhibitor of aggregation (sample 0 min is identical to CR). In contrast, M8 aggregated instantly in presence of CR (sample 0 min is identical to sample final). ATR-FTIR shows that the M8/CR complex is also an amyloid with the same antiparallel β-sheet content as that seen in the mature amyloid fibers. (C) All aggregates formed were stained by CR and showed birefringence under a polarized light microscope.
M8 shows visible intermediates
We then investigated fibrillogenesis of our proteins by TEM. Solid-state NMR has previously been used for the elegant analysis of WT fiber three-dimensional structure. The refined model shows that the WT fiber forms a left-handed β-solenoid (Fig. 3 A), with monomers composed of four β-strands (18); however, nothing is known about potential intermediates. For the WT protein, we were not able to detect any species other than protofibrils, larger fibers, and bundles of fibers. Bundles of WT fibers were present as soon as 30 min (Fig. 3 B). This may reflect either a very fast kinetics of amyloid formation that would lead to transient intermediates not detectable by such approach or by a real absence of any putative intermediates. This reproducible result was quite different with M8. In contrast, the assembly of M8 aggregates over time was markedly different (Fig. 3 C). First, we detected the appearance of spherical oligomers, ∼10 nm in diameter (Fig. 3 D), followed by the appearance of nanofibers of ∼10 × 100 nm, which we had previously observed (19). These fibers seemed to further assemble into larger aggregates by merging. This was confirmed by DLS (see Fig. 4). Unlike TEM, DLS allows the evaluation of the particle size distribution in situ, for the whole sample in solution. We did not detect any intermediate species for WT, at 37°C in the 1–1000-nm range of detection (data not shown). We slowed down the experiment by decreasing the temperature at 15°C, but only observed initial small species followed by huge species at a later stage (Fig. 4 A). M8 displayed a different pattern of behavior in solution: species in the nanometer order were detected initially, growing into aggregates of 100–200 nm, which then became larger, before precipitating out of solution (Fig. 4 B). The differences in findings between TEM and DLS can be explained by the different states of the samples (dried/stained and hydrated/dynamic). It would be of interest to determine the specificity of these intermediates, as this could also be a factor explaining M8 toxicity.
Figure 3.
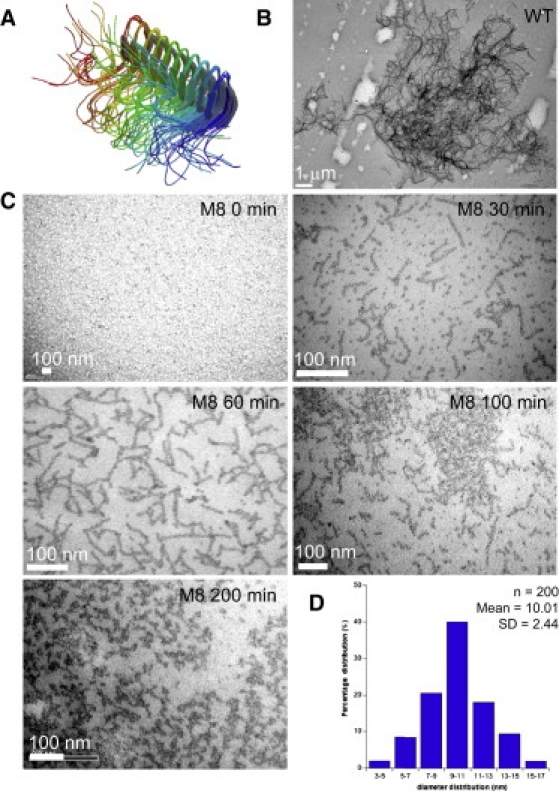
M8 presents several different intermediates during polymerization, which can be detected by transmission electronic microscopy (TEM). (A) Structure of HET-s(218-289) fibers obtained by solid-NMR in a previous study (18). (B) We did not observe any intermediates formed by the WT protein during the time period indicated, but detected rapidly the appearance of protofibrils of various sizes, fibers, bundles of fibers, or protein plaques of amorphous aggregates. The picture presents a large bundle at 30 min. (C) Analysis of M8 polymerization over time showed the appearance of several different intermediates characterized by round oligomers, followed by short fibers of ∼100 × 10 nm, which then seemed to fuse laterally to form larger aggregates that were able to precipitate. (D) Size of round oligomers was analyzed, and was ∼10 nm in diameter, consistent with the width of a basic M8 fiber.
Figure 4.
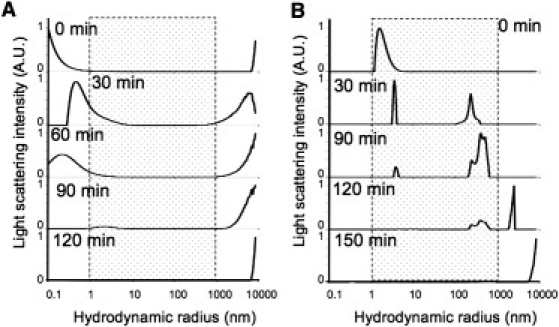
DLS size distribution graphs of WT and M8 during aggregation. (A) WT proteins did not form any intermediate species visible in the range of the DLS detection (from 1 to 1000 nm; gray shaded); we observed only smaller and then bigger species. (B) M8 formed a number of intermediates in the range of detection. Fibrilization experiments were carried out at a concentration of 10 μM, at 15°C and without agitation.
M8 is inherently structured in solution at pH 7.4
Before starting analyzing structures over time, we first needed to establish what the initial structuration of our proteins was after purification. To perform FTIR in solution, we could only do so by exchanging H2O/HCl by D2O/DCl to avoid a strong interference with the vibration mode of H2O (δO-H) at 1640 cm−1, in the amide I band range. At pH 2.0, just after desalting in 10 mM DCl/D2O, the amide I′ bands were observed at 1646 cm−1 and 1648 cm−1 for WT and M8, respectively (Fig. 5 A). These wavenumbers indicate that both WT and M8 at pD 2.0 were unstructured in a mainly random coil conformation. The exchange H2O/D2O induces a small shift of the wavenumber of the amide I, as expected (23,24). The area of amide II (1550 cm−1) permits quantification of percentage of unexchanged amide group, and the area of amide II′, measures the percentage of exchanged amides. Fig. 5 A reveals that for both WT and M8, only the amide II′ are observed, leading us to conclude to a complete exchange of the NH to ND in presence of D2O. The random coil conformations of WT and M8 favor this large exchange. Thus, at pD 2.0, the nontoxic WT and the toxic M8 are unstructured, according to the profile of the amide I′ and the complete exchange with D2O.
Figure 5.
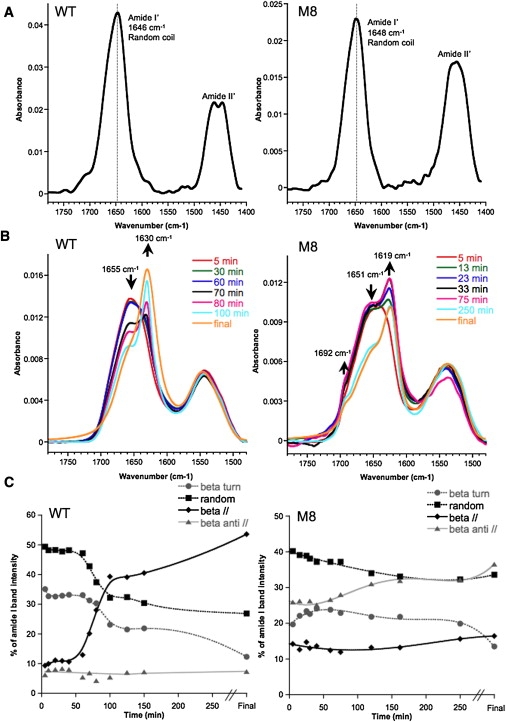
Structures of WT and M8 intermediates followed by FTIR and ATR-FTIR. (A) FTIR spectra of WT and M8 proteins at pH 2.0 in DCl. Maximum absorbance peaks were observed at 1646 nm for WT and 1648 nm for M8, characteristic of a random coil conformation in D2O. Proteins were desalted in 10 mM DCl in D2O and were used at 200 μM (WT) and 165 μM (M8), respectively. (B) Aggregation kinetics shown by infrared spectra of air-dried (5 min) WT and M8 displaying different structural transitions. WT undergoes a clear transition from random coil to a mainly parallel β-sheet structure, whereas M8 seems to instantly form antiparallel β-sheets, which are then modified over time. (C) Structural evolution of amyloid species over time for WT and M8 expressed as percentages of random coils, β-turns, and parallel and antiparallel β-sheets. Kinetics were performed at a concentration of 50 μM, pH 7.4 and 37°C without agitation. Smooth curves are presented.
Secondly, we used ATR-FTIR to investigate the changes in structure over time during amyloid polymerization after air-drying the sample. The amide I band recorded was deconvoluted into four main structures: random coils (1655 cm−1), β-turns (1669 cm−1), and parallel (1630 cm−1) and antiparallel β-sheets (1619 and 1692 cm−1), characteristic of amyloids (Table S1). As expected, WT underwent a molecular transition from a random coil organization (main peak at 1655 cm−1; Fig. 5 B), with a predominance of β-turns, to a mainly parallel β-sheet amyloid conformation as described by NMR (18). Under our experimental conditions, this transition occurred after a lag phase of ∼40 min (Fig. 5 C), possibly corresponding to a slow nucleation process. Surprisingly, M8 behaved very differently. M8 assembled immediately into antiparallel β-sheets when pH was raised to 7.4, as reflected by the characteristic 1619- and 1692-cm−1 peaks (Fig. 5 B). These contributions were observed on the first spectra after 5 min drying. We did not observe any lag phase, as if the monomer was instantly assembled into mainly antiparallel β-sheets (Fig. 5 C). During M8 fibrilization, we only observed a small change of structure, an increase of the percentage of amide group organized in antiparallel β-sheets (>10%) at the expense of the random coils and β-turns. M8 was therefore forming a more stable antiparallel β-sheet conformation.
Discussion
In terms of kinetics, it is truly impossible to compare the different approaches we chose to use, because of the restraints due to the technical approaches (concentrations, temperatures, etc.). Other conditions such as pH, ionic strength, oxidation, reacting volumes, test tube shape and composition, and shaking/mixing can also be key factors during polymerization (8,25). Moreover, surfaces and interfaces may influence greatly aggregation characteristics (26,27). Each technique was a powerful analysis method, but with resolution limits and inherent constraints. For example, TEM may provide partial or biased information, as samples may differently interact with the grid. In this case, the object under scrutiny is washed, dried, and stained, which could be very different from its bulk solution state. DLS is performed in solution, but only convenient for small assembly in the 1–1000-nm range. Big aggregates usually keep out of sight the smaller ones in the light scattering. Amyloid-dye (8-Anilino-1-naphthalenesulfonic acid, Thioflavine T, and Congo Red) staining is not very specific, as not all amyloids react the same way in presence of these dyes, and other polymers such as actin may also bind to CR (28). Because of high concentration detection limits and superposition of H20 vibration in the amide I band, it is almost impossible to work in solution with FTIR. The use of ATR is more appropriate to our samples but cannot exclude that amyloids get structured by contact or drying on the ATR crystal.
In any case, our work aimed to compare two amyloids: one toxic and the other not. And we clearly demonstrated here that WT and M8 amyloids display no similarities in aggregation kinetics or structure, aggregate morphology, or size. In our various experimental conditions, the two amyloids polymerized through apparently different pathways, involving different intermediates. We thus proposed two distinct models of amyloidogenesis (Fig. 6). The first pathway is that of the wild-type, which is similar to a nucleation-dependent fibrilization model previously proposed (29,30). This involves a nucleation step, which is usually quite slow (k1), and is followed by a quicker elongation of the fibril from the nucleus (k2). This model can also refer to other parameters in amyloid aggregation as secondary nucleation phenomena, seeding, or fragmentability (31,32). Curiously, during this lag-time, no changes in the level of aggregation could be detected whatever the technique used (DLS, TEM). Under our experimental conditions, and with the techniques we used, we could not detect the nucleus. This could be due to the smallness of the nucleus that would not be distinguished from the monomeric species. Then the fibril was assembled from the free native monomers and was elongated into a kind of template-assisted β-sheet structure. Protofibrils (5-nm large) formed from these WT monomers formed the basis of larger quaternary structures organized into a highly hydrated network, giving the aggregates the appearance of a gel (Fig. 2 C). These HET-s(218–289) fibril structures have been fully described by solid-state NMR, TEM, or the tilted-beam-TEM (18,33,34). The second pathway of M8 is clearly different and more straightforward; the nucleus is formed instantly (k3), then oligomers form and fibrils grow, probably through the fusion of 10-nm oligomers in diameter. In our particular case, the basic fiber growth is limited to ∼10 × 100 nm. These short fibers did not display any tendency to twist or interwind. Indeed, M8 fibers seemed to merge laterally to form larger aggregates that appeared as, and precipitated as, a finely granulated powder (Fig. 2 C).
Figure 6.
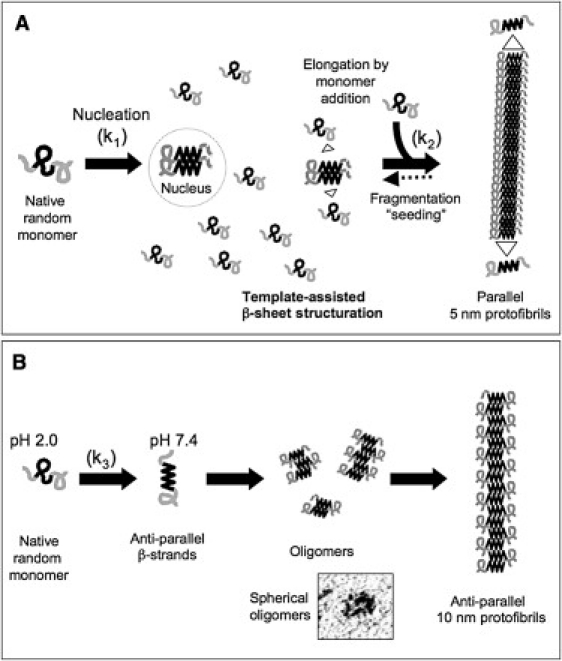
Two different models of amyloid polymerization. (A) Proposed proceeding of amyloidogenesis followed by HET-s(218-289). The soluble random coil monomer is subjected to a slow nucleation process (k1) and the nucleus formed is barely detectable. A rapid elongation then takes place (k2) by addition of unfolded monomers in a sort of template-assisted β-sheet structuration. The protofibril grew with the addition of soluble monomers, and then formed more complex and twisted fibers and bundles. (B) M8 amyloid followed a different pathway of aggregation. The soluble random coil form of M8 was only observed at pH 2.0. At pH 7.4, the process of polymerization started instantly (k3) by forming antiparallel β-sheet species. These species assembled into 10-nm-diameter oligomers and then into short fibers (∼10 × 100 nm).
Does size matter?
Amyloid polymerization is a complex process, involving several kinds of intermediates of various sizes and shapes. However, the role of intermediates in determining toxicity, particularly in neurodegenerative disorders, is highly contentious, probably due to their diversity. Indeed, the mechanism through which neuronal death is triggered remains unexplained. Several hypotheses have been proposed, all of which involve interactions with biological membranes. Most publications suggest that soluble oligomers and prefibrillar intermediates are the toxic species (3,5,35), but sometimes fibers or mature aggregates are also suspected. Amyloid intermediates may bind (36) or insert into membranes (37), and cause membrane permeabilization and leakage (15) or membrane disruption and damage (14). Some seem to be involved in the formation of channels or annular pores, which can modify cellular homeostasis (9,10,38). A recent study has also demonstrated that large fibrillar amyloids are able to cross a membrane barrier and may be internalized in the cytoplasm of mammalian cells (39). Additionally, some lipids may catalyze the fibrillogenesis pathway (40) and specific membrane components may trigger changes in the conformation of amyloids (41). Interestingly, lipids may resolubilize mature nontoxic amyloid fibers into toxic oligomers (16).
Our results show that highly similar sequences (86% identical) can lead to completely different ordered aggregates that display very different aggregation properties and cytotoxicity. Our evidences lead us to think that the kinetics of M8 aggregation is at the origin of its toxicity. The aggregation dynamics demonstrate the capacity of M8 to assemble into intermediates species that may be reminiscent of oligomeric toxic intermediates. Interestingly, the antiparallel signature of these toxic species is consistent with the antiparallel organization of Aβ(1–42) toxic oligomers recently published (42). In addition, it has been shown that fibril length is also correlated with the ability to disrupt membranes and to reduce cell viability (8). In parallel, we have carried out preliminary studies showing that M8 may clearly interfere with vesicular trafficking in vivo (43) and that it interacts in vitro specifically at the interface with various lipid monolayers (H. P. Ta, K. Berthelot, B. Desbat, J. Géan, L. Servant, C. Cullin, and S. Lecomte, unpublished data), which may explain its toxicity in yeast. It may now be of interest to investigate the fusogenic activity of our toxic amyloids. Such activity has already been suggested to play a role in PrP and amyloid-β toxicity (44,45). Our model based only on the gain of toxicity function is of general interest. Indeed, of the biochemical characteristics found for this particular toxic amyloid, we found specific features related to other amyloids involved in pathological events. Here, we have shown that in addition to a remarkable pattern of nanofibril formation, M8 displays a rapid kinetics with no lag-phase and forms highly structured oligomers. Our model opens up new perspectives to study the origins of the toxic properties of amyloids.
Acknowledgments
We thank Axel Edelman for English proofreading. We are grateful to Dr. Reiko Oda for kindly allowing us access to her equipment (dynamic light scattering, polarized microscope) at the European Institute of Chemistry and Biology, Pessac, France.
This work was supported by grants from the Centre National de la Recherche Scientifique (Programme Interdisciplinaire, Interface Physique Chimie Biologie, Soutien à la Prise de Risque) and a grant from the French National Agency for Research (project No. ANR-06-MRAR-011-01 “AMYLOI”). K.B. received fellowships from the French National Agency for Research and the Conseil Régional d'Aquitaine.
Supporting Material
References
- 1.Couthouis J., Rébora K., Cullin C. Screening for toxic amyloid in yeast exemplifies the role of alternative pathway responsible for cytotoxicity. PLoS ONE. 2009;4:e4539. doi: 10.1371/journal.pone.0004539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 4.Cleary J.P., Walsh D.M., Ashe K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 5.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 6.Kayed R., Pensalfini A., Glabe C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009;284:4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petkova A.T., Leapman R.D., Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 8.Xue W.F., Hellewell A.L., Radford S.E. Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 2009;284:34272–34282. doi: 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lashuel H.A., Hartley D., Lansbury P.T., Jr. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 10.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gharibyan A.L., Zamotin V., Morozova-Roche L.A. Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol. 2007;365:1337–1349. doi: 10.1016/j.jmb.2006.10.101. [DOI] [PubMed] [Google Scholar]
- 12.Meyer-Luehmann M., Spires-Jones T.L., Hyman B.T. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastor M.T., Kümmerer N., Serrano L. Amyloid toxicity is independent of polypeptide sequence, length and chirality. J. Mol. Biol. 2008;375:695–707. doi: 10.1016/j.jmb.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Engel M.F., Khemtémourian L., Höppener J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayed R., Sokolov Y., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 16.Martins I.C., Kuperstein I., Rousseau F. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Putchler H., Sweat F., Levine M. On the binding of Congo Red by amyloid. J. Histochem. Cytochem. 1962;10:355–364. [Google Scholar]
- 18.Wasmer C., Lange A., Meier B.H. Amyloid fibrils of the HET-s(218-289) prion form a β-solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 19.Berthelot K., Immel F., Cullin C. Driving amyloid toxicity in a yeast model by structural changes: a molecular approach. FASEB J. 2009;23:2254–2263. doi: 10.1096/fj.08-125724. [DOI] [PubMed] [Google Scholar]
- 20.Lorenzo A., Yankner B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo Red. Proc. Natl. Acad. Sci. USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lendel C., Bolognesi B., Gräslund A. Detergent-like interaction of Congo Red with the amyloid-β peptide. Biochemistry. 2010;49:1358–1360. doi: 10.1021/bi902005t. [DOI] [PubMed] [Google Scholar]
- 22.Bose P.P., Chatterjee U., Arvidsson P.I. Effects of Congo Red on Aβ1–40 fibril formation process and morphology. ACS Chem. Neurosci. 2010;1:315–324. doi: 10.1021/cn900041x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goormaghtigh E., Cabiaux V., Ruysschaert J.M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. I. Assignments and model compounds. Subcell. Biochem. 1994;23:329–362. doi: 10.1007/978-1-4615-1863-1_8. [DOI] [PubMed] [Google Scholar]
- 24.Goormaghtigh E., Cabiaux V., Ruysschaert J.M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. II. Experimental aspects, side chain structure, and H/D exchange. Subcell. Biochem. 1994;23:363–403. doi: 10.1007/978-1-4615-1863-1_9. [DOI] [PubMed] [Google Scholar]
- 25.Collins S.R., Douglass A., Weissman J.S. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jean L., Lee C.F., Vaux D.J. Competing discrete interfacial effects are critical for amyloidogenesis. FASEB J. 2010;24:309–317. doi: 10.1096/fj.09-137653. [DOI] [PubMed] [Google Scholar]
- 27.Kowalewski T., Holtzman D.M. In situ atomic force microscopy study of Alzheimer's β-amyloid peptide on different substrates: new insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bousset L., Redeker V., Melki R. Structural characterization of the fibrillar form of the yeast Saccharomyces cerevisiae prion Ure2p. Biochemistry. 2004;43:5022–5032. doi: 10.1021/bi049828e. [DOI] [PubMed] [Google Scholar]
- 29.Jarrett J.T., Lansbury P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 30.Serio T.R., Cashikar A.G., Lindquist S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 31.Knowles T.P., Waudby C.A., Dobson C.M. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka M., Collins S.R., Weissman J.S. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 33.Chen B., Thurber K.R., Tycko R. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc. Natl. Acad. Sci. USA. 2009;106:14339–14344. doi: 10.1073/pnas.0907821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabaté R., Baxa U., Saupe S.J. Prion and non-prion amyloids of the HET-s prion forming domain. J. Mol. Biol. 2007;370:768–783. doi: 10.1016/j.jmb.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Walsh D.M., Klyubin I., Selkoe D.J. Naturally secreted oligomers of amyloid-β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 36.Hertel C., Terzi E., Kemp J.A. Inhibition of the electrostatic interaction between β-amyloid peptide and membranes prevents β-amyloid-induced toxicity. Proc. Natl. Acad. Sci. USA. 1997;94:9412–9416. doi: 10.1073/pnas.94.17.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engel M.F., Yigittop H., Antoinette Killian J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006;356:783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 38.Jang H., Zheng J., Nussinov R. New structures help the modeling of toxic amyloid-β ion channels. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Ren P.H., Lauckner J.E., Kopito R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choo-Smith L.P., Garzon-Rodriguez W., Surewicz W.K. Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 1997;272:22987–22990. doi: 10.1074/jbc.272.37.22987. [DOI] [PubMed] [Google Scholar]
- 41.Elfrink K., Ollesch J., Gerwert K. Structural changes of membrane-anchored native PrP(C) Proc. Natl. Acad. Sci. USA. 2008;105:10815–10819. doi: 10.1073/pnas.0804721105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerf E., Sarroukh R., Raussens V. Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J. 2009;421:415–423. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 43.Couthouis J., Marchal F., Cullin C. The toxicity of an “artificial” amyloid is related to how it interacts with membranes. Prion. 2010 doi: 10.4161/pri.4.4.13126. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pillot T., Goethals M., Lins L. Fusogenic properties of the C-terminal domain of the Alzheimer β-amyloid peptide. J. Biol. Chem. 1996;271:28757–28765. doi: 10.1074/jbc.271.46.28757. [DOI] [PubMed] [Google Scholar]
- 45.Pillot T., Drouet B., Chambaz J. A nonfibrillar form of the fusogenic prion protein fragment [118–135] induces apoptotic cell death in rat cortical neurons. J. Neurochem. 2000;75:2298–2308. doi: 10.1046/j.1471-4159.2000.0752298.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.