

Abstract
Hyperhomocysteinemia, an increased level of plasma homocysteine, is an independent risk factor for the development of premature arterial fibrosis with peripheral and cerebro-vascular, neurogenic and hypertensive heart disease, coronary occlusion and myocardial infarction, as well as venous thromboembolism. It is reported that hyperhomocysteinemia causes vascular dysfunction by two major routes: (1) increasing blood pressure and, (2) impairing the vasorelaxation activity of endothelial-derived nitric oxide. The homocysteine activates metalloproteinases and induces collagen synthesis and causes imbalances of elastin/collagen ratio which compromise vascular elastance. The metabolites from hyperhomocysteinemic endothelium could modify components of the underlying muscle cells, leading to vascular dysfunction and hypertension. Homocysteine metabolizes in the body to produce H2S, which is a strong antioxidant and vasorelaxation factor. At an elevated level, homocysteine inactivates proteins by homocysteinylation including its endogenous metabolizing enzyme, cystathionine γ-lyase. Thus, reduced production of H2S during hyperhomocysteinemia exemplifies hypertension and vascular diseases. In light of the present information, this review focuses on the mechanism of hyperhomocysteinemia-associated hypertension and highlights the novel modulatory role of H2S to ameliorate hypertension.
Keywords: Homocysteine, Hypertension, Hydrogen sulfide, Vascular remodeling
Introduction
To accommodate pressure load, vessels undergo structural and functional adaptation. Elastic compliance consists of a vessel’s capacity to stretch on load and return to normal at normal load. Chronic changes in blood pressure, however, lead to a persistence of mechanical load on the vessel wall and induces resistance to stretch. The resistance to stretch to accommodate for load may lead to the development of hypertension. The development of hypertension in 95% of hypertensive patients is idiopathic and multifactorial [1]. Components such as circulating plasma factors, vascular endothelium, smooth muscle contractile apparatus, and homeostasis of extracellular matrix (ECM) surrounding the smooth muscle, in milieu, play a coordinated role to perform proper vascular function. Alteration in anyone of these components may result in impairment of vessel’s response to stress and may lead to hypertension.
One of such circulating factors is plasma homocysteine and elevated level of homocysteine is known as hyperhomocysteinemia. There are four ways by which hyperhomocysteinemia is developed (Fig. 1): (1) methionine-rich protein diet; (2) vitamin B12/folate deficiency; (3) heterozygous/homozygous cystathionine-β synthase (CBS) activity; (4) obstruction of renal clearance. Studies have demonstrated that a methionine-rich protein diet leads to increased levels of plasma homocysteine [2]. A diet of fruits and vegetables, which is low in methionine, leads to decreased hypertension [3] and improves vascular function [4]. Half of the dietary methionine is metabolically converted to homocysteine. Homocysteine is accumulated because the metabolic conversion to cysteine and their excretion are impaired [5]. This could lead to a decrease in the body’s ability to clear homocysteine and reduction in the levels of cysteine. Also, it is known that increased levels of homocysteine leads to reduced bioavailability of glutathione peroxidase activity [6]. This could lead to decreased redox of glutathione. In addition, the oxidative metal ion (Cu2+) concentration is elevated in hyperhomocysteinemic patients [7, 8]. Collectively, these studies suggest that increased plasma homocysteine is an important factor in causing the elevation of plasma redox stress. Therefore, body’s inability to clear metabolic by-product homocysteine could lead to hyperhomocysteinemia and redox stress.
Fig. 1.
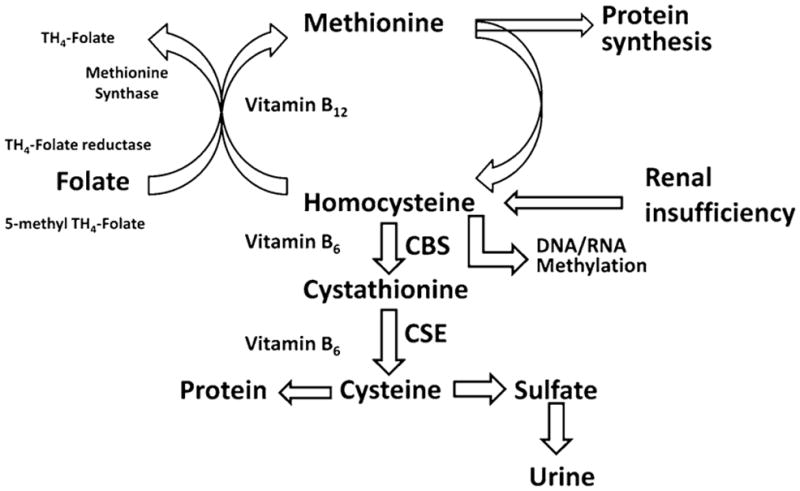
Schematic of methionine metabolism and development of hyperhomocysteinemia. Homocysteine in the body further metabolizes to produce sulfate and excretes through kidney
Arteriosclerosis is one of the primary causes of arterial hypertension. Acute/chronic inflammatory and redox processes facilitate atherosclerotic and arteriosclerotic lesion formation [9] and induce vasoconstriction and hypertension [10]. Although in hyperhomocysteinemia-associated hypertension, such as renovascular hypertension, endothelial dysfunction and vascular hypertrophy have been observed, the precise mechanism by which homocysteine causes vascular dysfunction and contributes to hypertension are largely unknown. Several mechanisms have been proposed, these include: (1) homocysteine causes endothelial injury and vascular hypertrophy by redox pathway; (2) this leads to increased blood pressure; (3) the molecular mechanism of endothelial dysfunction includes reduced bioavailability of nitric oxide (NO) due to elevated levels of homocysteine which causes nitration of tyrosine in proteins, such as actin and myosin; (4) homocysteine also activates certain metalloproteinases which can cause degradation of collagen and elastin leading to vascular hypertrophy.
Homocysteine is a Precursor for Endogenous Hydrogen Sulfide Generation
Homocysteine is a thiol-containing non-protein amino acid that is formed during the metabolism of the essential amino acid methionine and is recognized as an independent cardiovascular risk factor, such as arterial vascular disease [11]. An elevated plasma level of homocysteine known as hyperhomocysteinemia has been associated with hypertension [12–14]. Although several lines of evidences suggested the integrated physiological role of homocysteine to cause multi-organ damage, probably related to impair endothelial and smooth muscle function, the precise molecular mechanisms by which it mediates these adverse effects are still unknown. Under normal physiological conditions, homocysteine metabolizes to produce cysteine which is a substrate of two pyridoxol-5′-phosphate (PLP)-dependent enzymes—CBS and cystathionine-γ lyase (CSE) for endogenous production of hydrogen sulfide (H2S) (Fig. 2).
Fig. 2.
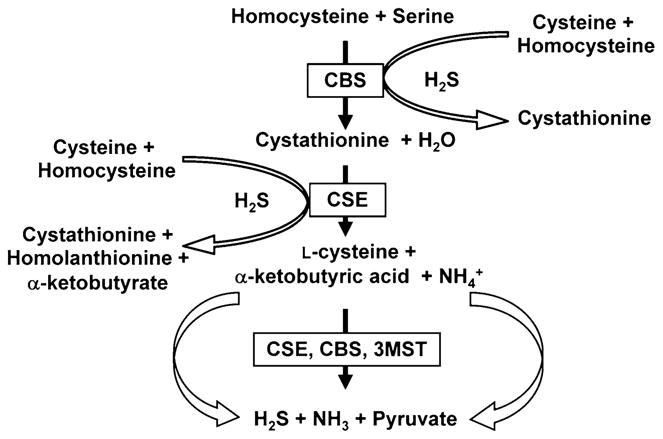
Schematic of homocysteine metabolism and formation of endogenous hydrogen sulfide (H2S). CBS cystathionine β-synthase, CSE cystathionine γ-lyase, 3MST 3-mercaptopyruvate sulfurtransferase
Hydrogen sulfide (H2S) is known for decades as a toxic gas which has intoxication effect on central nervous system; however, very recently H2S has been recognized as a key vasorelaxant gaseous molecule [15]. Physiologically, as stated earlier, H2S generates from L-cysteine, catalyzed by either CBS and/or CSE [16]. Recently, a PLP-independent enzyme, 3-mercaptopyruvate sulfurtransferase (3-MST), has been reported as a possible candidate for H2S production [17, 18] (Fig. 2). Unlike CBS and CSE, 3-MST uses 3-mercaptopyruvate as a substrate, which is a metabolite of cysteine and α-ketoglutarate (α-KG) by cysteine aminotransferase (CAT), to produce H2S [18]. The expression of CBS, CSE, and 3-MST are tissue specific. In some tissues CBS is the main H2S-generating enzyme, while CSE and 3-MST on the others. For example, CBS is highly expressed in the hippocampus and cerebellum and is a predominant H2S-generating enzyme in the brain and nervous tissues [19]. On the other hand, CSE expression in the vascular smooth muscle was initially demonstrated by Hosoki et al. [20]. Later, it was reported to be expressed mainly in the liver, kidney, and vascular smooth muscle [21, 22] with a major H2S producing enzyme in the cardiovascular system [22]. Recent evidence suggested that CSE is also expressed in the vascular endothelial cells and capable of producing H2S [15]. Unlike CBS and CSE, to date, 3-MST has only been reported as a H2S producing enzyme in the vascular endothelium [17, 18]. H2S, however, can also be generated by a condensation reaction between cysteine and homocysteine [23] (Fig. 2).
Homocysteine at higher level has been reported to inhibit CSE activity in the rat liver [24] and at pathological condition, hyperhomocysteinemia alter the transulfuration pathway by inhibiting CSE enzyme activity [25]; thereby reduce endogenous production of H2S in the body. Accumulating evidences indicated that H2S is a physiological vasorelaxant and reduced production of H2S in the vascular tissue leads to hypertension [15, 26]. Thus, the role of hyperhomocysteinemia in essential hypertension is not surprising.
Cysteine Versus Homocysteine and Hypertension
Compared with the levels of cysteine (~100 μM) in normal individuals, the levels of homocysteine (~20 μM), even in homocysteinemia patients, are much lower. Therefore, the question arises regarding the contribution of homocysteine to the production of H2S. It is interesting to point out that homocysteine and cysteine both are substrates for H2S generation; and CSE enzyme mainly uses cysteine to produce H2S endogenously. Biochemically during hyperhomocysteinemia, homocysteine competes for binding to CSE with cysteine; therefore, increases in homocysteine will decrease H2S production from cysteine through substrate inhibition [25, 27]. The reaction occurs as follows:
![]()
Furthermore, protein homocysteinylation is a major reaction in the presence of thiolactone and homocysteinylation led to protein damage; this was manifested as multimerization and precipitation of extremely modified proteins [28]. Although there is no direct evidence, the activity of CSE may be severely impaired during hyperhomocysteinemia due to damage and precipitation of modified-CSE that results in attenuated H2S generation from both cysteine and homocysteine leading to hypertension. However, at present this mechanism, if exists at all, is not elucidated and warrants investigation.
Hyperhomocysteinemia, Vascular Dysfunction, and Hypertension: Role of H2S
Elevated levels of plasma homocysteine, known as hyperhomocysteinemia, is recognized as an independent risk factor for the development of premature arterial fibrosis with peripheral and cerebrovascular, neurogenic, hypertensive heart disease, coronary occlusion and myocardial infarction, as well as venous thromboembolism [12, 29–32]. Compared to low homocysteine, high plasma levels of homocysteine show increase in the chance of death due to heart disease [33]. Furthermore, levels of homocysteine greater than 20 μM in plasma of patients with coronary heart disease were associated with a 35% increase in mortality [34].
Several studies have implicated hyperhomocysteinemia as a risk factor for hypertension. In the “Hordaland Homocysteine Study” of about 16,000 people of 40–67 years old with no history of hypertension, diabetes or coronary vascular disease, the plasma homocysteine levels were positively related to blood pressure [35]. Similarly, Malinow et al. [36] found that hypertensive men with no history of atherosclerotic disease had higher homocysteine levels than non-hypertensive men. Sutton-Tyrrell et al. [12] also found a significant association of homocysteine levels with systolic hypertension.
Vascular dysfunction, such as endothelial dysfunction and vascular hypertrophy, is a hallmark of hypertension and several lines of evidences support the role of homocysteine causing vascular dysfunction [37, 38]. Studies have also indicated that a number of forms causing hypertension are associated with elevated plasma levels of homocysteine. Although homocysteine has been documented to affect endothelial and vascular smooth muscle function, the importance of homocysteine in mediating the vascular dysfunction and hypertension is still incompletely defined. Furthermore, the molecular mechanisms whereby hyperhomocysteinemia results in vascular dysfunction are still unclear. In addition to vascular dysfunction by nitration of contractile (actin and myosin) and ECM proteins, homocysteine causes vascular dysfunction by reducing the bioavailability of endothelial NO. Also, homocysteine activates matrix metalloproteinases (MMPs) and collagenolysis, leading to vascular hypertrophy.
To understand the mechanism by which homocysteine causes hypertension, we studied the effects of hyperhomocysteinemia on endothelial endocardium and human coronary arteries. The results demonstrated that endothelium is impaired in hyperhomocysteinemic endocardium and vessels [39, 40]. The homocysteine activates metalloproteinases and induces collagenolysis [39]. The metabolites from hyperhomocysteinemic endothelium could modify components of the underlying muscle cells, leading to vascular dysfunction and hypertension [13]. In the rat model of hyperhomocysteinemia, a recent study suggests that H2S levels and the H2S-generating enzyme CSE activity in the myocardium were decreased [25]. Also, the activities of myocardial mitochondrial respiratory enzymes related to reactive oxygen species (ROS) metabolism were significantly dysfunctional in hyperhomocysteinemic rats [25]. Interestingly, this study demonstrated that intraperitoneal administration of H2S restored the enzymatic activities and reduced oxidative stress by scavenging H2O2 and superoxide ( ) generated by Hcy in isolated myocardial mitochondria. The expression of CSE enzyme in the portal vein was initially reported by Hosoki et al. [20] and inhibition of this enzyme blocked H2S production. Also, in vitro relaxation of norepinephrine precontracted portal vein by NaHS (a H2S donor) was documented by the same group [20]. Later, in a rat model of hyperhomocysteinemia, Distrutti et al. [41] reported that homocysteine caused reduced NO release from sinusoidal endothelial cells resulting in hepatic stellate cell contraction, whereas perfusion of the liver with sodium sulfide (a H2S donor) resulted in vasodilation, suggesting the portal hypertension caused by hyperhomocysteinemia was being counteracted by H2S. Thus, these findings clearly indicate that hyperhomocysteinemia not only impairs the endothelium through oxidative stress but also impairs CSE/H2S pathway causing hypertension and H2S plays a significant role to normalize the blood pressure caused by hyperhomocysteinemia.
Hyperhomocysteinemia, Formation of Nitrotyrosine, and Hypertension
Homocysteine causes endothelial dysfunction [42] and is associated with hypertension in humans [12]; but, the mechanisms remain poorly understood. It is known that homocysteine inhibits growth, reduces cell density, and causes a dose dependent decrease in DNA synthesis in vascular endothelial cells [43]. Additionally, it reduces bioavailability of endothelial-derived NO [44] and induces iNOS [45]. The mechanism underlying reduced bioavailability of NO includes: (1) homocysteine reduces thioredoxin and increases NAD(P)H oxidase resulting in generation of superoxide ( ); (2) elevated level of homocysteine also reduces endogenous generation of H2S, therefore increases oxidative stress; (3) the superoxide reacts with NO to form peroxynitrite (ONOO−); (4) peroxynitrite reacts with protein tyrosine residues resulting in protein modification (please see Fig. 3). We demonstrated that homocysteine activated protease-activated receptor-4, which induces production of ROS by increasing NAD(P)H oxidase and decreasing thioredoxin expression and reduces NO bioavailability in cultured cardiac microvascular endothelial cell [46]. Lentz et al. [47] have reported that diet-induced moderate hyperhomocysteinemia in monkeys exhibited increased platelet-mediated vasoconstriction, impaired endothelial-dependent vasodilation, and decreased thrombomodulin-dependent activation of protein C. We have demonstrated that normal human coronary artery is responsive to homocysteine, whereas the atheroscleroticartery is not [39]. The tissue levels of homocysteine in atherosclerotic artery were 10-fold higher than normal artery [39] and homocysteine treatment induces hypertension in pigs and rats [2, 48]. The ex vivo treatment of aortas with homocysteine for 14 days in tissue culture conditions produces endothelium disruption and arteriosclerotic lesions, similar to in vivo [49]. These studies suggested a role of homocysteine in vascular structure and function which may be due to protein modification by peroxynitrite that generated from homocysteine-induced production.
Fig. 3.
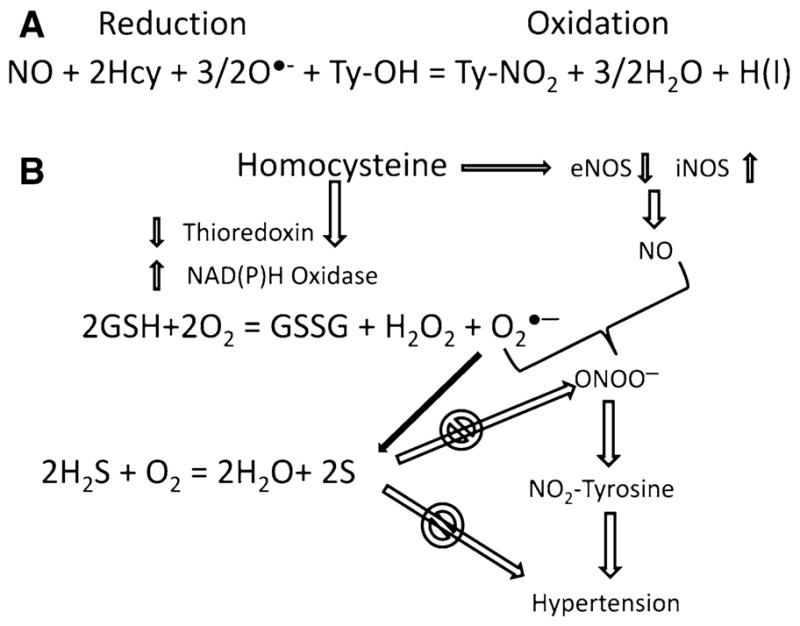
a Oxidation reduction and formation of nitrotyrosine by homocysteine. b Homocysteine causes reduction of thioredoxin and increases superoxide production by inducing NAD(P)H oxidase. Homocysteine also induces eNOS and iNOS to produce NO. Reaction of NO and tyrosine forms peroxynitrite and causes nitrosylation of protein tyrosine residues, such as actin and myosin. This leads to impairment of contractility and resulting in hypertension. H2S scavenges superoxide; therefore, reduces hypertension
Homocysteine, Vascular Smooth Muscle Cell (VSMC) Proliferation, and Hypertension: Modulatory Role of H2S
Homocysteine is known to diminish bioavailability of NO and increases oxidative stress. This inhibits vasodilation and causes hypertension. Vascular relaxation is very much dependent on proper coordination between endothelial and smooth muscle cell (SMC) and arterial resistance increases with the proliferation of these cells. In an attempt to examine the effect of homocystine on human VSMC, we isolated VSMC from idiopathic dilated cardiomyopathic hearts [50]. Coronaries in these hearts were apparently normal and homocysteine found to induce SMC proliferation in vitro [50]. Similar results were previously reported by Tsai et al. [51]. One of the pathophysiological mechanisms of hyperhomocysteinemia-associated hypertension underlies on the SMC proliferation which alters the elastic properties of the vascular wall [52]. As homocysteine competes with cysteine to bind CSE [25, 27] and hyperhomocysteinemia decreases the CSE activity [25], it could be speculated that during hyperhomocysteinemia H2S production will be diminished. Therefore, it not surprising that genetic manipulation to increase endogenous production of H2S will inhibit SMC proliferation and will maintain vascular elastic compliance to retain normal blood pressure during hyperhomocysteinemia. In fact, in vitro experimental evidence suggested that overexpression of H2S forming enzyme, CSE inhibited cell proliferation primarily through increased H2S production [53]. Homocystine induces collagen synthesis and expression and increases intimal–medial thickness [48, 54]. This consequently reduces elastin/collagen ratio which impairs vascular compliance resulting in increased systemic vascular resistance and sustained arterial hypertension.
Calcium (Ca2+) Handling, Collagen Expression, and Hypertension
Regulation of intracellular calcium plays a key role in hypertension. Acute intracellular calcium overload in VSMCs increases peripheral vascular resistance, produces hypercontractility, and elevates blood pressure [55]. It is reported that progressive elevation of calcium destroys the structural integrity of the artery and arteriolar walls [56]. Calcium has also been shown to promote for the synthesis and deposition of collagen in the vascular wall, because calcium channel blockers reduce ECM collagen synthesis [57, 58]. The collagen during hyperhomocysteinemia can oxidatively modify and deposit in the ECM. Collagen and elastin are two major components of connective tissue and homeostasis of these two proteins in the vascular bed regulate proper elasticity of blood flow through vessels. While elastin allows tissues to resume their shape after stretching or contracting, the collagen of the ECM supports most tissues and gives cells structure from the outside. The imbalance between elastin and collagen destroys proper elasticity of the vessel and excessive collagen deposition causes vascular stiffness and fibrosis. This creates vascular resistance for proper blood flow that results in hypertension. We have reported that homocysteine induces ECM production via intracellular calcium release [59]. Depleting extracellular calcium did not alter homocysteine-effect on intracellular calcium; however, thapsigargin pretreatment, which depletes intracellular Ca2+ stores, abolished the homocysteine effect demonstrating its dependence on intracellular Ca2+ stores [59]. Therefore, regulation of intracellular calcium seems to play a role in ECM remodeling during hyperhomocysteinemia, although regulation of intracellular calcium by H2S has been shown with conflicting results. For example, Nagai et al. [60] have shown that H2S increases intracellular Ca2+ and induces calcium waves in astrocytes and this increase was largely by inducing Ca2+influx rather than through release from intracellular Ca2+stores; whereas Pan et al. [61] reported that H2S-preconditioning regulated intracellular calcium handling which facilitated intracellular Ca2+ removal via both accelerating uptake of Ca2+into sarcoplasmic reticulum and enhancing Ca2+ extrusion through Na+/Ca2+ exchanger in a PKC-dependent manner in cardiomyocytes. While H2S activated Ca2+ channels to induce Ca2+influx to mediate signals between neurons and glia, the intracellular Ca2+removal in H2S preconditioning cardiomyocyte was against myocyte hypercontracture that protected heart against ischemia–reperfusion insult. Thus, H2S played a differential role of intracellular Ca2+regulation in neuronal cells where Ca2+ influx mediates signals between neighboring cells and in cardiomyocytes where H2S protected heart against Ca2+ overload and hypercontracture. We demonstrated that pretreatment of VSMC with homocysteine increases the ability to react with potent agonist, such as angiotensin II, which normally has no effect on intra-cellular Ca2+. After homocysteine pretreatment, VSMCs were extremely responsive to angiotensin II at concentrations well below the physiologic range. This result suggested that an initial effect of homocysteine is to induce release of intracellular Ca2+ in VSMC and may induce vascular reactivity. The transient in Ca2+correlates with the effect on ECM associated with homocysteine [59]. The effects of homocysteine on collagen production correlated with its effect in intracellular Ca2+ and were mediated by multiple intracellular signaling pathways in VSMC [59]. These include protein kinase C, nitric oxide synthase (NOS), phospholipase A2, HMG co-A reductase, tyrosine kinase, and calcium channel [59]. A possible mechanism of homocysteine-induced ECM production and hypertension in VSMC has been depicted in Fig. 4.
Fig. 4.
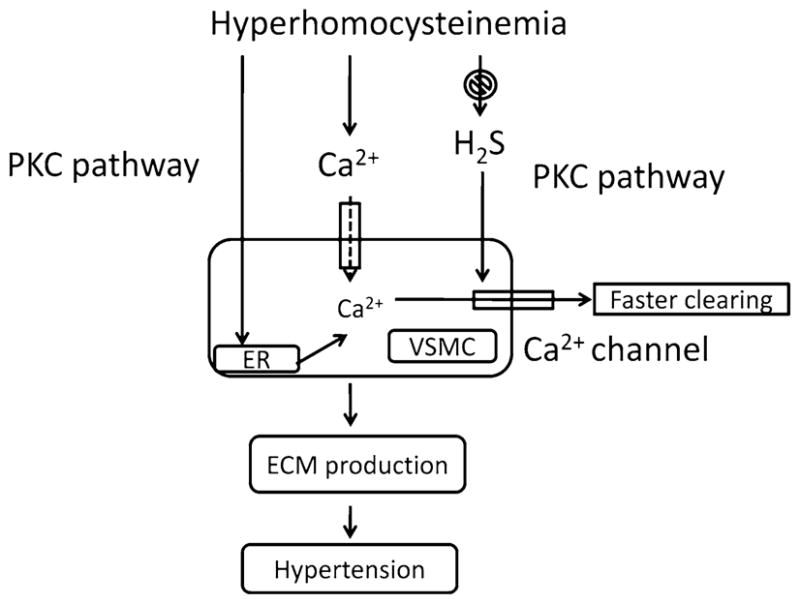
Homocysteine induces intracellular Ca2+ release and ECM production which results in vascular contraction and stiffness. H2S helps faster release of intracellular calcium thereby prevents contractility and ECM formation
Contractile Proteins and Hypertension: H2S is a Key Player
Alterations in the composition and distribution of isoforms of contractile proteins in hypertension have been demonstrated [62, 63]. However, little has been reported on either the modification or the changes in the composition of these proteins in hyperhomocysteinemia. Several consequences are observed leading to vascular damage by homocysteine. These include endothelial cell desquamation [64], oxidative modification of low density lipoproteins [65], increased adhesion of monocytes to the vessel wall [66], and impaired vascular response to the endothelium-dependent relaxing factor (NO) [47]. Impaired flow-mediated vasodilation has been demonstrated in healthy humans after an acute increase in plasma homocysteine [42]. Although flow-mediated vasodilation is largely dependent on the release of NO, it focuses only on the vasomotor response of the endothelium. However, due to its connection in controlling underlying VSMC function, it is plausible that it also modifies the underlying contractile apparatus. In an aortic banding and two kidney one clip Goldblatt hypertension, formation of nitrotyrosine in the aorta and kidney was enhanced [67, 68]. However, in these studies the role of protein labeled with nitrotyrosine in vasoconstriction was not addressed. Homocysteine induces proliferation in VSMCs [43, 50] and H2S has been shown to inhibit rat aortic VSMC proliferation [69]. However, the link between endothelial dysfunction and proliferation of VSMCs is unclear. We also demonstrated that factors released from endothelium inhibit homocysteine-induced contraction of endocardium [40], while others have demonstrated that H2S potentiates NO production via enhancement of extracellular signal-regulated kinase activation in rat VSMC [70]. Thus, the homocysteine-mediated vascular contraction may be mediated through decreased production of H2S in the VSMCs in hyperhomocysteinemia. We also demonstrated that de-endothelialized endocardium enhanced contraction to homocysteine [40]. These results suggest plausible modification (nitrotyrosine) of contractile apparatus in underlying muscle cells in myocardium [40]. Based on the current literature, we conclude that homocysteine neutralizes endothelial NO and modifies underlying smooth muscle actin and myosin by redox mediated peroxynitrite generation and nitrotyrosine formation. This initiates the cascades of vascular ECM remodeling, fibrosis, and vasoconstriction. H2S, being an scavenger neutralizes oxidative stress, thus either endogenous activation of H2S producing enzymes and/or supplementation of H2S may protects smooth muscle protein from being oxidative modification and reduce hypertension in hyperhomocysteinemia. Future studies are required to confirm or cancel this concept.
ECM, Protein Homocysteinylation, and Hypertension: Importance of H2S for Vascular Elastance
The vascular fibrosis, stiffness, atherosclerosis, and arteriosclerosis are associated with hypertension [31]. Studies in animal models of hyperhomocysteinemia suggested that hyperhomocysteinemia develops more like arteriosclerotic/prothrombotic than like atherosclerotic lesions [71–73]. Homocysteine induces ECM fibrillar collagen [50, 74] and elastinolytic proteinase in VSMCs [75]. A high-protein diet causes abdominal aortic dysfunction by reducing elastic compliance [76]. Considerable endothelial damage and loss of endothelium-derived NO [77], collagen synthesis and deposition [74], and increase expression of connective tissue growth factor (CTGF) in VSMC both in vivo and in vitro [78] were observed in hyperhomocysteinemic condition. These findings suggest that homocysteine contribute to progression of atherosclerosis that may lead to hypertension. A possible mechanism is shown in Fig. 5.
Fig. 5.
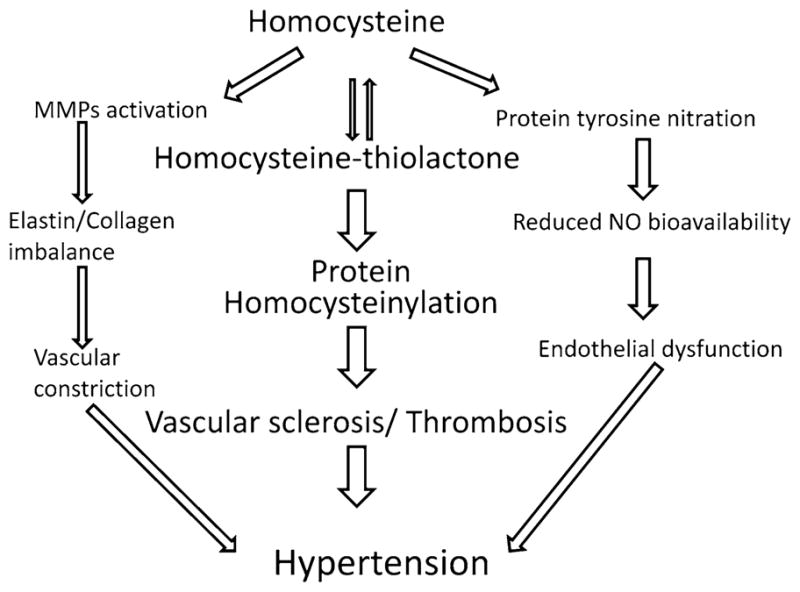
Schematic of homocysteine-induced MMP activation, protein homocysteinylation and endothelial dysfunction that causes hypertension
Hypertension results from persistent vasoconstriction, smooth muscle growth, and ECM remodeling of arteries. The dynamics of elastin and collagen are physiological processes in the normal arteries and imbalances of elastin/collagen ratio cause vascular dysfunction. For example, excessive collagen deposition or oxidative modification of collagen (glycated collagen) in the basement membrane can cause vascular stiffness and hypertension. MMPs degrade extracellular protein collagen and elastin, but the turnover of collagen is faster than elastin, therefore, during oxidative stress glycated collagen deposits in the ECM causing vascular stiffness and hypertension. Homocysteine has been reported to cause arterial stiffness by modulating elastin/collagen ratio [79] resulting in hypertension. On the other hand hyperhomocysteinemia has been shown to decrease H2S [25] and decrease in plasma H2S has been reported to cause hypertension [15], whereas NaHS (a H2S donor) significantly increased plasma H2S, decreased mean pulmonary arterial pressure in rats [80]. NaHS also inhibited the proliferation of smooth muscle cells in the pulmonary artery wall. The expression of collagen I and III were decreased by NaHS in the pulmonary arteries of rats under hypoxia suggesting that H2S played an important role in the development of hypoxic pulmonary vascular structural remodeling resulting in reduced arterial pressure [80]. MMPs are matrix-degrading enzymes involved in ECM turnover and promotes smooth muscle cell (SMC) and endothelial cell proliferation and migration. Tissue inhibitors of metalloproteinases (TIMPs) are natural inhibitors of MMPs and imbalance of MMPs/TIMPs axis could lead to abnormal ECM deposition and form tissue fibrosis. We have reported that homocysteine activates latent resident tissue MMPs [39, 81] and inhibition of MMPs triggers fibrosis [82]; therefore reduces hypertension [83]. In a one kidney hypertensive mouse model we also demonstrated that hyperhomocysteinemia induced MMP-2 and -9 activation was normalized with H2S supplementation that prevented renal damage [84]. Renal insufficiency due to reno-vascular damage is linked to hypertension; therefore, in hyperhomocysteinemia, treatment with H2S could be a therapeutic approach to prevent deleterious vascular remodeling and hypertension.
Hyperhomocysteinemia, Angiotensin, and Hypertension
Angiotensin II is the main peptide of rennin–angiotensin system and activation of angiotensin type 1 (AT1) receptor leads to the production of ROS. Although studies have demonstrated AT1 receptor-mediated production of ECM components, very little is known about AT1 receptor regulation and its consequences in hyperhomocysteinemia. We have demonstrated that homocysteine induced the AT1-receptor induced MMP-9 and collagen synthesis in vascular endothelial cells [85]. Laggner et al. [86] showed that H2S inhibited angiotensin-converting enzyme (ACE) activity of endothelial cells. Therefore, it is possible that during hyperhomocysteinemia reduced-H2S will promote ACE activity that may lead to upregulation of angiotensin II and subsequently hypertension. However, this mechanism needs to be investigated thoroughly before a conclusion can be drawn. A possible pathway of angiotensin II modulation by H2S during hyperhomocysteinemia resulting in vascular fibrosis and hypertension has been shown in Fig. 6.
Fig. 6.
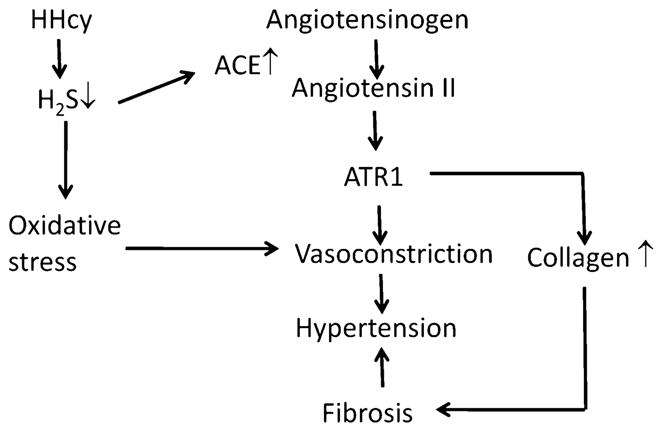
Schematic relationship of hyperhomocysteinemia, reduced H2S and upregulation of angiotensin that may develop hypertension
Concluding Remarks and Perspective
To understand the cause and effect of relationship between homocysteine-mediated nitrotyrosine formation and vascular dysfunction, it is essential to determine whether NOS is induced in hyperhomocysteinemic subjects and H2S therapy can modulate this effect. We have demonstrated that homocysteine induces iNOS and reduces eNOS in endothelial cells [45, 46] and reduces bioavailability of NO through the formation of nitrotyrosine. However, whether this mechanism is related to reduce plasma H2S content that may further exacerbates protein modification through nitration of tyrosine residue and complement hyperhomocysteinemia-associated hypertension needs to be determined. Actin and myosin are the primary components of vascular smooth muscle proteins associated with contractile function and have abundant exposed tyrosine in them. Together, the formation of nitrotyrosine in actin and myosin and induction of NOS may demonstrate that these components are affected by homocysteine. Although many ECM components such as elastin, collagen, and proteoglycans also contain exposed tyrosine, their modification may affect primarily the structure. However, tyrosine modification in TIMP-4, which regulates the vascular ECM remodeling, may reduce its ability to regulate MMP and causes adverse ECM remodeling, impairs vascular function and may develop hypertension.
Acknowledgments
This research was supported, in part, by the National Institutes of Health grants, HL-75185, HL-71010 and NS-51568.
Abbreviations
- AT1
Angiotensin type 1
- CBS
Cystathionine-β synthase
- CSE
Cystathionine-γ lyase
- CTGF
Connective tissue growth factor
- ECM
Extracellular matrix
- H2S
Hydrogen sulfide
- MMP
Matrix metalloproteinase
- NO
Nitric oxide
- NOS
Nitric oxide synthase
Superoxide
- ROS
Reactive oxygen species
- TIMP
Tissue inhibitor of metalloproteinase
- VSMC
Vascular smooth muscle cell
Footnotes
Disclosures No competing financial interests exist.
References
- 1.Oparil S, Zaman MA, Calhoun DA. Pathogenesis of hypertension. Annals of Internal Medicine. 2003;139:761–776. doi: 10.7326/0003-4819-139-9-200311040-00011. [DOI] [PubMed] [Google Scholar]
- 2.Rolland PH, Friggi A, Barlatier A, Piquet P, Latrille V, Faye MM, et al. Hyperhomocysteinemia-induced vascular damage in the minipig. Captopril-hydrochlorothiazide combination prevents elastic alterations. Circulation. 1995;91:1161–1174. doi: 10.1161/01.cir.91.4.1161. [DOI] [PubMed] [Google Scholar]
- 3.Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. New England Journal of Medicine. 1997;336:1117–1124. doi: 10.1056/NEJM199704173361601. [DOI] [PubMed] [Google Scholar]
- 4.Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. Journal of the American College of Cardiology. 1999;34:2002–2006. doi: 10.1016/s0735-1097(99)00469-6. [DOI] [PubMed] [Google Scholar]
- 5.Wollesen F, Brattstrom L, Refsum H, Ueland PM, Berglund L, Berne C. Plasma total homocysteine and cysteine in relation to glomerular filtration rate in diabetes mellitus. Kidney International. 1999;55:1028–1035. doi: 10.1046/j.1523-1755.1999.0550031028.x. [DOI] [PubMed] [Google Scholar]
- 6.Upchurch GR, Jr, Welch GN, Fabian AJ, Freedman JE, Johnson JL, Keaney JF, Jr, et al. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. Journal of Biological Chemistry. 1997;272:17012–17017. doi: 10.1074/jbc.272.27.17012. [DOI] [PubMed] [Google Scholar]
- 7.Dudman NP, Wilcken DE. Increased plasma copper in patients with homocystinuria due to cystathionine beta-synthase deficiency. Clinica Chimica Acta. 1983;127:105–113. doi: 10.1016/0009-8981(83)90080-3. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida Y, Nakano A, Hamada R, Kamitsuchibashi H, Yamamoto K, Akagi H, et al. Patients with homocystinuria: High metal concentrations in hair, blood and urine. Acta Neurologica Scandinavica. 1992;86:490–495. doi: 10.1111/j.1600-0404.1992.tb05130.x. [DOI] [PubMed] [Google Scholar]
- 9.Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 10.Lamping KG. Hypercontractility of vascular muscle in atherosclerosis. Circulation. 1997;96:4131–4132. [PubMed] [Google Scholar]
- 11.Boers GH. Mild hyperhomocysteinemia is an independent risk factor of arterial vascular disease. Seminars in Thrombosis and Hemostasis. 2000;26:291–295. doi: 10.1055/s-2000-8096. [DOI] [PubMed] [Google Scholar]
- 12.Sutton-Tyrrell K, Bostom A, Selhub J, Zeigler-Johnson C. High homocysteine levels are independently related to isolated systolic hypertension in older adults. Circulation. 1997;96:1745–1749. doi: 10.1161/01.cir.96.6.1745. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigo R, Passalacqua W, Araya J, Orellana M, Rivera G. Homocysteine and essential hypertension. Journal of Clinical Pharmacology. 2003;43:1299–1306. doi: 10.1177/0091270003258190. [DOI] [PubMed] [Google Scholar]
- 14.Lip GY, Edmunds E, Martin SC, Jones AF, Blann AD, Beevers DG. A pilot study of homocyst(e)ine levels in essential hypertension: Relationship to von Willebrand factor, an index of endothelial damage. American Journal of Hypertension. 2001;14:627–631. doi: 10.1016/s0895-7061(00)01321-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? The FASEB Journal. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. Journal of Biochemistry. 2009;146:623–626. doi: 10.1093/jb/mvp111. [DOI] [PubMed] [Google Scholar]
- 18.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxidants and Redox Signaling. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 19.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. Journal of Neuroscience. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochemical and Biophysical Research Communications. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 21.Barber T, Triguero A, Martinez-Lopez I, Torres L, Garcia C, Miralles VJ, et al. Elevated expression of liver gamma-cystathionase is required for the maintenance of lactation in rats. Journal of Nutrition. 1999;129:928–933. doi: 10.1093/jn/129.5.928. [DOI] [PubMed] [Google Scholar]
- 22.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO Journal. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative transsulfuration reactions. Journal of Biological Chemistry. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao K. Effects of several unusual sulfur-containing amino acids on rat liver cystathionine-gamma-lyase. Physiological Chemistry and Physics. 1975;7:401–408. [PubMed] [Google Scholar]
- 25.Chang L, Geng B, Yu F, Zhao J, Jiang H, Du J, et al. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids. 2008;34:573–585. doi: 10.1007/s00726-007-0011-8. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. American Journal of Physiology. Heart and Circulatory Physiology. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 27.Stabler SP, Steegborn C, Wahl MC, Oliveriusova J, Kraus JP, Allen RH, et al. Elevated plasma total homocysteine in severe methionine adenosyltransferase I/III deficiency. Metabolism. 2002;51:981–988. doi: 10.1053/meta.2002.34017. [DOI] [PubMed] [Google Scholar]
- 28.Jakubowski H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. The FASEB Journal. 1999;13:2277–2283. [PubMed] [Google Scholar]
- 29.McCully KS. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. American Journal of Pathology. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- 30.McCully KS. Homocysteine and vascular disease. Nature Medicine. 1996;2:386–389. doi: 10.1038/nm0496-386. [DOI] [PubMed] [Google Scholar]
- 31.Tyagi SC. Homocyst(e)ine and heart disease: Pathophysiology of extracellular matrix. Clinical and Experimental Hypertension. 1999;21:181–198. doi: 10.3109/10641969909068660. [DOI] [PubMed] [Google Scholar]
- 32.Boers GH, Smals AG, Trijbels FJ, Fowler B, Bakkeren JA, Schoonderwaldt HC, et al. Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. New England Journal of Medicine. 1985;313:709–715. doi: 10.1056/NEJM198509193131201. [DOI] [PubMed] [Google Scholar]
- 33.Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. BMJ. 2002;325:1202. doi: 10.1136/bmj.325.7374.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nygard O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. Plasma homocysteine levels and mortality in patients with coronary artery disease. New England Journal of Medicine. 1997;337:230–236. doi: 10.1056/NEJM199707243370403. [DOI] [PubMed] [Google Scholar]
- 35.Nygard O, Vollset SE, Refsum H, Stensvold I, Tverdal A, Nordrehaug JE, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA. 1995;274:1526–1533. doi: 10.1001/jama.1995.03530190040032. [DOI] [PubMed] [Google Scholar]
- 36.Malinow MR, Levenson J, Giral P, Nieto FJ, Razavian M, Segond P, et al. Role of blood pressure, uric acid, and hemorheological parameters on plasma homocyst(e)ine concentration. Atherosclerosis. 1995;114:175–183. doi: 10.1016/0021-9150(94)05481-w. [DOI] [PubMed] [Google Scholar]
- 37.Jakubowski H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. Journal of Physiology and Pharmacology. 2008;59(Suppl 9):155–167. [PubMed] [Google Scholar]
- 38.Dayal S, Lentz SR. Murine models of hyperhomocysteinemia and their vascular phenotypes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:1596–1605. doi: 10.1161/ATVBAHA.108.166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyagi SC, Smiley LM, Mujumdar VS, Clonts B, Parker JL. Reduction-oxidation (Redox) and vascular tissue level of homocyst(e)ine in human coronary atherosclerotic lesions and role in extracellular matrix remodeling and vascular tone. Molecular and Cellular Biochemistry. 1998;181:107–116. doi: 10.1023/a:1006882014593. [DOI] [PubMed] [Google Scholar]
- 40.Tyagi SC, Smiley LM, Mujumdar VS. Homocyst(e)ine impairs endocardial endothelial function. Canadian Journal of Physiology and Pharmacology. 1999;77:950–957. [PubMed] [Google Scholar]
- 41.Distrutti E, Mencarelli A, Santucci L, Renga B, Orlandi S, Donini A, et al. The methionine connection: Homocysteine and hydrogen sulfide exert opposite effects on hepatic microcirculation in rats. Hepatology. 2008;47:659–667. doi: 10.1002/hep.22037. [DOI] [PubMed] [Google Scholar]
- 42.Chambers JC, McGregor A, Jean-Marie J, Kooner JS. Acute hyperhomocysteinaemia and endothelial dysfunction. Lancet. 1998;351:36–37. doi: 10.1016/S0140-6736(05)78090-9. [DOI] [PubMed] [Google Scholar]
- 43.Tsai JC, Perrella MA, Yoshizumi M, Hsieh CM, Haber E, Schlegel R, et al. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6369–6373. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Upchurch GR, Jr, Welch GN, Fabian AJ, Pigazzi A, Keaney JF, Jr, Loscalzo J. Stimulation of endothelial nitric oxide production by homocyst(e)ine. Atherosclerosis. 1997;132:177–185. doi: 10.1016/s0021-9150(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 45.Tyagi N, Gillespie W, Vacek JC, Sen U, Tyagi SC, Lominadze D. Activation of GABA-A receptor ameliorates homocysteine-induced MMP-9 activation by ERK pathway. Journal of Cellular Physiology. 2009;220:257–266. doi: 10.1002/jcp.21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. American Journal of Physiology. Heart and Circulatory Physiology. 2005;289:H2649–H2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- 47.Lentz SR, Sobey CG, Piegors DJ, Bhopatkar MY, Faraci FM, Malinow MR, et al. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. Journal of Clinical Investigation. 1996;98:24–29. doi: 10.1172/JCI118771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller A, Mujumdar V, Shek E, Guillot J, Angelo M, Palmer L, et al. Hyperhomocyst(e)inemia induces multiorgan damage. Heart and Vessels. 2000;15:135–143. doi: 10.1007/s003800070030. [DOI] [PubMed] [Google Scholar]
- 49.Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. International Journal of Cardiology. 2001;79:277–286. doi: 10.1016/s0167-5273(01)00449-1. [DOI] [PubMed] [Google Scholar]
- 50.Tyagi SC. Homocysteine redox receptor and regulation of extracellular matrix components in vascular cells. American Journal of Physiology. 1998;274:C396–C405. doi: 10.1152/ajpcell.1998.274.2.C396. [DOI] [PubMed] [Google Scholar]
- 51.Tsai JC, Wang H, Perrella MA, Yoshizumi M, Sibinga NE, Tan LC, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. Journal of Clinical Investigation. 1996;97:146–153. doi: 10.1172/JCI118383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Guldener C, Stehouwer CD. Hyperhomocysteinemia, vascular pathology, and endothelial dysfunction. Seminars in Thrombosis and Hemostasis. 2000;26:281–289. doi: 10.1055/s-2000-8472. [DOI] [PubMed] [Google Scholar]
- 53.Yang G, Cao K, Wu L, Wang R. Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. Journal of Biological Chemistry. 2004;279:49199–49205. doi: 10.1074/jbc.M408997200. [DOI] [PubMed] [Google Scholar]
- 54.Raziel A, Kornberg Y, Friedler S, Schachter M, Sela BA, Ron-El R. Hypercoagulable thrombophilic defects and hyperhomocysteinemia in patients with recurrent pregnancy loss. American Journal of Reproductive Immunology. 2001;45:65–71. doi: 10.1111/j.8755-8920.2001.450201.x. [DOI] [PubMed] [Google Scholar]
- 55.Zemel MB. Calcium modulation of hypertension and obesity: Mechanisms and implications. Journal of the American College of Nutrition. 2001;20:428S–435S. doi: 10.1080/07315724.2001.10719180. (discussion 440S–442S) [DOI] [PubMed] [Google Scholar]
- 56.Fleckenstein-Grun G, Frey M, Thimm F, Hofgartner W, Fleckenstein A. Calcium overload—an important cellular mechanism in hypertension and arteriosclerosis. Drugs. 1992;44(Suppl 1):23–30. doi: 10.2165/00003495-199200441-00005. [DOI] [PubMed] [Google Scholar]
- 57.Vijayagopal P, Subramaniam P. Effect of calcium channel blockers on proteoglycan synthesis by vascular smooth muscle cells and low density lipoprotein–proteoglycan interaction. Atherosclerosis. 2001;157:353–360. doi: 10.1016/s0021-9150(00)00742-5. [DOI] [PubMed] [Google Scholar]
- 58.Schachter M. Vascular smooth muscle cell migration, atherosclerosis, and calcium channel blockers. International Journal of Cardiology. 1997;62(Suppl 2):S85–S90. doi: 10.1016/s0167-5273(97)00245-3. [DOI] [PubMed] [Google Scholar]
- 59.Mujumdar VS, Hayden MR, Tyagi SC. Homocyst(e)ine induces calcium second messenger in vascular smooth muscle cells. Journal of Cellular Physiology. 2000;183:28–36. doi: 10.1002/(SICI)1097-4652(200004)183:1<28::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 60.Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. The FASEB Journal. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- 61.Pan TT, Neo KL, Hu LF, Yong QC, Bian JS. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. American Journal of Physiology. Cell Physiology. 2008;294:C169–C177. doi: 10.1152/ajpcell.00282.2007. [DOI] [PubMed] [Google Scholar]
- 62.Sen S, Young D. Effect of sodium deprivation on cardiac hypertrophy in spontaneously hypertensive rats: Influence of aging. Journal of Molecular and Cellular Cardiology. 1991;23:695–704. doi: 10.1016/0022-2828(91)90979-v. [DOI] [PubMed] [Google Scholar]
- 63.Packer CS, Roepke JE, Oberlies NH, Rhoades RA. Myosin isoform shifts and decreased reactivity in hypoxia-induced hypertensive pulmonary arterial muscle. American Journal of Physiology. 1998;274:L775–785. doi: 10.1152/ajplung.1998.274.5.L775. [DOI] [PubMed] [Google Scholar]
- 64.Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. Journal of Clinical Investigation. 1986;77:1370–1376. doi: 10.1172/JCI112442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinecke JW, Kawamura M, Suzuki L, Chait A. Oxidation of low density lipoprotein by thiols: Superoxide-dependent and -independent mechanisms. Journal of Lipid Research. 1993;34:2051–2061. [PubMed] [Google Scholar]
- 66.Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 67.Bouloumie A, Bauersachs J, Linz W, Scholkens BA, Wiemer G, Fleming I, et al. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension. 1997;30:934–941. doi: 10.1161/01.hyp.30.4.934. [DOI] [PubMed] [Google Scholar]
- 68.Bosse HM, Bachmann S. Immunohistochemically detected protein nitration indicates sites of renal nitric oxide release in Goldblatt hypertension. Hypertension. 1997;30:948–952. doi: 10.1161/01.hyp.30.4.948. [DOI] [PubMed] [Google Scholar]
- 69.Du J, Hui Y, Cheung Y, Bin G, Jiang H, Chen X, et al. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart and Vessels. 2004;19:75–80. doi: 10.1007/s00380-003-0743-7. [DOI] [PubMed] [Google Scholar]
- 70.Jeong SO, Pae HO, Oh GS, Jeong GS, Lee BS, Lee S, et al. Hydrogen sulfide potentiates interleukin-1beta-induced nitric oxide production via enhancement of extracellular signal-regulated kinase activation in rat vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2006;345:938–944. doi: 10.1016/j.bbrc.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 71.Jovin IS, Wolfe ML, Gefter WB, Miller WT, Aron-chick JM, Flannery DT, et al. Plasma homocysteine levels are not associated with extent of coronary artery calcification in asymptomatic persons. Circulation. 1999;100:25–25. [Google Scholar]
- 72.Hofmann MA, Lu Y, Ferran L, Kohl B, Schmidt AM. Homocysteine induces vascular activation in vitro and in vivo: Accelerated atherosclerosis develops in apo E null mice with hyperhomocysteinemia. Circulation. 1999;100:44–44. [Google Scholar]
- 73.Matthias D, Becker CH, Riezler R, Kindling PH. Homocysteine induced arteriosclerosis-like alterations of the aorta in normotensive and hypertensive rats following application of high doses of methionine. Atherosclerosis. 1996;122:201–216. doi: 10.1016/0021-9150(95)05740-4. [DOI] [PubMed] [Google Scholar]
- 74.Majors A, Ehrhart LA, Pezacka EH. Homocysteine as a risk factor for vascular disease. Enhanced collagen production and accumulation by smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:2074–2081. doi: 10.1161/01.atv.17.10.2074. [DOI] [PubMed] [Google Scholar]
- 75.Jourdheuil-Rahmani D, Rolland PH, Rosset E, Branchereau A, Garcon D. Homocysteine induces synthesis of a serine elastase in arterial smooth muscle cells from multi-organ donors. Cardiovascular Research. 1997;34:597–602. doi: 10.1016/s0008-6363(97)00048-5. [DOI] [PubMed] [Google Scholar]
- 76.Hodgkin DD, Gilbert RD, Roos PJ, Sandberg LB, Boucek RJ. Dietary lipid modulation of connective tissue matrix in rat abdominal aorta. American Journal of Physiology. 1992;262:R389–394. doi: 10.1152/ajpregu.1992.262.3.R389. [DOI] [PubMed] [Google Scholar]
- 77.Dayal S, Lentz SR. Role of redox reactions in the vascular phenotype of hyperhomocysteinemic animals. Antioxidants and Redox Signaling. 2007;9:1899–1909. doi: 10.1089/ars.2007.1806. [DOI] [PubMed] [Google Scholar]
- 78.Liu X, Luo F, Li J, Wu W, Li L, Chen H. Homocysteine induces connective tissue growth factor expression in vascular smooth muscle cells. Journal of Thrombosis and Haemostasis. 2008;6:184–192. doi: 10.1111/j.1538-7836.2007.02801.x. [DOI] [PubMed] [Google Scholar]
- 79.Mayer O, Filipovsky J, Dolejsova M, Cifkova R, Simon J, Bolek L. Mild hyperhomocysteinaemia is associated with increased aortic stiffness in general population. Journal of Human Hypertension. 2006;20:267–271. doi: 10.1038/sj.jhh.1001983. [DOI] [PubMed] [Google Scholar]
- 80.Hongfang J, Cong B, Zhao B, Zhang C, Liu X, Zhou W, et al. Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sciences. 2006;78:1299–1309. doi: 10.1016/j.lfs.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 81.Moshal KS, Sen U, Tyagi N, Henderson B, Steed M, Ovechkin AV, et al. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. American Journal of Physiology. Cell Physiology. 2006;290:C883–C891. doi: 10.1152/ajpcell.00359.2005. [DOI] [PubMed] [Google Scholar]
- 82.Corbel M, Caulet-Maugendre S, Germain N, Molet S, Lagente V, Boichot E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. Journal of Pathology. 2001;193:538–545. doi: 10.1002/path.826. [DOI] [PubMed] [Google Scholar]
- 83.Martinez ML, Castro MM, Rizzi E, Fernandes K, Demacq C, Bendhack LM, et al. Lercanidipine reduces matrix metalloproteinase-2 activity and reverses vascular dysfunction in renovascular hypertensive rats. European Journal of Pharmacology. 2008;591:224–230. doi: 10.1016/j.ejphar.2008.06.096. [DOI] [PubMed] [Google Scholar]
- 84.Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, et al. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. American Journal of Physiology. Renal Physiology. 2009;297:F410–F419. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sen U, Herrmann M, Herrmann W, Tyagi SC. Synergism between AT1 receptor and hyperhomocysteinemia during vascular remodeling. Clinical Chemistry and Laboratory Medicine. 2007;45:1771–1776. doi: 10.1515/CCLM.2007.354. [DOI] [PubMed] [Google Scholar]
- 86.Laggner H, Hermann M, Esterbauer H, Muellner MK, Exner M, Gmeiner BM, et al. The novel gaseous vasorelaxant hydrogen sulfide inhibits angiotensin-converting enzyme activity of endothelial cells. Journal of Hypertension. 2007;25:2100–2104. doi: 10.1097/HJH.0b013e32829b8fd0. [DOI] [PubMed] [Google Scholar]