

Abstract
Circularly permuted fluorescent proteins (FPs) have a growing number of uses in live cell fluorescence biosensing applications. Most notably, they enable the construction of single fluorescent protein-based biosensors for Ca2+ and other analytes of interest. Circularly permuted FPs are also of great utility in the optimization of fluorescence resonance energy transfer (FRET)-based biosensors by providing a means for varying the critical dipole–dipole orientation. We have previously reported on our efforts to create circularly permuted variants of a monomeric red FP (RFP) known as mCherry. In our previous work, we had identified six distinct locations within mCherry that tolerated the insertion of a short peptide sequence. Creation of circularly permuted variants with new termini at the locations corresponding to the sites of insertion led to the discovery of three permuted variants that retained no more than 18% of the brightness of mCherry. We now report the extensive directed evolution of the variant with new termini at position 193 of the protein sequence for improved fluorescent brightness. The resulting variant, known as cp193g7, has 61% of the intrinsic brightness of mCherry and was found to be highly tolerant of circular permutation at other locations within the sequence. We have exploited this property to engineer an expanded series of circularly permuted variants with new termini located along the length of the 10th β-strand of mCherry. These new variants may ultimately prove useful for the creation of single FP-based Ca2+ biosensors.
Keywords: red fluorescent protein, mCherry, protein engineering, circular permutation, directed evolution, fluorescence imaging, biosensing
Introduction
A variety of intrinsically fluorescent proteins (FP) from the superfamily of Aequorea victoria green FP (avGFP)-like proteins have proven to be popular and powerful tools for live cell fluorescence imaging applications. Their usefulness stems from the fact that these proteins are self-sufficient to form a brightly fluorescent chromophore within the confines of their own tertiary structure.1 Accordingly, expression of a FP gene chimera in a wide variety of cell-types and tissue-types can provide a researcher with a noninvasive fluorescent marker for imaging of fusion protein localization and dynamics.
For many applications of FPs, the fact that the FP chromophore is fully encapsulated inside an 11-stranded ‘β-can’ tertiary structure [Fig. 1(A)],2 and thus effectively isolated from the environment of the bulk solvent, is advantageous. As the chromophore is isolated, its inherent fluorescent properties (e.g., quantum yield, extinction coefficient, lifetime, and rate of photobleaching) are presumably less susceptible to changes in the protein environment (e.g., changes in pH, viscosity, and concentration of quenchers of fluorescence) than if the chromophore was exposed to the bulk solvent. The relative steadfastness of the intrinsic fluorescent properties of the FP allow researchers to confidently assume that fluorescent intensity is directly proportional to protein concentration in live cell imaging experiments.
Figure 1.
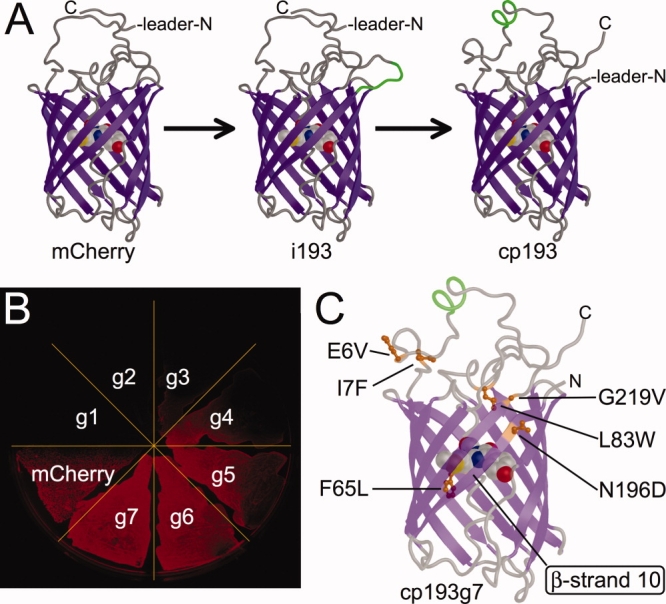
Engineering of a bright circularly permuted mCherry variant. (A) Representation of the steps of modification that produced the original cp193 variant from the insertional variant i193.21 (B) Fluorescence image of a streak plate of E. coli expressing each of the seven generations of cp193 generated during the directed evolution process. (C) Location of amino acid substitutions in the cp193g7 variant following seven rounds of directed evolution. The ribbon representation is rendered as partially transparent to facilitate visualization of all the substitutions. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
There are some applications of FPs for which the protected nature of the chromophore is a distinct disadvantage. For example, there is substantial interest in using induced changes in the fluorescent properties of the FP chromophore to report on specific changes in the intracellular environment.3 FP constructs in which the fluorescent intensity or hue is dependent upon an external parameter are commonly referred to as single FP-type biosensors. An alternative strategy for creating FP-based biosensors is to exploit the modulation of fluorescence resonance energy transfer (FRET) between two different hues of FP.4 The advantages and disadvantages of each of these distinct strategies has previously been discussed.5
In designing and constructing single FP-type biosensors, an effective ‘line of communication’ between the external variable of interest and the fluorescent properties of the chromophore must be created. That is, the external change in the environmental variable must produce a physical change in the chromophore environment that modulates its fluorescent hue, intensity, or lifetime. One of the simplest examples of an environmental variable that can be ‘sensed’ and imaged in live cells using FPs is pH. With few exceptions, the fluorescence of FPs is pH dependent and a number of variants that change fluorescence intensity at near-physiological pH values have been reported.6 Some FPs also exhibit an intrinsic sensitivity towards halide ions and have been used to image physiologically-relevant changes in chloride ion concentrations.7
The creation of single FP-based biosensors that specifically respond to environmental variables other than pH or halide ions has required some relatively sophisticated engineering of avGFP chimeras. Specifically, researchers have found means of fusing avGFP with a second protein that provides the molecular recognition specificity for the target analyte of interest. Upon binding to (or being modified by) the target, the molecular recognition domain undergoes a conformational change that alters the chromophore environment and the fluorescent properties of the chromophore. However, the N- and C-termini of the FP, which are the most accessible locations for fusion of the molecular recognition domain, are distant from the chromophore. Accordingly, a conformational change in a protein fused to one of these termini is expected to have no substantial effect on the chromophore properties. One solution to this fundamental problem is to insert the molecular recognition domain into the FP at a location near the chromophore.8 Examples of this strategy include the insertion of calmodulin8 or a single EF-hand domain9 into avGFP to create Ca2+ biosensors. Similarly, biosensors for the protease trypsin have been created by introducing substrate sequence containing loops at various locations within an avGFP variant.10
A second solution is to create a circularly permuted variant of avGFP in which the new N- and C- termini are located in close proximity to the chromophore.8,11 Fusion of peptides or protein domains (that undergo a conformational change or an interaction in the presence of the analyte of interest) to the new termini can produce highly effective single FP-based biosensors.12,13 Perhaps the most important example of this second type of biosensor design are the Ca2+ biosensors,12–14 in which calmodulin and the M13 peptide are fused to the termini of a circularly permuted avGFP variant. X-ray crystal structures of one of the most promising single FP-based Ca2+ biosensors, known as G-CaMP, have recently been reported.15,16 Yet another variation on this design involves the insertion of a circularly permuted FP into a second protein that undergoes a conformational change upon binding to its target analyte. For example, a circularly permuted version of the red fluorescent protein mKate17 has recently been converted into a voltage-sensitive fluorescent protein by insertion into the voltage-sensitive domain of a membrane protein.18
A potentially valuable application of single FP-based biosensors would be to use them in pair wise combinations for multiparameter live cell imaging.19 However, as almost all of the single FP-based biosensors reported to date are based on either green or yellow variants of avGFP, it is not possible to image multiple biosensors in a single cell. Single FP-type biosensors are better suited to multiparameter imaging than FRET-based biosensors which each require two distinct hues of FP.20 With the goal of eventually producing an expanded repertoire of fluorescent hues for FP-based biosensors, we have undertaken the development of circularly permuted monomeric red FPs (RFPs).21 Our starting template is the engineered variant of Discosoma RFP22 known as mCherry.23
We have previously reported the discovery of three variants of mCherry (cp22, cp184, and cp193) that were permuted such that the new termini were located in loop regions at either end of the β-barrel and that retained detectable red fluorescence when expressed in colonies of E. coli.21 We had employed a strategy in which we first identified positions within the protein that tolerated insertion of a peptide, and then created the corresponding circular permutation at that position [Fig. 1(A)]. The greatly diminished brightness of these variants was attributed to a diminished ability to fold and undergo the post-translational modifications necessary for formation of the red fluorescent chromophore. The intrinsic brightness of the chromophore (i.e., the product of quantum yield and extinction coefficient for the fraction of proteins that did form the chromophore) was essentially unperturbed in the circularly permuted variants. We now report the directed evolution of the most compliant of these circularly permuted variants (cp193) for improved folding and chromophore maturation efficiency. This effort has led to the discovery of a number of new permutation sites, including some that are in within the β-sheet region of the β-barrel and in relatively close proximity to the chromophore. These sites represent promising locations for the insertion of binding domains with the aim of constructing single RFP-type biosensors.
Results
Directed evolution of cp193 for brighter fluorescence
Libraries of randomly mutated genes derived from the previously reported21 circularly permuted mCherry variants cp22, cp26, cp136, cp183, and cp193, were expressed in E. coli and colonies were screened for improvements in the brightness of the red fluorescence. The brightest variant that was obtained after the first round of screening was the N196D variant of cp193 (cp193g1). Our subsequent directed evolution efforts focused on this lineage, as variants of comparable brightness were not found after one round of screening libraries based on the other four permutation sites. An additional six generations of directed evolution were undertaken, with the brightest variant identified in each generation serving as the template for the subsequent cycle of library creation by error prone PCR [Fig. 1(B)]. The directed evolution process was stopped at the seventh generation, as no substantial improvements arose in subsequent efforts. DNA sequencing revealed that the seventh generation variant (cp193g7) had six substitutions relative to the original variant and was equivalent to cp193 + E6V, I7F, F65L, L83W, G219V, N196D [Fig. 1(C) and 2]. The cp193g7 variant has 61% of the intrinsic fluorescent brightness of mCherry and 69% of the in vivo brightness following expression for 24 h in colonies of E. coli (Table I). The absorbance and fluorescence emission maxima of cp193g7 (λabs, max = 580 nm, λem, max = 602 nm), are slightly blue-shifted relative to mCherry (λabs, max = 587 nm, λem, max = 610 nm).23
Figure 2.

Sequence alignment of mCherry, cp193, cp193g1, and cp193g7. The sequences of the circularly permuted variants have been rearranged to maintain residue number consistency with the original mCherry sequence. Amino acid substitutions are highlighted with white text on a shaded background. The chromophore forming residues are enclosed in the red box. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Table I.
Properties of Improved cp193 Variants Identified During Directed Evolution
| Protein | φ | ɛ (M−1 cm−1) | Fluorescent brightnessa (mM−1 cm−1) | Relative brightness in E. coli after 24 h at 37°C (%) | Relative brightness in E. coli after 72 h at 4°C (%) |
|---|---|---|---|---|---|
| mCherryb | 0.22 | 72000 | 16 | 100 | 100 |
| cp193 | 0.20 | 12000 | 2.4 | 4 | 8 |
| cp193g1 | 0.20 | 29000 | 5.8 | 2 | 47 |
| cp193g7 | 0.23 | 42000 | 9.7 | 69 | 100 |
| cp193g7N | 0.23 | 41000 | 9.4 | 81 | 100 |
| cp193g7C | 0.21 | 18000 | 3.8 | 3 | 22 |
The product of φ and ɛ.
Previously reported values.21
In the original cp193 variant, three residues (AYN) had been repeated at the N- and C-termini (Fig. 2). These were originally included to increase the odds of success for identifying brightly fluorescent circularly permuted variants. To further explore the role of the overlapping AYN sequence in cp193g7, we created two additional variants that retained the AYN sequence at either the N- or C-terminus, but not both. These variants are designated as cp193g7N (with permutation site at 191/192) and cp193g7C (with permutation site at 194/195), respectively. In vitro characterization of these variants revealed that cp193g7N retained 97% of the intrinsic fluorescent brightness of cp193g7, but cp193g7C retained only 39% (Table I). This result indicates that it is the N-terminal repeat of the AYN sequence in cp193g7 that is participating in stabilizing interactions and most likely present in a conformation that mimics the same sequence of residues in nonpermuted mCherry. In contrast, the C-terminal repeat is likely displaced from the folded structure and not participating in stabilizing interactions.
Identification of new circular permutation sites in β-strand 10
Starting from the cp193g7 template, we created all possible circular permutations with new termini between each pair of consecutive residues from positions 192 through 207 (15 in total) [Fig. 3(A)]. To minimize ambiguity, no overlapping sequences were included at the termini of the new circularly permuted variants. To increase the odds of success, we created a series of libraries in which the two residues immediately flanking the ‘break’ in the circularly permuted variants (i.e., the first and last residues of the protein) were both randomized to all possible amino acid residues. Libraries were expressed in E. coli and, for each permutation, thousands of colonies were screened for bright red fluorescence. Overall, we identified red fluorescent variants for 10 of the 15 permutation sites [Fig. 3(B)]. Of the five permutation sites that did not result in any red fluorescent variants, four of them were the sites in closest proximity to the chromophore (196/197, 197/198, 198/199, and 199/200). The fifth site was 193/194, which is only two residues removed from the presumed 191/192 permutation location in the optimized template cp193g7.
Figure 3.
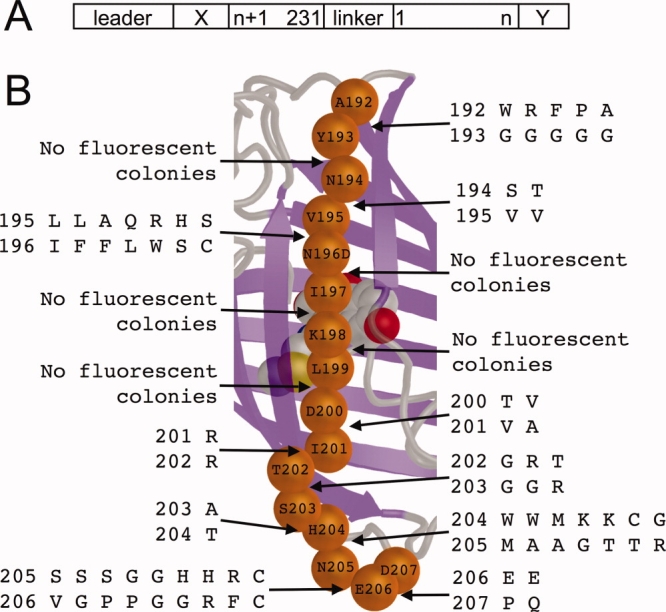
Identification of new circular permutation sites along β-strand 10 [refer to Fig. 1(C)] of mCherry. (A) Schematic view of the protein structure with a circular permutation site introduced between residues n and n + 1. ‘Leader’ is the purification and epitope tag sequence of the pBAD/His B vector (MGGSHHHHHHGMASMTGGQQMGRDLYDDDDKDPSSR); ‘X’ is the sequence SSGALGG; ‘linker’ is the amino acid sequence GGTGGS; and ‘Y’ is the sequence GGDQLTEE. (B) Overview of new permutations attempted in this work. The α-carbons of residues 192–207 are represented as orange spheres. For each consecutive pair of residues shown, a library of circularly permuted variants was created in which the chain was split at the peptide bond and both residues were randomized with the codon NNK (1024 members in each library). Pairs of residues that produced appreciable red fluorescence when expressed in E. coli are shown for each position in vertical alignment. For example, for n = 192 [refer to (A)] the brightest RFPs had the Y193G substitution at the N-terminal end and the A192W, R, F, P, or A substitution at the C-terminal end. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
For each permutation site that produced red fluorescent colonies, individual red fluorescent clones were cultured and the plasmid DNA isolated. DNA sequencing of the permuted RFP genes was used to determine the identity of the residues immediately flanking the permutation site. These results are shown in Figure 3(B). For each permutation site, the variants that exhibited the brightest fluorescence in colonies were purified and their quantum yields (φ) and extinction coefficients (ɛ) determined (Table II). This characterization revealed a general trend that positions closer to the loops at each end of β-strand 10 tended to be more tolerant of introduction of new termini. With respect to both in vitro and in vivo brightness, the four brightest variants had new termini either close to the N-terminal end of β-strand 10 (192/193) or close to the C-terminal end of β-strand 10 (203/204, 205/206, and 206/207). Variants with new termini closer to the middle of β-strand 10, and thus closer to the chromophore, tended to be substantially dimmer (i.e., 194/195, 195/196, 200/201, 201/202, and 202/203).
Table II.
Properties of Circularly Permuted mCherry Variants with New Termini Between Residues 192 and 207
| N-terminal residuea | C-terminal residuea | φ | ɛ (M−1 cm−1) | Fluorescent brightnessb (mM−1 cm−1) | Relative brightness in E. coli after 24 h at 37°C (%) | Relative brightness in E. coli after 72 h at 4°C (%) |
|---|---|---|---|---|---|---|
| Y193G | A192F | 0.23 | 20000 | 4.6 | 2 | 13 |
| V195V | N194S | 0.23 | 6300 | 1.4 | 3 | 8 |
| D196F | V195A | 0.15 | 4600 | 0.78 | 2 | 2 |
| I201V | D200T | 0.22 | 2600 | 0.57 | 2 | 6 |
| T202R | I201R | 0.24 | 3500 | 0.86 | 2 | 4 |
| S203G | T202G | 0.20 | 1500 | 0.3 | 2 | 4 |
| H204T | S203A | 0.21 | 54000 | 11 | 9 | 100 |
| N205M | H204W | 0.22 | 9300 | 2.0 | 2 | 9 |
| E206G | N205G | 0.21 | 47000 | 9.9 | 39 | 70 |
| D207Q | E206E | 0.20 | 19000 | 3.8 | 3 | 10 |
For each position, only the brightest variant was characterized.
Defined in Table I.
Discussion
Analysis of beneficial substitutions identified in cp193g7
A key assumption in our previous efforts to generate circularly permuted variants of mCherry was that the linking of the C- to N- termini was a relatively benign modification compared to the introduction of the new termini elsewhere in the protein.21 Accordingly, we had expected that the amino acid substitutions that would ‘rescue’ the properties of the cp193 variant would tend to cluster in close proximity to the new termini. This assumption was thought to be valid due to the seemingly generous length of polypeptide sequence joining the last and first residues to be resolved in the x-ray crystal structure of mCherry.24 Indeed, models of the cp193 circular permutation loop with reasonable conformations [i.e., as shown in Fig. 1(A)] reveal that the circularly permutation loop region extends well away from the β-can portion of the protein. The role of circular permutation linker length has previously been investigated for avGFP.25 The researchers found a minimal linker length of three residues was sufficient and longer linker did not provide further substantial advantageous or deleterious effects.
Our expectation that beneficial substitutions would occur near to the site of the new termini was, ostensibly, borne out in the first generation of directed evolution in which a substitution (N196D) at a residue just a few positions removed from the location of the new termini resulted in substantially brighter red fluorescence in colonies of E. coli [Fig. 1(C)]. The overall effect of this substitution is to place a negative charge into a patch on the surface dominated by basic amino acids including Lys198, Arg 216, and Arg220. In the x-ray crystal structure of mCherry,24 the basic guanidinium group of Arg 220 is just 3.3 Å from the amide group of the side chain of Asn196. We speculate that the carboxylate side chain of the Asp is participating in a salt-bridge interaction with the Arg 220 guanidinium group. It is interesting to note that Arg 220 is located within the extended loop that links together the 11th and 1st strands of the protein. This loop is composed of the original C- and N- terminal sequences fused by the GGTGGS linker sequence.
Of the five additional substitutions accumulated during the remaining rounds of the directed evolution process, three of them (E6V, I7F, and G219V) are also located on the extended loop that connects the 11th and 1st strands of the protein [Fig. 1(C)]. Notable among these is G219V that is immediately adjacent to Arg 220, the residue discussed in the previous paragraph. Yet another of the five substitutions, L83W, lies on the loop between the central helix and the 4th strand of the protein, and is effectively sandwiched between the circular permutation loop and the bulk of the β-can structure. Overall, it is apparent that there is a clustering of the beneficial mutations either on or near the introduced circular permutation linker that ties together the original C- and N-termini. These results suggest that, in contradiction to our original expectations, mutations in the vicinity of the circular permutation loop were of greater importance than mutations in the vicinity of the newly introduced termini for rescuing the fluorescence of cp193. We speculate that these mutations are helping to stabilize the circular permutation loop in a conformation that is more favorable for overall protein folding which in turn leads to a greater proportion of the proteins having a properly formed chromophore.
The last of the five substitutions that arose during the directed evolution process is F65L, which is structurally analogous to the F64L substitution that dramatically improves the folding efficiency of avGFP.26 Of all the substitutions in cp193g7, F65L is in the closest proximity to the chromophore and is the most likely candidate for altering the chromophore environment to produce the observed blue-shift in the absorbance and emission maxima.
Analysis of circular permutation sites identified in β-strand 10
It has previously been demonstrated that, when starting with template FPs with destabilizing loop insertions, directed evolution for efficient folding can produce FPs with extreme thermal stability.27 Furthermore, FPs optimized for folding efficiency are more tolerant of circular permutation.28,29 Similarly, we found that the optimized cp193g7 is generally more tolerant of circular permutation than the progenitor protein, mCherry. We suggest two possible explanations for the greater tolerance of cp193g7 to circular permutation. The first explanation is that, due to the additional rounds of optimization, cp193g7 has improved folding efficiency and overall stability relative to mCherry. The second explanation is that the original introduction of the loop that connected the C- and N-termini in circularly permuted mCherry had a destabilizing effect that had prevented the discovery of all but the most favorable variants. In cp193g7, point mutations have compensated for the destabilizing effect of the circular permutation linker and thus it is possible to generate additional permuted variants with new termini in less-favored sites.
We interpret the observation that the beneficial substitutions in cp193g7 tended to occur in close proximity to the circular permutation linker region as evidence in favor the second explanation. If these substitutions were generally beneficial to folding efficiency, as opposed to specifically compensating for the circular permutation linker, no particular bias would be expected in terms of their localization within the protein structure. Furthermore, as mCherry is the result of extensive optimization for folding efficiency,23 it is unlikely that the substitutions identified in this work (which did not require the screening of particularly large libraries), could have produced a protein with an overall improvement in folding efficiency. Indeed, cp193g7 has an ensemble ɛ that is only 58% of mCherry (Table I), indicating it is less efficient at folding into the exact tertiary structure required for chromophore formation. We have previously demonstrated that the inherent spectral properties of the fraction of FP molecules that form chromophores are essentially identical, and differences in ensemble ɛ are attributable to differences in chromophore formation efficiency.21
We attribute the high rate at which we successfully identified circularly permuted RFPs derived from cp193g7 to the fact that we generated libraries of all possible residues at the two positions immediately flanking the site of the new permutation. As shown in Figure 3(B), in no case did we identify the combination of both wild-type residues as being among the preferred combinations at the randomized positions. We did observe that wild-type residue V195 was strictly required when 195 was the N-terminus of the protein, but not when 195 was the C-terminus of the protein. Similarly, wild-type residue E206 was strictly required when 206 was the C-terminus of the protein, but not the N-terminus. Yet other circular permutation positions showed a strong requirement for one particular non-wild-type residue type at either the N-terminus (192/193 and 194/195) or both termini (201/202 and 203/204). For some permutation sites (192/193, 195/196, 204/205, and 205/206), five or more different combinations of flanking residues were observed, suggesting that these locations do not have stringent requirements regarding the identity of the flanking residues. For other permutation sites, no red fluorescent colonies were observed, indicating that no combination of substitutions at the flanking residues could rescue the protein folding and/or chromophore formation.
The cp193g7C variant has an in colony brightness (Table I) that is practically identical to the brightest 194/195 variants identified by library screening (Table II). However, the combination of N- and C-terminal residues in cp193g7C (Asn (N) at the C-terminus and Val (V) at the N-terminus), was not identified among the brightest variants in our library screening. A key difference between cp193g7C and the 194/195 variants is that cp193g7C had no additional residues appended at the C-terminus, whereas the 194/195 variants had an additional eight residues (GGDQLTEE) appended. To determine if the appended residues had an adverse effect on the brightness of cp193g7C, we constructed a 194/195 variant with N- and C-terminal residues that corresponded to cp193g7C. We found that that this variant had only 70% of the brightness of the 194/195 variant (Table II) that had been determined to be the brightest within the library.
In the absence of further structural characterization of these circular permutation variants, we are unable to rationalize these amino acid preferences. However, these results do suggest that other efforts to circularly permute proteins could benefit from the creation and screening of libraries, in which the first and last residues of the protein are randomized.
One important application of circularly permuted FPs is in the optimization of FRET-based biosensors, where swapping a donor and/or acceptor FP with a variety of circularly permuted variants can often produce a more favorable FRET-response due to the fortuitous changes of distance or orientation between the chromophores.30 Considering only the brightest circularly permuted variants described here, we now have two new mCherry topologies (i.e., cp193g7 and S203A/H204T) with spatially distinct termini and relatively high values of ɛ and could likely be useful for optimization of FRET-based biosensors based on donor lifetime or intensity quench. Two drawbacks would be their relatively slow maturation times and their low φ values that would, like with mCherry itself, lead to poor sensitized emission. Directed evolution to improve the brightness of these and other variants is ongoing in our lab.
In conclusion, we engineered a brightly fluorescent circularly permuted variant of mCherry with new termini located at the N-terminal end of β-strand 10. With this optimized variant in hand, we created a series of circularly permuted variants with new termini along the length of β-strand 10. Of those positions within the strand that tolerated the introduction of the new termini, most resulted in variants that were quite dim relative to mCherry. However, one variant with new termini between residues 203 and 204 did retain 67% of the inherent fluorescent brightness of mCherry itself. Our next goal is to fuse calmodulin and M13 to the N- and C-termini of each of these variants in an attempt to create red fluorescent Ca2+ biosensors analogous to the existing Pericam12 and GCaMP13 type biosensors.
Materials and Methods
General methods and materials
PCR products and products of restriction digestion were routinely purified using agarose gel electrophoresis and the GenCatch kit method (Epoch Biolabs). Plasmids were purified using GeneJet plasmid miniprep kit (Fermentas) according to the manufacturer's protocol. Restriction enzymes and Taq DNA polymerase were purchased from New England Biolabs and used with the supplied buffers. Ligations were performed using T4 DNA ligase (Invitrogen) according to the manufacturer's instructions. All FP genes were cloned into the Xho1/EcoR1 sites of pBAD/His B (Invitrogen) for expression in E.coli. Synthetic DNA oligonucleotides were purchased from Integrated DNA Technologies. DNA was sequenced by dye terminator cycle sequencing using the DYEnamic ET kit (Amersham Biosciences). Sequencing reactions were analyzed at the University of Alberta Molecular Biology Service Unit. Protein samples for in vitro spectral characterization experiments were first dialyzed into 50 mM Tris, pH 7.5.
Construction of cp193 libraries by error prone PCR
Error prone PCR was performed using primers that annealed to the pBAD/His B plasmid outside of the cp193 gene sequence. Error prone PCR reactions were done using Taq polymerase (New England Biolabs) and with a deficit of each one of the four dNTPs as previously reported.31 DNA was purified using argarose gel electrophoresis, doubly digested using the restriction enzymes XhoI and HindIII (New England Biolabs), and ligated into appropriately digested pBAD/His B.
Construction of cp193g7N and cp193g7C
The two variants cp193g7N and cp193g7C with the AYN amino acid sequence at the N or C-termini, respectively, were constructed with cp193g7 as template. The primers used for cp193g7N deleted the AYN sequence at the C-terminus, while the cp193g7C primers deleted AYN at the N-terminus. Similar to the β10 variants, XhoI and EcoRI restriction sites were added to the 5′ and 3′ ends of the DNA constructs, respectively. The two variants were ligated into pBAD/His B after digestion with the appropriate restriction enzymes. The correct construct was confirmed with DNA sequencing.
Construction of libraries of FP variants with termini in β-strand 10
Circularly permuted variants of mCherry were constructed with new termini at each peptide bond between positions 192 and 207. The starting template DNA was the cp193g7 construct that resulted from the directed evolution of cp193. Primers were designed such that the first and last residues of each circularly permuted variant were encoded by a degenerate codon (NNK) either preceded by (at the N-terminus), or followed by (at the C-terminus), a GG linker. Accordingly, a library of 1024 genetically distinct variants was generated for each circular permutation. Furthermore, gene sequence corresponding to the last five residues of M13 (SSGAL) were appended before the GG linker at the N-terminus and the first six residues of CaM (DQLTEE) was appended immediately after the GG sequence at the C-terminus. XhoI and EcoRI restriction sites were introduced at the 5′ and 3′ ends of the DNA construct, respectively. The PCR reactions were carried out in series to limit the length of the primers. That is, the cp193 variant served as the template for cp194, and cp194 was the template for cp195, and so on. After digestion with XhoI and EcoRI the gene libraries were ligated into pBAD/His B that had been digested with the same two restriction enzymes.
Plasmid library screening
Ligation products were introduced into E. coli DH10B cells via electroporation and then plated out on LB/agar plates supplemented with 0.01% ampicillin and 0.02% arabinose and then left to incubate at 37°C overnight. Colonies were screen for red fluorescence using a previously described colony-imaging system.32 Briefly, plates were illuminated with 535–550 nm excitation light and emission was collected by a monochromatic CCD camera and a 630–660 nm emission filter. If no red colonies were observed, the plates were allowed to sit on the bench top or 4°C fridge for several days. Colonies that exhibited red fluorescence were picked and used to inoculate 5 mL volumes of LB media supplemented with ampicillin (amp). After overnight growth, 100 μL of the culture was pipetted into a microwell plate. The fluorescence emission of each well was measured over 560 to 700 nm using at an excitation wavelength of 540 ± 5 nm in a Safire II microplate reader (Tecan). The plasmid DNA for the most brightly fluorescent variants was purified and sequenced as described above. For most permutation positions there were several red variants found. To determine the brightest, streak plates were made of each variant and the appearance of red fluorescence was monitored using the same imaging system described above. Only the brightest variant at each position was subsequently purified and characterized.
Protein expression and purification
For all protein production, E. coli strain DH10B was first transformed with the gene of interest in the pBAD/His B vector by electroporation. A single colony was used to inoculate 5 ml LB/amp cultured overnight (37°C, 250 rpm) before being diluted into 500 mL of LB/amp supplemented with arabinose (0.02%). Again the culture was shaken overnight before harvesting the cells (37°C, 250 rpm). For the slowly maturing constructs, the temperature was decreased to 28°C once the culture reached an OD of 0.6, and protein production was induced with 0.2% arabinose and shaken overnight at 250 rpm. The bacteria were pelleted by centrifugation and resuspended in PBS before being lysed by a cell disrupter (Constant Systems). The lysates were then centrifuged at 14,000 g for 45 min, and the proteins were purified from the supernatants using Ni-NTA chromatography (Amersham). Purified proteins were then dialyzed into 10 mM Tris-HCl, pH 7.4. Proteins were stored at 4°C spectral characterization.
Protein characterization
Absorbance spectra were recorded on a DU-800 UV-visible spectrophotometer (Beckman). Fluorescence emission and excitation spectra were acquired on a Quantamaster spectrofluorometer (Photon Technology International). Determination of quantum yields (φ) for the circularly permuted mCherry variants utilized mCherry as the reference standard.23 Extinction coefficients (ɛ) were measured by UV-visible absorbance spectroscopy on purified proteins. The ensemble ɛ of the proteins was determined by comparing the intensity of the 587 nm absorbance peak for each protein to that of a solution of mCherry with matched absorbance at 280 nm. The ɛ was calculated by multiplying the ratio of absorbance intensities at 587 nm by the ɛ of mCherry (72 000 M cm−1).23
Molecular modeling and figures
Molecular models of mCherry24 variants with complete termini, insertion, or circular permutation [as shown in Fig. 1(A)] were assembled using the Modloop server33 and the loop modeling application of Rosetta 3.0.34 MacPyMOL35 was used for molecular rendering and structure viewing.
Acknowledgments
The authors thank the University of Alberta and the Canadian Foundation for Innovation for infrastructure support. R. E. Campbell holds a Canada Research Chair in Bioanalytical Chemistry.
Glossary
Abbreviations
- FP
fluorescent protein
- avGFP
Aequorea victoria green FP
- RFP
red FP
- i
insertion
- cp
circularly permuted
- g
generation
- m
monomeric.
References
- 1.Campbell RE. Fluorescent proteins. Scholarpedia J. 2008;3:5410. [Google Scholar]
- 2.Yang F, Moss LG, Phillips GNJ. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 3.VanEngelenburg SB, Palmer AE. Fluorescent biosensors of protein function. Curr Opin Chem Biol. 2008;12:60–65. doi: 10.1016/j.cbpa.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Campbell RE. Fluorescent-protein-based biosensors: modulation of energy transfer as a design principle. Anal Chem. 2009;81:5972–5979. doi: 10.1021/ac802613w. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DE, Ai HW, Wong P, Young JD, Campbell RE, Casey JR. Red fluorescent protein pH biosensor to detect concentrative nucleoside transport. J Biol Chem. 2009;284:20499–20511. doi: 10.1074/jbc.M109.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jayaraman S, Haggie P, Wachter RM, Remington SJ, Verkman AS. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J Biol Chem. 2000;275:6047–6050. doi: 10.1074/jbc.275.9.6047. [DOI] [PubMed] [Google Scholar]
- 8.Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou J, Hofer AM, Lurtz MM, Gadda G, Ellis AL, Chen N, Huang Y, Holder A, Ye Y, Louis CF, Welshhans K, Rehder V, Yang JJ. Developing sensors for real-time measurement of high Ca2+ concentrations. Biochemistry. 2007;46:12275–12288. doi: 10.1021/bi7007307. [DOI] [PubMed] [Google Scholar]
- 10.Chen N, Zou J, Wang S, Ye Y, Huang Y, Gadda G, Yang JJ. Designing protease sensors for real-time imaging of trypsin activation in pancreatic cancer cells. Biochemistry. 2009;48:3519–3526. doi: 10.1021/bi802289v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topell S, Hennecke J, Glockshuber R. Circularly permuted variants of the green fluorescent protein. FEBS Lett. 1999;457:283–289. doi: 10.1016/s0014-5793(99)01044-3. [DOI] [PubMed] [Google Scholar]
- 12.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci USA. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- 14.Souslova EA, Belousov VV, Lock JG, Strömblad S, Kasparov S, Bolshakov AP, Pinelis VG, Labas YA, Lukyanov S, Mayr LM, Chudakov DM. Single fluorescent protein-based Ca2+ sensors with increased dynamic range. BMC Biotechnol. 2007;7:37. doi: 10.1186/1472-6750-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Shui B, Kotlikoff MI, Sondermann H. Structural basis for calcium sensing by GCaMP2. Structure. 2008;16:1817–1827. doi: 10.1016/j.str.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akerboom J, Rivera JD, Guilbe MM, Malavé EC, Hernandez HH, Tian L, Hires SA, Marvin JS, Looger LL, Schreiter ER. Crystal structures of the GCaMP calcium sensor reveal the mechanism of fluorescence signal change and aid rational design. J Biol Chem. 2009;284:6455–6464. doi: 10.1074/jbc.M807657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 18.Gautam SG, Perron A, Mutoh H, Knöpfel T. Exploration of fluorescent protein voltage probes based on circularly permuted fluorescent proteins. Front Neuroengineering. 2009;2:14. doi: 10.3389/neuro.16.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlson HJ, Campbell RE. Genetically encoded FRET-based biosensors for multiparameter fluorescence imaging. Curr Opin Biotechnol. 2009;20:19–27. doi: 10.1016/j.copbio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Ai H, Hazelwood KL, Davidson MW, Campbell RE. Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nat Methods. 2008;5:401–403. doi: 10.1038/nmeth.1207. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Sierra AM, Ai HW, Campbell RE. Identification of sites within a monomeric red fluorescent protein that tolerate peptide insertion and testing of corresponding circular permutations. Photochem Photobiol. 2008;84:111–119. doi: 10.1111/j.1751-1097.2007.00206.x. [DOI] [PubMed] [Google Scholar]
- 22.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 23.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 24.Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel chromophores and buried charges control color in mFruits. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- 25.Akemann W, Raj CD, Knopfel T. Functional characterization of permuted enhanced green fluorescent proteins comprising varying linker peptides. Photochem Photobiol. 2001;74:356–363. doi: 10.1562/0031-8655(2001)074<0356:fcopeg>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 26.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 27.Kiss C, Temirov J, Chasteen L, Waldo GS, Bradbury AR. Directed evolution of an extremely stable fluorescent protein. Protein Eng Des Sel. 2009;22:313–323. doi: 10.1093/protein/gzp006. [DOI] [PubMed] [Google Scholar]
- 28.Zapata-Hommer O, Griesbeck O. Efficiently folding and circularly permuted variants of the Sapphire mutant of GFP. BMC Biotechnol. 2003;3:5. doi: 10.1186/1472-6750-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 30.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Z, Campbell RE. Assessing the structural stability of designed beta-hairpin peptides in the cytoplasm of live cells. ChemBioChem. 2006;7:1147–1150. doi: 10.1002/cbic.200500540. [DOI] [PubMed] [Google Scholar]
- 33.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 34.Rohl CA, Strauss CE, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL. 2008. Available at http://www.pymol.org The PyMOL Molecular Graphics System. [accessed May 10, 2010]