

Abstract
We report efficient syntheses of axially chiral biaryl amides in yields ranging from 80–92%, and with enantioselectivity in the range 88–94% ee employing an asymmetric Suzuki-Miyaura process with Pd(OAc)2 and KenPhos as ligand. These studies demonstrate that electron-rich and electron-deficient o-halobenzamides can be efficiently coupled with 2-methyl-1-naphthylboronic acid and 2-ethoxy-1-naphthylboronic acid. The yields and selectivities of the reactions are independent of the nature of halogen substituent on the benzamide coupling partner. Our investigations demonstrate that axially chiral heterocyclic and biphenyl compounds can also be synthesized with this methodology. We also report computational studies used to determine the origin of stereoselectivity during the selectivity-determining reductive elimination step of the related coupling of tolyl boronic acid with naphthylphosphonate bromide that was reported in a previous publication (J. Am. Chem. Soc. 2000, 122, 12051–12052). These studies indicate that the stereoselectivity arises from a combination of weak -(C)H•••O interactions as well as steric interactions between the tolyl and naphthylphosphonate addends in the transition state for C-C coupling.
Introduction
Many reports of synthetic protocols that provide access to biaryl compounds with a high degree of axial stereocontrol have appeared in recent years.1–6 The axially chiral biaryl structural motif is a prominent feature of a number of biologically active natural products such as vancomycin7–9 and korupensamine A10–12 (Figure 1). Biaryls that exhibit axial chirality appear in ligands such as BINOL13,14 and BINAP15,16 (Figure 1) have had a profound impact on asymmetric synthesis17,18 and in organocatalysis.19,20 There has also been interest in exploiting this feature in nanoscience applications such as molecular switches.21
Figure 1.

Ligands and biologically active natural products containing the axially chiral biaryl structural motif.
A number of synthetic approaches to this motif have been explored including metal-mediated diastereoselective reactions of substrates with chiral tethering units,22–24 auxiliaries25–28 or leaving groups.29–32 Enantioselective metal-catalyzed reactions including the oxidative dimerization of phenols4,33–37 and metal-catalyzed [2+2+2] formal cycloadditions have also been reported.5,38–40 Palladium-catalyzed reactions involving Kumada, Negishi, Hiyama and Suzuki-Miyaura cross-coupling have emerged as one of the most powerful and general means of constructing biaryl bonds.41–44 There has been growing interest in implementing enantioselective variants of these reactions in order to access enantiopure, axially chiral biaryls. Generally, in order for a biaryl compound to exhibit atropisomerism there must be at least three ortho substituents about the biaryl axis,45 this high degree of steric hindrance presents a serious challenge to many cross-coupling transformations.
Hayashi reported the first use of a chiral ligand in redox-neutral asymmetric biaryl coupling reactions with a transition metal catalyst. It was demonstrated that axially chiral binaphthyl compounds could be formed via the Ni-catalyzed Kumada cross-coupling reaction between aryl bromides and aryl Grignard reagents in the presence of a chiral ferrocenylphosphine ligand.46,47 Espinet later demonstrated that Pd-catalyzed Negishi reactions could be used to synthesize axially chiral binaphthyl compounds in the presence of a chiral ferrocene ligand.48,49
The Suzuki-Miyaura cross-coupling of aryl boronic acids with aryl halides offers a number of advantages over Negishi and Kumada cross-coupling methods to form biaryl bonds, especially in terms of functional group tolerance and the stability and lack of air sensitivity of the boronic acid coupling partner.50,51 Uemura,52–54 Nelson,55 Colobert56 and Lipshutz57 demonstrated that axially chiral biaryl products are formed in high yields and excellent selectivities in Pd-catalyzed Suzuki-Miyaura reactions with aryl halides bearing chiral auxiliaries. Later Nicolaou employed chiral phosphines to modulate the selectivity of a diastereoselective Suzuki-Miyaura reaction used in the synthesis of vancomycin.9
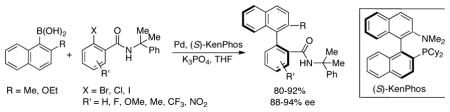
Our group reported one of the earliest procedures for the preparation of functionalized biaryl compounds via the asymmetric Suzuki-Miyaura coupling reaction.58 A number of enantioenriched biaryl phosphonates could be synthesized in yields of 74% or greater and enantiomeric excesses up to 92% with the use of catalytic amounts of a Pd source and the (S)-KenPhos ligand (Scheme 1). At around the same time, Cammidge disclosed the Pd-catalyzed synthesis of axially chiral binaphthalene compounds with the use of a chiral ferrocene-derived monophosphine ligand.59 Since then, a variety of ligands48,60–66 have been examined for the asymmetric Pd-catalyzed Suzuki coupling reaction. Recent noteworthy examples have separately shown that a chiral bis-hydrazone ligand67 and a chiral resin-supported phosphine ligand68 can effectively catalyze the synthesis of axially chiral biaryl compounds with excellent selectivities in some cases.
Scheme 1.

Synthesis of enantioenriched biaryl phosphonates by Suzuki-Miyaura coupling
Despite considerable effort in this area, a number of limitations remain. The coupling of functionalized substrates remains challenging and there are no reports of heteroaryl substrates being employed in these reactions. These issues are important to consider if this method is to find application in the synthesis of natural products or pharmaceuticals. Furthermore, previous studies have largely been limited to the synthesis of chiral binaphthyls; there are no reports of the synthesis of chiral biphenyls by the Suzuki-Miyaura reaction. Previous studies have also been largely empirical and there have been no detailed efforts to rationalize the observed selectivity in these reactions.
Herein we report our efforts to expand the substrate scope of our previously reported method. Further, we describe combined experimental and computational studies that attempt to rationalize the observed selectivities. DFT calculations indicate that the combination of hydrogen-bonding interactions and steric interactions between the addends and the ligand are responsible for stereoinduction during the reductive elimination step of these reactions.
Results and Discussion
Synthesis of Axially Chiral Biaryl Compounds
In our previous report on asymmetric Suzuki-Miyaura coupling with the Pd(KenPhos) system,58 we found that aryl halides bearing an ortho phosphonate group were the most efficient substrates (in terms of ee of the product) for the reaction. We have attempted to gain some insight into the source of enantioselectivity in these reactions by examining X-ray crystal structures for key compounds in a prototypical reaction from that study, but unfortunately, we could not obtain crystals suitable for X-ray crystallographic studies. However, we were able to study a closely related reaction by reacting in situ-generated Pd(KenPhos) with (1-chloro-2-naphthyl)diisopropylphosphine oxide to generate an oxidative addition complex which was subsequently treated with o-tolylboronic acid to afford the axially chiral biaryl phosphine oxide in 90% ee (Figure 2). Single crystals of the major product A were obtained after recrystallization from hexane. X-ray diffraction studies were used to determine the absolute configuration (S stereochemistry) of the product (Figure 3b).
Figure 2.

The reaction of Pd(KenPhos) with (1-chloro-2-naphthyl)diisopropylphosphine oxide and subsequent reaction of the oxidative addition complex with o-tolylboronic acid.
Figure 3.

Crystallographic structures for the (a) oxidative addition complex of Pd(KenPhos) with (1-chloro-2-naphthyl)diisopropylphosphine oxide. (Hydrogen atoms have been omitted and atoms belonging to the KenPhos ligand have been faded for clarity.) (b) coupled biaryl product from the reaction of (1-chloro-2-naphthyl)diisopropylphosphine oxide and o-tolylboronic acid.
We were able to obtain an X-ray crystal structure for the oxidative addition complex B which reveals a putative interaction between the palladium (II) atom and the oxygen atom (Pd-O bond distance = 2.77 Å) belonging to the phosphine oxide (Figure 3a). We speculated that this feature could be responsible for the induction of stereoselectivity by operating as an “anchor” for the naphthylphosphonate addend during the reductive elimination process. We thus began to examine other functional groups bearing coordinating atoms that could serve as a source of stereoinduction in these types of reactions.
We initiated these investigations by examining reactions of 2-methyl-1-naphthylboronic acid with aryl halide coupling partners incorporating ester, phosphonate and phosphine oxide substituents in the ortho position. Products arising from ester-functionalized aryl halides were obtained in greater than 80% yield but showed only moderate ee values (40–45%) (Scheme 2a, 2b). All attempts to improve the enantioselectivity of these reactions with the use of alternative Pd sources, solvents, bases or ligands were unsuccessful. In contrast, reactions involving aryl halide substrates incorporating phosphonate and phosphine oxide substituents, diphenyl-(1-bromo-2-naphthyl) phosphonate and (1-chloro-2-naphthyl)diisopropylphosphine oxide, respectively, afforded desired products 4 and 5 in yields similar to those found with the aryl esters (>76%), but in 84–85% ee (Scheme 2c, 2d). The latter results are comparable with the use of diethyl-(1-bromo-2-naphthyl)phosphonate as the aryl halide substrate in the coupling with toluene boronic acid, which provides coupled product in 87% ee.
Scheme 2.

Synthesis of chiral biaryl compounds containing esters and phosphonate
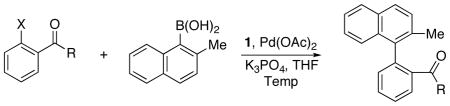
Encouraged by these results, we turned our attention to aryl bromides possessing amide groups ortho to the halide substituent, types of substrates that had not been previously been utilized in asymmetric Suzuki-Miyaura reactions. We hypothesized that the Pd•••O interaction observed with in the X-ray structure of B might exist with amides as well. Aryl bromides containing a variety of ortho amide substituents were subjected to Suzuki-Miyaura coupling with 2-methyl-1-naphthylboronic acid under reaction conditions similar to those described previously for ortho halo arylphosphonates (Table 1). The enantioselectivity increased as the steric demand of the amide unit did. For example, when aryl bromides with the more sterically demanding diisopropylamido substituent (9, 82% ee) was used instead of a diethylamido (6, 75% ee) substituent (cf. Table 1, entries d and a).
Table 1.
Effect of functional group located ortho to the halide substituent on the enantioselectivity.
 | ||||
|---|---|---|---|---|
| R = NEt2, X = Br (6) | R = N(Me)tBu, X = Br (10) | |||
| R = NEt2, X = I (7) | R = N(OMe)Me, X = Br (11) | |||
| R = NEt2, X = OTf (8) | R = N(H)tBu, X = Br (12) | |||
| R = NiPr2, X = Br (9) | R = N(H)C(Me)2Ph, X = Br (13) | |||
| Entry | ArX | Temp (°C) | Yield %b | ee %c |
| a | 6 | rt | 80 | 75 |
| b | 7 | rt | 78 | 73 |
| c | 8 | rt | 85 | 71 |
| d | 9 | rt | 76 | 82 |
| e | 10 | rt | 82 | 75 |
| f | 11 | rt | 75 | 80 |
| g | 12 | 50 | 68 | 87 |
| h | 13 | 50 | 81 | 93 |
Reaction conditions: 1.0 equiv of aryl halide, 2.0 equiv of boronic acid, 5 mol% Pd, 6 mol% (S)-1, 3 equiv of K3PO4, THF (2.5–3 mL/mmol of halide), 24–40 h.
Isolated yield.
The ee values were determined by chiral HPLC on a Chiralcel OD-H or AD-H.
The reaction was also effective when an aryl triflate was used as substrate (Table 1, entry c) in the first demonstration of this type of substrate in asymmetric Suzuki-Miyaura coupling reactions. An aryl bromide bearing an ortho Weinreb amide substituent (11) formed product with 80% ee (Table 1, entry f), a useful result due to the ease of further synthetic manipulation of this functional group. Significantly, changing the nitrogen substituent to a monoalkylamido group resulted in reactions with even greater degrees of selectivity - the product obtained from the coupling of 2-bromo-N-(tert-butyl)benzamide (12) was formed in 87% ee (Table 1, entry g). The enantioselectivity could be further increased to 93% when the amide was protected with the cumyl group69 as shown in the reaction involving 2-bromo-N-(2-phenyl-2-propyl)benzamide (13) (Table 1, entry h).
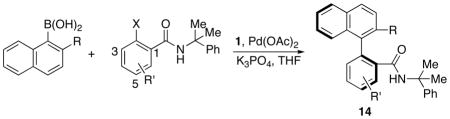
Due to the ease of removal of the cumyl protecting group to reveal the free primary amide for further synthetic manipulation and the high selectivity of these reactions (vide infra), we elected to further explore the scope of reactions involving these types of substrates. Table 2 summarizes results obtained upon coupling of cumyl amide-substituted aryl halides with naphthylboronic acids. Products are obtained in good to excellent yields and with ee values typically being 88–94%.
Table 2.
Asymmetric synthesis of biaryls by enantioselective Suzuki-Miyaura coupling.a
 | |||||
|---|---|---|---|---|---|
| Entry | R | R′ | X | Yield %b | ee %c |
| a | Me | H | Cl | 87 | 93(R) |
| b | Me | H | Br | 83 | 93(R) |
| c | Me | H | I | 81 | 94(R) |
| d | OEt | H | Cl | 81 | 91(S) |
| e | OEt | H | Br | 80 | 91(S) |
| f | Me | 4-F | Br | 83 | 93(R) |
| g | OEt | 4-F | Br | 81 | 90(S) |
| h | Me | 4-Me | Br | 82 | 94(R) |
| i | OEt | 4-Me | Br | 81 | 92(S) |
| j | Me | 4-NO2 | Br | 92 | 88(R) |
| k | Me | 5-OMe | Br | 83 | 94(R) |
| l | OEt | 5-OMe | Br | 84 | 91(S) |
| m | Me | 5-CF3 | Br | 87 | 89(R) |
| n | Me | 5-F | Br | 85 | 92(R) |
| o | Me | 6-Me | Br | 80 | 48(R) |
| p | Me | 6-F | Br | 46 | 90(R) |
Reaction conditions: 1.0 equiv of aryl halide, 1.5–2.0 equiv of boronic acid, (S)-1/Pd(OAc)2 = 1.2, 3 equiv of K3PO4, THF (2.5–3 mL/mmol of halide).
Isolated yield (average of two runs).
The ee values were determined by chiral HPLC on a Chiralcel OD-H or AD-H.
The absolute configuration of 14h was determined by X-ray crystallography. The configurations of other biaryl compounds were assigned by analogy.
As shown, both the yields and enantioselectivities of biaryl compounds are similar when either 2-ethoxy-1-naphthylboronic acid (Table 2, entries d, e, g, i, l) or 2-methyl-1-naphthylboronic acid (Table 2, entries a, b, c, f, h, j, k, m, n) are used as the boronic acid coupling partners. The yields and enantioselectivities of these reactions are only slightly affected by the use of aryl chloride or aryl iodide substrates instead of aryl bromides (Table 2. entries a, b, c); these range from 81–87% and 93–94%, respectively.
Substitution at the 4 or 5 position of the aryl bromide with electron-donating or electron-withdrawing groups had little effect on the yield or ee values (Table 2, entries b, e-n). Substitution at the 6 position with fluorine also gave a selective reaction (Table 2, entry p), however, a methyl group in this position resulted in a significant drop in selectivity (Table 2, entry o). It is tempting to speculate that this large substituent disrupts the Pd•••O interaction that we hypothesize is key to observing high levels of enantioselectivity. The complementary mode of coupling is also successful; α-phenylnaphthyl amides may be synthesized by reaction of arylboronic acids with α-bromonaphthyl amides (Scheme 3a), the biaryl product 15 is formed in 80% yield and 88% ee which is comparable to the yields and selectivities found for reactions of naphthylboronic acids with phenyl amides shown in Table 2.
Scheme 3.

Synthesis of axially chiral aromatic compounds.
Notably, an axially chiral biaryl pyridine derivative could be synthesized in good yield and enantioselectivity if the reaction temperature were increased to 80 °C (Scheme 3b). To the best of our knowledge, this represents the first synthesis of enantioenriched heterocyclic biaryl compounds by asymmetric Suzuki-Miyaura coupling. Moreover, axially chiral biphenyl amides can be synthesized in good yields and enantioselectivities (Scheme 3c, 3d). For example, at room temperature and at 5 mol% of Pd(OAc)2, biphenyl amides 17 and 18 could be obtained in greater than 70% yield and 80% ee through the coupling of 2-cumyl-6-methylphenylbromide with 2-ethylphenylboronic acid and 2-methylphenylboronic acid, respectively. These are the first reported example of the enantioselective synthesis of a biphenyl compound using Pd-catalyzed asymmetric Suzuki-Miyaura coupling.
The cumyl protecting group was readily removed to reveal the corresponding primary amide via a known procedure involving treatment of biaryl 14a (eg. Table 2, entry a) with neat TFA at ambient temperature (Figure 4).69 The deprotected naphthamide 19a was obtained in 95% yield and 93% ee; recrystallization increased the ee of the isolated compound to 99%.
Figure 4.

Acid-catalyzed deprotection of a cumylamide.
Computational Studies on the Origin of Enantioselectivities in Suzuki-Miyaura Reactions
Despite increasing interest in the enantioselective Pd-catalyzed Suzuki-Miyaura coupling reaction with a range of ligands, computational studies to understand the origin of the selectivity in these processes are limited to a single molecular mechanics study by Baudoin.70 We have performed computational studies to understand why aryl halides bearing an ortho functional group with a potentially coordinating oxygen atom on either a phosphonate or amide substituent provide the highest ee values in the reaction and to understand how enantioselectivity is induced by the ligand.
Computational Methodology
All calculations were carried out with the Gausssian0371 suite of computational programs. The B3LYP72–74 density functional method was used to optimize transition state geometries and obtain free energies. The 6–31G(d) basis set was employed for the C, H, N, and O atoms while the LANL2DZ effective core potentials of Hay and Wadt with double-ζ basis sets were employed for the Pd and P atoms.
Transition State Geometries and Energies
As a starting point for our computational studies, we began with information from X-ray crystallographic data (described above and in the supporting information) that would be employed in the optimization of transition state geometries. Because we could obtain more meaningful X-ray crystallographic data from the reaction of (1-chloro-2-naphthyl)diisopropylphosphine oxide and Pd(KenPhos) (cf. Figures 2 and 3) than from reactions involving amides, we proceeded to carry out detailed computational investigations of the closely related reaction of dimethyl-(1-bromo-2-naphthyl)phosphonate with o-tolylboronic acid catalyzed by the Pd(KenPhos) complex (Figure 5).75 The biaryl phosphonate is produced in 91% yield and 84% ee at a temperature of 60 °C.58
Figure 5.

The Pd(KenPhos)-catalyzed reaction of dimethyl-(1-bromo-2-naphthyl)phosphonate with o-tolylboronic acid.
Although the X-ray crystal structure of the oxidative addition complex formed from the reaction of Pd(KenPhos) with (1-chloro-2-naphthyl)diisopropylphosphine oxide revealed a single geometrical isomer (cf. Figure 3), we hypothesized that other geometrical isomers can be formed after transmetalation. Indeed, computational results presented in the Supporting Information indicate that the bound tolyl and naphthylphosphonate addends can adopt a number of geometrical configurations and conformations that can all lead to distinct transition structures for reductive elimination. One must assume that these intermediates are capable of interconversion in order for all of these transition states to be accessed. In accordance with this, we have proposed a mechanism for the interconversion of the intermediates formed after transmetalation in the Supporting Information.
Scheme 4 shows the structure for a prototypical transition state for the reaction of interest involving the reductive elimination of a coupled biaryl product from a Pd(KenPhos) complex; the tolyl and naphthylphosphonate addends replace the chlorine atom and phosphine oxide addends, respectively, shown in Figure 3b. The naphthylphosphonate and tolyl addends in the transition states can occupy either of the coordination sites on palladium. In addition, the methyl and phosphonate substituents on the respective tolyl and naphthylphosphonate addends can be either above or below the plane defined by palladium and the bound carbon atoms belonging to the addends. Finally, the aromatic rings of the naphthylphosphonate and tolyl addends in the transition structures are not aligned due to the inclination of the tolyl addend during reductive elimination. Therefore, the methyl group on the tolyl addend can be inclined away from, or towards, the palladium atom. Consequently, a total of sixteen transition structures can be generated from these orientations.
Scheme 4.

Possible orientations of addends in transition state geometries for reductive elimination. (Some atoms have been omitted and atoms belonging to the KenPhos ligand have been faded for clarity.)
We were able to optimize fourteen of the sixteen possible transition structures for the reductive elimination of 20 from the Pd(KenPhos) complex (Figure 6, TS1-TS14). Transition state pairs TS1/TS2, TS3/TS4, TS5/TS6, TS7/TS8, TS9/TS10 and TS11/TS12 which lead to products with opposite stereochemistries, were formed by twisting the tolyl addend in both directions (see below for further details). Transition state analogues of TS13 and TS14 with twisted tolyl addends could not be located. The lowest energy transition structure is TS1 in which the phosphonate substituent belonging to the naphthylphosphonate addend and the methyl group belonging to the tolyl group are both above the plane of palladium and the attached ligands. TS3, TS5 and TS8 are 1.8, 0.9 and 1.9 kcal/mol higher in energy than TS1, respectively; all other transition structures are at least 2 kcal/mol higher in energy than TS1.
Figure 6.


Transition structures and relative free energies of activation in kcal/mol for reductive elimination of biaryl phosphines from Pd(KenPhos) complexes formed after transmetalation. Atoms participating in H-bonding interactions are highlighted in green. The stereochemistry of the product formed after reductive elimination is denoted in brackets. Relevant O-H bond distances in Å are shown on the pictures; H-O-P bond angles in degrees are shown in parentheses. Chemdraw representations of the transition structures are shown below the pictures; the KenPhos ligand in these drawings has been replaced by the symbol P and the naphthylphosphonate addend has been truncated for clarity. Transition structures and relative free energies of activation in kcal/mol for reductive elimination of biaryl phosphines from Pd(KenPhos) complexes formed after transmetalation. Atoms participating in H-bonding interactions are highlighted in green. The stereochemistry of the product formed after reductive elimination is denoted in brackets. Relevant O-H bond distances in Å are shown on the pictures; H-O-P bond angles in degrees are shown in parentheses. Chemdraw representations of the transition structures are shown below the pictures; the KenPhos ligand in these drawings has been replaced by the symbol P and the naphthylphosphonate addend has been truncated for clarity.
The stereochemistry of the product formed in the reaction results directly from the geometry of the addends in the transition state for reductive elimination. We were able to predict the stereochemistry of the biaryl product formed from each transition structure by taking advantage of the observation that the tolyl addends in each transition state can be inclined in one of two ways with respect to the naphthylphosphonate addend. The direction in which the tolyl addend twists defines the stereochemistry of the product (this was confirmed with IRC calculations, see below for details). From this analysis we determined that TS1, TS4, TS6, TS7, TS10, TS11 and TS13 reductively eliminate to form the Pd(KenPhos) complex and stereochemically equivalent biaryl products with S stereochemistry. Eyring distribution analysis based on these results predict that the biaryl product is formed in 50% ee in favor of S stereochemistry at 25 °C; significantly, this is the configuration found by X-ray crystallography for the major product formed in the closely related Pd(KenPhos) catalyzed reaction of (1-chloro-2-naphthyl)diisopropylphosphine oxide with o-tolylboronic acid (cf. Figures 2 and 3) and the predicted selectivity is in reasonable agreement with the selectivity found by experiment of 84% ee at 60 °C.76
Hydrogen-Bonding Interactions in Transition States
Notably, the naphthylphosphonate addend is cis to the phosphine ligand in the four most stable transition structures predicted by our calculations (Figure 6, TS1 which gives rise to a product with S stereochemistry, and TS3, TS5 and TS8 which give rise to products with R stereochemistry). This geometric preference is presumably due to the trans influence and could be related to the fact that the naphthylphosphonate addend prefers to be cis to the phosphine ligand in the oxidative addition complex formed from the reaction of Pd(KenPhos) with (1-chloro-2-naphthyl)diisopropylphosphine oxide (cf. Figure 3).
Close examination of the geometries of TS1, TS3, TS5 and TS8 reveals interactions between the phosphonate substituents on the naphthylphosphonate addends and weak C–H donors on the ligand backbone and the tolyl addends that appear to contribute to the observed energy differences. The bond distances for relevant O-H interactions range from 2.2–2.8 Å and relevant C-H-O bond angles atoms range from 84–161°; these values are typical of weak hydrogen bonds.77–81 Mulliken charge analysis provides additional indications that (-C)H•••O interactions are important features of these transition states. The charges on hydrogen atoms that interact with phosphonate substituents range from 0.20–0.25e. In contrast, all other hydrogen atoms have charges ranging from 0.12–0.18e.82
The phosphonate group in TS1 interacts closely with two hydrogen atoms located separately on the cyclohexyl group and the aromatic binaphthyl portion of the ligand backbone, and with another hydrogen atom on the tolyl addend. TS3 is destabilized by 1.8 kcal/mol in comparison with TS1, even though similar H-bonding interactions are present in both transition structures; the phosphonate substituent presumably interacts more favorably with the hydrogen atom on an sp2-hybridized carbon atom in TS1 than with the hydrogen atom on an sp3-hybridized carbon atom on the tolyl addend in TS3. One expects hydrogen atoms on sp2-hybridized atoms to be stronger donors than hydrogen atoms on sp3-hybridized atoms, which could account for the greater stability of TS1. In addition, the aromatic proton in TS1 is sterically less demanding than the methyl group in TS3.
The phosphonate groups are located above the planes formed by the palladium atom and the bound atoms of the attached ligands in TS5 and TS8, and only interact with hydrogen atoms on the cyclohexyl substituents on the ligand backbone, as well as with hydrogen atoms on the tolyl addends. The phosphonate group in TS5 interacts with a hydrogen atom belonging to the methyl group on the tolyl addend as well as with one belonging to the cyclohexyl group, and is 0.9 kcal/mol less stable than TS1. The phosphonate group in TS8 interacts with hydrogen atoms on the tolyl addend and a cyclohexyl substituent on the ligand backbone; TS7 is 1.9 kcal/mol less stable than TS1 (i.e. 1.0 kcal/mol less stable than TS5). The methyl group on the tolyl addend in TS7 is presumably sterically encumbering and, as a result, this transition structure is destabilized in comparison to TS5.
TS1 and TS5 are the two transition states that contribute most to the calculated stereoselectivity. These reductively eliminate to give S and R biaryl products, respectively. Although both transition states form interactions with hydrogen atoms on both sp2 and sp3-hybridized carbon atoms, TS1 is 0.9 more stable than TS5. This is presumably due to the fact that the phosphonate group in TS1 is better aligned for interaction with a hydrogen atom on an sp2-hybridized carbon (∠P-O•••H = 158°), but this alignment is poor in TS5 (∠P-O•••H = 90°).
Overall, the electronic property of the phosphonate group strongly influences the geometric arrangement of the addends in low energy transition states. The phosphonate group is also involved in the formation of weak (-C)H•••O interactions; subtle differences in these interactions contribute to the relative energies of the transition states. The presence of (-C)H•••O interactions in these transition states rationalizes the need for carbonyl or phosphonate groups located ortho to the aryl halide for stereoinduction.
Stereoselectivity Due to “Twisting” in Transition States
The stereochemistry of coupled biaryl products were determined by examining the inclination of the tolyl addend in each transition state with respect to the naphthylphosphonate ligand (Scheme 5). The angle between the aromatic planes is defined by C1-C2-C3-C4. As exemplified by the drawing in Scheme 4 with the methyl substituent attached to C1, the coupled product is formed with R stereochemistry if the angle between the aromatic planes lies between −1° and −89°. If the tolyl addend is tilted in the opposite direction the transition state gives a product with the opposite, S, stereochemistry. Products with opposite stereochemistry are obtained if the H atom is attached to C1. Thus, twisting the tolyl addend in both directions leads to the formation of pairs of transition states which form products with opposite stereochemistries as exemplified by the transition state pairs TS1/TS2, TS3/TS4, TS5/TS6, TS7/TS8, TS9/TS10 and TS11/TS12.83 Although the reductive elimination step of Pd-catalyzed Suzuki-Miyaura coupling reactions has previously been examined by DFT methods,84 the interaction of the ligand with the addends and the effects on the outcome of the reaction has not been examined.
Scheme 5.

Determination of the stereochemistry of coupled biaryl products based on transition state geometries.
An example illustrating how twisting the tolyl addend destabilizes the transition state is provided by comparison of the lowest energy transition state, TS1, with the higher energy analogue, TS2 (Figure 7). These transition structures are geometrically similar except that the tolyl addends are inclined in opposite directions, and consequently the products formed from these transition states have opposite absolute stereochemistry. Whereas the tolyl addend in TS1 is twisted so that the aromatic proton is inclined away from the sterically encumbering OMe moiety of the phosphonate substituent, the aromatic proton comes into close contact with this moiety upon twisting to form TS2. Moreover, twisting causes the interaction between the aromatic proton and the phosphonate oxygen to be broken, and causes the methyl group on the aromatic ring to interact with the dimethylamino group located on the ligand backbone.
Figure 7.

Comparison of the lowest energy transition structure with the “twisted” tolyl; addend analogue. Atoms participating in H-bonding interactions are highlighted in green. The stereochemistry of the product formed after reductive elimination is denoted in brackets.
Examination of the other pairs of closely related transition states reveals that twisting the tolyl addends causes the methyl substituents or the aryl protons to interact with the phosphonate which destabilizes the transition states by 2–3 kcal/mol. This destabilization is presumably due to increased steric interactions between the tolyl addend and the phosphonate substituent as well as the loss of hydrogen-bonding interactions between hydrogen atoms on the tolyl addend and the phosphonate group. The transition state pairs TS7/TS8 and TS11/TS12 are exceptions to this observation; here apparent increased interactions between the tolyl addend and the phosphonate substituent actually stabilizes the transition state. The small relative energy differences between these pairs of transition states is presumably due to the fact that hydrogen-bonding interactions are preserved after twisting.
In closing, we emphasize that although hydrogen-bonding interactions are partially responsible for the relative stabilities observed for transition states involved in the coupling of naphthylphosphonate and tolyl addends attached to the Pd(KenPhos) complex, we recognize that similar interactions may not be involved in all of the transition states in reactions such as those involved in the coupling of phenylamides and naphthyl addends. Nevertheless, our computational results illustrate that the combination of interactions such as hydrogen-bonding and steric effects stabilize or destabilize transition states and lead to enantioselectivities observed experimentally; we believe that a similar combination of interactions are operative in the closely related coupling of phenylamides with naphthyl addends by Pd(KenPhos).
Conclusion
In summary, this manuscript discloses the enantioselective syntheses of axially chiral biaryl amides by Suzuki-Miyaura reactions catalyzed by the combination of 1 and Pd(OAc)2. These reactions typically form compounds in yields greater than 80% and typical enantioselectivities range from 80–94%. Chloro-, bromo- and iodophenylamides form products with similar yields and selectivities. Both electron-rich and electron-poor phenylamides are well tolerated in reactions with naphthylboronic acids.
Computational studies on a reaction involving naphthylphosphonate and tolyl addends bound to the Pd(KenPhos) complex reveal fourteen transition states for the selectivity-determining reductive elimination step. The predicted selectivity of this reaction is 50% ee, in reasonable agreement with the selectivity found by experiment. Close examination reveals that the relative energies of these transition states is dependent on a combination of hydrogen-bonding interactions between the phosphonate acceptor and hydrogen atoms attached to the tolyl addend and the ligand backbone. The stereochemistry of the products arising from the transition states is greatly influenced by the direction in which the tolyl addend twists and the relative energies of closely related transition state analogues is therefore dependent on the interaction of methyl or proton substituents on the tolyl addend with the phosphonate substituent.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (NIH) for financial support (Grant GM-46059), and to the National Science Foundation via the National Center for Supercomputing Applications (NCSA) for supercomputing resources. The NMR instruments used for this study were furnished by funds from the National Institutes of Health (GM-1S10RR13886-01). We are grateful to Dr. David Surry and Dr. Tom Kinzel for comments and help with this manuscript.
Footnotes
Supporting Information Available. Detailed experimental procedures and compound characterization data. Energies and Cartesian coordinates of stationary points from B3LYP calculations. Complete citation of reference 71. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kozlowski MC, Morgan BJ, Linton EC. Chem Soc Rev. 2009;38:3193–3207. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace TW. Org Biomol Chem. 2006;4:3197–3210. doi: 10.1039/b608470m. [DOI] [PubMed] [Google Scholar]
- 3.Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew Chem Int Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- 4.Baudoin O. Eur J Org Chem. 2005:4223–4229. [Google Scholar]
- 5.Tanaka K. Chem Asian J. 2009;4:508–18. doi: 10.1002/asia.200800378. [DOI] [PubMed] [Google Scholar]
- 6.Ogasawara M, Watanabe S. Synthesis. 2009:1761–1785. [Google Scholar]
- 7.Hubbard BK, Walsh CT. Angew Chem Int Ed. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 8.Williams DH, Bardsley B. Angew Chem Int Ed. 1999;38:1173–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 9.Nicolaou KC, Boddy CNC, Brase S, Winssinger N. Angew Chem Int Ed. 1999;38:2097–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe T, Tanaka Y, Shoda R, Sakamoto R, Kamikawa K, Uemura M. J Org Chem. 2004;69:4152–4158. doi: 10.1021/jo049600x. [DOI] [PubMed] [Google Scholar]
- 11.Bringmann G, Ochse M, Gotz R. J Org Chem. 2000;65:2069–2077. doi: 10.1021/jo991634v. [DOI] [PubMed] [Google Scholar]
- 12.Hoye TR, Chen MZ. J Org Chem. 1996;61:7940–7942. doi: 10.1021/jo960882d. [DOI] [PubMed] [Google Scholar]
- 13.Brunel JM. Chem Rev. 2005;105:4233–4233. doi: 10.1021/cr040079g. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Yekta S, Yudin AK. Chem Rev. 2003;103:3155–3211. doi: 10.1021/cr020025b. [DOI] [PubMed] [Google Scholar]
- 15.Berthod M, Mignani G, Woodward G, Lemaire M. Chem Rev. 2005;105:1801–1836. doi: 10.1021/cr040652w. [DOI] [PubMed] [Google Scholar]
- 16.Noyori R, Takaya H. Accounts Chem Res. 1990;23:345–350. [Google Scholar]
- 17.Jacobsen ENPA, Yamamoto H. Comprehensive Asymmetric Catalysis. Springer; New York: 2004. [Google Scholar]
- 18.Ojima I. Catalytic Asymmetric Synthesis. 2. John Wiley and Sons; New York: 2000. [Google Scholar]
- 19.Cheon CH, Yamamoto H. J Am Chem Soc. 2008;130:9246–9247. doi: 10.1021/ja8041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akiyama T. Chem Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- 21.Zehm D, Fudickar W, Hans M, Schilde U, Kelling A, Linker T. Chem Eur J. 2008;14:11429–11441. doi: 10.1002/chem.200801355. [DOI] [PubMed] [Google Scholar]
- 22.Miyano S, Tobita M, Hashimoto H. Bull Chem Soc Jpn. 1981;54:3522–3526. [Google Scholar]
- 23.Lipshutz BH, Kayser F, Liu ZP. Angew Chem Int Ed Engl. 1994;33:1842–1844. [Google Scholar]
- 24.Bringmann G, Breuning M, Endress H, Vitt D, Peters K, Peters EM. Tetrahedron. 1998;54:10677–10690. [Google Scholar]
- 25.Meyers AI, Lutomski KA. J Am Chem Soc. 1982;104:879–881. [Google Scholar]
- 26.Meyers AI, Himmelsbach RJ. J Am Chem Soc. 1985;107:682–685. [Google Scholar]
- 27.Warshawsky AM, Meyers AI. J Am Chem Soc. 1990;112:8090–8099. [Google Scholar]
- 28.Meyers AI, Nelson TD, Moorlag H, Rawson DJ, Meier A. Tetrahedron. 2004;60:4459–4473. [Google Scholar]
- 29.Wilson JM, Cram DJ. J Am Chem Soc. 1982;104:881–884. [Google Scholar]
- 30.Wilson JM, Cram DJ. J Org Chem. 1984;49:4930–4943. [Google Scholar]
- 31.Suzuki T, Hotta H, Hattori T, Miyano S. Chem Lett. 1990:807–810. [Google Scholar]
- 32.Baker RW, Pocock GR, Sargent MV, Twiss E. Tetrahedron: Asymmetry. 1993;4:2423–2426. [Google Scholar]
- 33.Smrcina M, Polakova J, Vyskocil S, Kocovsky P. J Org Chem. 1993;58:4534–4538. [Google Scholar]
- 34.Nakajima M, Miyoshi I, Kanayama K, Hashimoto S, Noji M, Koga K. J Org Chem. 1999;64:2264–2271. [Google Scholar]
- 35.Irie R, Masutani K, Katsuki T. Synlett. 2000:1433–1436. [Google Scholar]
- 36.Li XL, Yang J, Kozlowski MC. Org Lett. 2001;3:1137–1140. doi: 10.1021/ol015595x. [DOI] [PubMed] [Google Scholar]
- 37.Luo ZB, Liu QZ, Gong LZ, Cui X, Mi AQ, Jiang YZ. Angew Chem Int Ed. 2002;41:4532–4535. doi: 10.1002/1521-3773(20021202)41:23<4532::AID-ANIE4532>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Gutnov A, Heller B, Fischer C, Drexler HJ, Spannenberg A, Sundermann B, Sundermann C. Angew Chem Int Ed. 2004;43:3795–3797. doi: 10.1002/anie.200454164. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka K, Nishida G, Wada A, Noguchi K. Angew Chem Int Ed. 2004;43:6510–6512. doi: 10.1002/anie.200461533. [DOI] [PubMed] [Google Scholar]
- 40.Shibata T, Fujimoto T, Yokota K, Takagi K. J Am Chem Soc. 2004;126:8382–8383. doi: 10.1021/ja048131d. [DOI] [PubMed] [Google Scholar]
- 41.Bringmann G, Walter R, Weirich R. Angew Chem Int Ed Engl. 1990;29:977–991. [Google Scholar]
- 42.Stanforth SP. Tetrahedron. 1998;54:263–303. [Google Scholar]
- 43.Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Chem Rev. 2002;102:1359–1469. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 44.Denmark SE, Sweis RF. Accounts Chem Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]
- 45.Eliel ELWSH. Stereochemistry of Organic Compounds. John Wiley and Sons; New York: 1994. [Google Scholar]
- 46.Hayashi T, Hayashizaki K, Kiyoi T, Ito Y. J Am Chem Soc. 1988;110:8153–8156. [Google Scholar]
- 47.Hayashi T, Niizuma S, Kamikawa T, Suzuki N, Uozumi Y. J Am Chem Soc. 1995;117:9101–9102. [Google Scholar]
- 48.Genov M, Almorin A, Espinet P. Chem Eur J. 2006;12:9346–9352. doi: 10.1002/chem.200600616. [DOI] [PubMed] [Google Scholar]
- 49.Genov M, Almorin A, Espinet P. Tetrahedron: Asymmetry. 2007;18:625–627. [Google Scholar]
- 50.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 51.Hall DG. Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 52.Uemura M, Kamikawa K. J Chem Soc, Chem Commun. 1994:2697–2698. [Google Scholar]
- 53.Kamikawa K, Uemura M. Tetrahedron Lett. 1996;37:6359–6362. [Google Scholar]
- 54.Kamikawa K, Uemura M. Synlett. 2000:938–949. [Google Scholar]
- 55.Nelson SG, Hilfiker MA. Org Lett. 1999;1:1379–1382. [Google Scholar]
- 56.Broutin PE, Colobert F. Org Lett. 2003;5:3281–3284. doi: 10.1021/ol0351908. [DOI] [PubMed] [Google Scholar]
- 57.Lipshutz BH, Keith JM. Angew Chem Int Ed. 1999;38:3530–3533. [PubMed] [Google Scholar]
- 58.Yin JJ, Buchwald SL. J Am Chem Soc. 2000;122:12051–12052. [Google Scholar]
- 59.Cammidge AN, Crepy KVL. Chem Comm. 2000:1723–1724. [Google Scholar]
- 60.Bronger RPJ, Guiry PJ. Tetrahedron: Asymmetry. 2007;18:1094–1102. [Google Scholar]
- 61.Castanet AS, Colobert F, Broutin PE, Obringer M. Tetrahedron: Asymmetry. 2002;13:659–665. [Google Scholar]
- 62.Herrbach A, Marinetti A, Baudoin O, Guenard D, Gueritte F. J Org Chem. 2003;68:4897–4905. doi: 10.1021/jo034298y. [DOI] [PubMed] [Google Scholar]
- 63.Jensen JF, Johannsen M. Org Lett. 2003;5:3025–3028. doi: 10.1021/ol034943n. [DOI] [PubMed] [Google Scholar]
- 64.Mikami K, Miyamoto T, Hatano M. Chem Comm. 2004:2082–2083. doi: 10.1039/b407250b. [DOI] [PubMed] [Google Scholar]
- 65.Takemoto T, Iwasa S, Hamada H, Shibatomi K, Kameyama M, Motoyama Y, Nishiyama H. Tetrahedron Lett. 2007;48:3397–3401. [Google Scholar]
- 66.Sawai K, Tatumi R, Nakahodo T, Fujihara H. Angew Chem Int Ed. 2008;47:6917–6919. doi: 10.1002/anie.200802174. [DOI] [PubMed] [Google Scholar]
- 67.Bermejo A, Ros A, Fernandez R, Lassaletta JM. J Am Chem Soc. 2008;130:15798–15799. doi: 10.1021/ja8074693. [DOI] [PubMed] [Google Scholar]
- 68.Uozumi Y, Matsuura Y, Arakawa T, Yamada YMA. Angew Chem Int Ed. 2009;48:2708–2710. doi: 10.1002/anie.200900469. [DOI] [PubMed] [Google Scholar]
- 69.Metallinos C, Nerdinger S, Snieckus V. Org Lett. 1999;1:1183–1186. [Google Scholar]
- 70.Joncour A, Decor A, Liu JM, Dau M, Baudoin O. Chem Eur J. 2007;13:5450–5465. doi: 10.1002/chem.200601764. [DOI] [PubMed] [Google Scholar]
- 71.Frisch MJ. Gaussian 03. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 72.Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 73.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 74.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 75.In addition to the constraint imposed by the limited availability of appropriate crystallographic data for reactions involving phenylamides, a preliminary analysis of transition state geometries revealed that the cumylamide substituents are not aligned with the aromatic plane of the phenyl ring. Therefore, twice as many calculations would be required for reactions involving the coupling of phenylamide with naphthylphosphonate addends in comparison to investigations involving the coupling of naphthylphosphonate and tolyl addends. Because of these factors, we therefore decided to restrict our calculations to reactions involving the coupling of naphthylphosphonate and tolyl addends.
- 76.If we consider enantioselectivity provided by two transition states, a selectivity of 50% ee at 25 ºC corresponds to an energy difference of 0.7 kcal/mol and 84% ee at 60 ºC corresponds to an energy difference of 1.6 kcal/mol. The difference between these energies is within the error limits of the B3LYP method.
- 77.Desiraju GRST. The Weak Hydrogen Bond In Structural Chemistry and Biology. Oxford University Press; New York, NY: 1999. [Google Scholar]
- 78.Steiner T, Desiraju GR. Chem Comm. 1998:891–892. [Google Scholar]
- 79.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford University Press; New York, NY: 1997. [Google Scholar]
- 80.Taylor R, Kennard O. J Am Chem Soc. 1982;104:5063–5070. [Google Scholar]
- 81.Jeffrey GA, Maluszynska H. Int J Biol Macromol. 1982;4:173–185. [Google Scholar]
- 82.It should be noted that the compression of the ligand and the tolyl and naphthylphosphonate addends during the transition state for C-C bond formation could be responsible for these forced (-C)H•••O interactions; as a result, these interactions may not stabilize the transition states. Note, however, that the observed enantioselectivity does not depend on whether these interactions stabilize or destabilize the transition state. Instead, the degree of enantioselectivity depends solely on energy differences among the observed interactions since all transition states contain one or more of these types of interactions.
- 83.Additional evidence for the stereochemistry of the coupled biaryl compounds was provided by integration of intrinsic reaction coordinates (IRCs) to obtain reaction paths for each transition state. Fifty steps were calculated for each IRC calculation thereby allowing for almost complete cleavage of the bonds to the Pd atom and formation of the new C-C bond. The tolyl addend becomes further inclined away from the bound naphthylphosphonate ligand as the new C-C bond is formed; the direction in which it twists ultimately defines the stereochemistry of the product. The results obtained from the IRC study were in agreement with results obtained by visual inspection of the transition states.
- 84.Braga AAC, Ujaque G, Maseras F. Organometallics. 2006;25:3658–3658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.