

Abstract
Fragile X syndrome (FXS) is an inherited neurologic disease caused by loss of fragile X mental retardation protein (FMRP), which is hypothesized to mediate negative regulation of mRNA translation at synapses. A prominent feature of FXS animal models is exaggerated signaling through group 1 metabotropic glutamate receptors (gp1 mGluRs), and therapeutic strategies to treat FXS are targeted mainly at gp1 mGluRs. Recent studies, however, indicate that a variety of receptor-mediated signal transduction pathways are dysregulated in FXS, suggesting that FMRP acts on a common downstream signaling molecule. Here, we show that deficiency of FMRP results in excess activity of phosphoinositide 3-kinase (PI3K), a downstream signaling molecule of many cell surface receptors. In Fmr1 knock-out neurons, excess synaptic PI3K activity can be reduced by perturbation of gp1 mGluR-mediated signaling. Remarkably, increased PI3K activity was also observed in FMRP-deficient non-neuronal cells in the absence of gp1 mGluRs. Here, we show that FMRP regulates the synthesis and synaptic localization of p110β, the catalytic subunit of PI3K. In wild type, gp1 mGluR activation induces p110β translation, p110β protein expression, and PI3K activity. In contrast, both p110β protein synthesis and PI3K activity are elevated and insensitive to gp1 mGluR stimulation in Fmr1 knock-out. This suggests that dysregulated PI3K signaling may underlie the synaptic impairments in FXS. In support of this hypothesis, we show that PI3K antagonists rescue three FXS-associated phenotypes: dysregulated synaptic protein synthesis, excess AMPA receptor internalization, and increased spine density. Targeting excessive PI3K activity might thus be a potent therapeutic strategy for FXS.
Introduction
Fragile X syndrome (FXS) is the most frequent inherited form of intellectual disability. It is caused by loss of function of fragile X mental retardation protein (FMRP), an mRNA binding protein involved in the regulation of translation, transport, and stability of target mRNAs (Bassell and Warren, 2008; De Rubeis and Bagni, 2010). FMRP has been shown to act as a translational inhibitor of a few select target mRNAs in vitro (Laggerbauer et al., 2001; Li et al., 2001) and in vivo (Zalfa et al., 2003; Muddashetty et al., 2007). However, these results are insufficient to explain the global dysregulation of basal and neurotransmitter-induced protein synthesis (Weiler et al., 2004; Dolen et al., 2007; Muddashetty et al., 2007) or the loss of a protein synthesis requirement for group I metabotropic glutamate receptor (gp1 mGluR)-induced long-term depression (LTD) observed in the absence of FMRP (Nosyreva and Huber, 2006). The molecular mechanism whereby FMRP couples the activation of cell surface receptors to protein synthesis regulation is unclear.
Several studies have shown exaggerated gp1 mGluR signaling in FXS animal models (Pfeiffer and Huber, 2009). The correction of FXS phenotypes by genetic or pharmacologic reduction of mGluR5 signaling (Dolen et al., 2007; D'Hulst and Kooy, 2009) supports the widely accepted “mGluR theory of FXS,” which postulates that excessive signaling through gp1 mGluRs underlies synaptic defects observed in the absence of FMRP (Bear et al., 2004). Targeting excess gp1 mGluR signaling is therefore a promising therapeutic strategy (Bear et al., 2008). However, none of these studies reveal how FMRP limits gp1 mGluR-induced signal transduction. Although loss of FMRP-mediated regulation of protein synthesis is believed to be the cause, there is little experimental evidence in support of an underlying molecular mechanism.
Other signal transduction pathways besides gp1 mGluR signaling have been reported recently to be dysregulated in the absence of FMRP (Volk et al., 2007; Wang et al., 2008; Weinshenker and Warren, 2008). Thus, FMRP might not exclusively target gp1 mGluRs but rather regulate a common signaling molecule downstream of multiple membrane receptors. A few studies describe aberrant gp1 mGluR-regulated downstream signaling in FXS (Hou et al., 2006; Ronesi and Huber, 2008a; Sharma et al., 2010); the underlying molecular mechanisms, however, are unknown. Furthermore, the therapeutic value of targeting the intracellular signaling pathway instead of a surface receptor has not been addressed in detail.
These previous studies imply that FMRP may directly regulate the translation and synaptic localization of a downstream signaling molecule regulating protein synthesis. Here, we show that loss of FMRP leads to excess mRNA translation and synaptic protein expression of p110β, a catalytic subunit of phosphoinositide 3-kinase (PI3K), which is a key signaling molecule downstream of gp1 mGluRs and other membrane receptors. Remarkably, the basal enzymatic activity of PI3K increased threefold at FXS synapses, and gp1 mGluR-mediated induction of p110β translation, protein levels, and activity at synapses are occluded in Fmr1 knock-out (KO). Antagonizing PI3K can correct three FXS phenotypes: aberrant synaptic translation, enhanced AMPA receptor (AMPAR) endocytosis, and excess spine density. Our results broaden the mGluR theory of FXS and suggest aberrant PI3K activity as a promising therapeutic target for FXS.
Materials and Methods
Drugs, chemicals, and peptides.
Puromycin was obtained from Sigma-Aldrich; all other drugs were purchased from Tocris Bioscience. Radiolabeled amino acids and trinucleotides were purchased from GE Healthcare and PerkinElmer Life and Analytical Sciences. l-α-Phosphatidylinositol (liver, bovine; sodium salt) was obtained from Avanti Polar Lipids. All other chemicals were purchased from Sigma-Aldrich. Tat–mGluR5–CT or tat–mGluR5–MUT peptides were synthesized by Invitrogen [sequence was described previously (Mao et al., 2005)]. Stealth small interfering RNA (siRNA) (Invitrogen) was designed using BLOCK-iT RNAi Designer (Invitrogen) (sequences: for sifmr1: NM_008031_stealth_298, sense, UGG CGC UUU CUA CAA GGC AUU UGU A; sictr: NM_008031_stealth_control_298, sense, UGG UUU CCA UCG GAA UUA CUG CGU A). siRNA targeting p110β was purchased from Santa Cruz Biotechnology (sc-29447).
Primers for quantitative real-time PCR.
Sequences of primers for quantitative real-time (qRT)-PCR were as follows: p110β forward, 5′-ttcttttcagtgttgtgaccaag-3′ and reverse, 5′-gccccgaatgtggtaagtt-3′; NR1 forward, 5′-ggcagtaaaccaggccaata-3′ and reverse, 5′-agcagagccgtcacattctt-3′; postsynaptic density protein 95 (PSD95) forward, 5′-ctatgagacggtgacgcaga-3′ and reverse, 5′-cgggaggagacaaagtggta-3′; enhanced green fluorescent protein (EGFP) forward, 5′-aaggacgacggcaactacaag-3′ and reverse, 5′-atgccgttcttctgcttgtcg-3′; phosphoinositide 3-kinase enhancer, long isoform (PIKE-L) forward, 5′-tgtccacaccatctaagactgaa-3′ and reverse, 5′- aggattttagtttccacattttgc-3′; and β-actin forward, 5′-gcactcttccagccttcc-3′ and reverse, 5′-ccgctcaggagcaat-3′.
Antibodies.
Mouse monoclonal anti-tubulin, anti-PSD95, anti-GFAP, anti-MAP2, and anti-synaptophysin antibodies, as well as rabbit polyclonal anti-mGluR5 antibodies, were purchased from Millipore Corporation; mouse monoclonal anti-mGluR1 antibody was purchased from BD Biosciences. Rabbit polyclonal antibody against p110β was purchased from Santa Cruz Biotechnology. Rabbit monoclonal antibodies specific for phosphor-Akt(Thr308), Akt(pan), and ERK1/2, as well as mouse monoclonal anti-phospho-ERK1/2(Thr202/Tyr204) antibody, were purchased from Cell Signaling Technology. Anti-PIKE-L antibody was described previously (Tang et al., 2008). Mouse monoclonal anti-FMRP antibody 7G1-1 (Brown et al., 2001) was obtained from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Mouse monoclonal anti-FMRP antibody 2F5-1 (Gabel et al., 2004) (used for Western blot analyses) was a kind gift from Jennifer C. Darnell (The Rockefeller University, New York, NY). A rabbit polyclonal antibody raised against the C terminus of FMRP (as described by Ferrari et al., 2007) was used for Western blot analysis of polysomal gradients in supplemental Fig. S3F (available at www.jneurosci.org as supplemental material).
cDNA constructs and plasmids.
Murine flag–mCherry–FMRP (open reading frame) was subcloned from EGFP–FMRP (Antar et al., 2005), and murine EGFP–p110β–3′ untranslated region (UTR) (GenBank accession number NM_029094.2; nucleotides 3401-4649) and murine EGFP–β-actin–3′ UTR (GenBank accession number NM_007393.3; nucleotides 1208-1860) were generated by PCR from mouse cDNA and subcloned into pEGFP–C1 (Clontech). The cDNA encoding the Pleckstrin homology (PH) domain of mouse Akt1 was generated by PCR from mouse cDNA (GenBank accession number NM_009652.2; nucleotides 285-725) and cloned into a plasmid containing cDNA encoding monomeric red fluorescent protein (RFP) (Campbell et al., 2002). GFP–Lifeact was a kind gift from Roland Wedlich-Soeldner (Max Planck Institute for Biochemistry, Martinsried, Germany) (Riedl et al., 2008).
Kinase activity assays.
In vitro PI3K assays were performed as described previously (Ye et al., 2000) using p110β antibody. In vitro extracellular signal-regulated kinase (ERK) activity assays were performed using the p44/42 MAP Kinase Assay kit from Cell Signaling Technology according to the manual. HEK293T cells were cultured in DMEM supplemented with 10% FBS. Fmr1-specific siRNA was transfected using LF2000 (Invitrogen) according to the manual. At 48 h after transfection, cells were scraped from the dish with PBS, divided into two equal parts for PI3K and ERK activity assays, respectively. For treatment with PI3K antagonist, cells were incubated with 40 μm LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] or LY303511 (2-(1-piperazinyl)-8-phenyl-4H-1-benzopyran-4-one, inactive analog) for 15 min before further processing for PI3K assay. Cortical synaptoneurosomes (SNS) were prepared as described previously (Muddashetty et al., 2007). SNS were split in two equal parts for PI3K and ERK activity assays. Cells and SNS were lysed in kinase assay buffers (for PI3K assay: 50 mm Tris-HCl, pH 7.4, 40 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 1.5 mm Na3VO4, 50 mm NaF, 10 mm sodium pyrophosphate, and 10 mm sodium β-glycerol phosphate, supplemented with proteinase inhibitors; for ERK activity assay: lysis buffer provided by Cell Signaling Technology). Lysates were cleared by centrifugation, and protein concentrations were determined by the Bradford method. For both kinase assays, the same amount of protein was used for wild-type (WT) and Fmr1 KO SNS or sifmr1 knockdown (KD) and sictr HEK293T cells, respectively. For measuring PI3K activity following various treatments, SNS were incubated for 10 min at 37°C with either 100 μm (S)-3,5-dihydroxyphenylglycine (DHPG) or 10 μm 2-methyl-6-(phenylethynyl)-pyridine (MPEP) or for 1 h at 37°C with tat–mGluR5–CT or tat–mGluR5–MUT peptides (5 μm) before lysis (Mao et al., 2005; Ronesi and Huber, 2008a). For PI3K assays with cortical neurons, cells at 11 d in vitro (DIV) were transfected with 10 μl of control adenovirus or adenovirus carrying short-hairpin PIKE-L (as described previously by Tang et al., 2008) for 48 h. PI3K assays were performed with 300 μg of protein as described above. To confirm PIKE-L knockdown, 80 μg of protein was used for Western blot analyses using anti-PIKE-L antibody (Tang et al., 2008).
Quantification of Western blots.
SDS-PAGE and Western blots were performed as described previously (Muddashetty et al., 2007) and quantified using NIH ImageJ. Signal intensities of p110β- and FMRP-specific bands were normalized to tubulin signal on the same blot. Signal intensities of phospho-proteins were normalized to total levels of the respective protein on the same blot. For this purpose, Western blots were stripped after detection with the phospho-specific antibodies using 100 mm β-mercaptoethanol and 2% SDS in 62.5 mm Tris-HCl, pH 6.8, at 50°C for 30 min and, after extensive washing, detected with the respective antibody specific for the total protein.
Cell culture.
Hippocampal primary neurons were dissected from WT and Fmr1 KO mice at embryonic day 17 and cultured as described previously (Muddashetty et al., 2007). Neurons were transfected with RFP–PH(Akt) at 9 DIV using Lipofectamine 2000 (LF2000; Invitrogen) according to the manual. Six hours after transfection, neurons were fixed with 4% paraformaldehyde (PFA) and further processed for immunocytochemistry.
Immunohistochemistry.
For p110β-specific staining, neurons were fixed at 17 DIV. Cells were permeabilized with 0.3% Triton X-100 and blocked with 2% normal donkey serum and 4% BSA. Primary antibodies were incubated at room temperature for 1 h (anti-p110β at 1:50; anti-synaptophysin at 1:300) and detected with cyanine dye 3-coupled donkey anti-rabbit and cyanine dye 2-coupled donkey anti-mouse antibodies, respectively.
Imaging and processing.
Images of immunocytochemistry on cultured neurons were acquired using a wide-field fluorescent Nikon Eclipse inverted microscope with cooled CCD camera and built-in Z-drives. Stacks were deconvolved with AutoQuant X (Media Cybernetics). For quantification of signal intensities, as well as colocalization analyses, a region of interest along a dendrite at least 50 μm from the cell body was chosen. Mean fluorescent intensities were determined using IPlab software (BD Biosciences) or Imaris software (Bitplane) and normalized to background staining of the same area on the coverslip. Colocalization analyses were performed using Imaris Coloc software (Bitplane). Images of fluorescent in situ hybridization experiments on tissue were acquired using a Carl Zeiss LSM510 confocal microscope, deconvolved with AutoQuant X (Media Cybernetics), and are displayed as flattened z-stacks (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material) or as single slices (supplemental Fig. S4B,C, available at www.jneurosci.org as supplemental material).
RNA coimmunoprecipitation.
Lysates for RNA coimmunoprecipitations were prepared by homogenizing cortices from mice (C57BL/6 and Fmr1 KO, backcrossed in C57BL/6J, postnatal day 17–21) in lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, and 1% NP-40, supplemented with proteinase and RNase inhibitors) on ice. RNA coimmunoprecipitations and subsequent qRT-PCR were performed as described previously (Muddashetty et al., 2007) using the mouse monoclonal 7G1-1 antibody.
Anti-flag pulldown.
HEK293T cells were transfected with flag–mCherry–FMRP and EGFP–p110β–3′ UTR or EGFP–β-actin–3′ UTR using Lipofectamine 2000 (Invitrogen) according to the manual. For controls, EGFP constructs were cotransfected with flag–mCherry. At 24 h after transfection, cells were lysed with coimmunoprecipitation lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, and 1% NP-40, supplemented with proteinase and RNase inhibitors) on ice. Anti-flag pulldowns were performed with anti-flag M2 affinity gel (Sigma) and analyzed with EGFP-specific qRT-PCRs as described previously (Muddashetty et al., 2007).
Linear sucrose gradients.
Gradient preparation, centrifugation, fractionation, RNA purification, and qRT-PCRs were performed as described previously (Muddashetty et al., 2007).
Synaptoneurosome preparation, treatment, and metabolic labeling.
Cortical SNS were prepared as described previously (Muddashetty et al., 2007) and treated with the indicated antagonists or solvent for 5 min at 37°C, for some experiments followed by DHPG (100 μm) for 15 min at 37°C as indicated, and with puromycin (2 mm) for 30 min at 37°C. Metabolic labeling was performed with 100 μCi Pro-Mix, L-(35S) in vitro cell labeling mix (GE Healthcare) for 5 min. Triplicate samples from time point 0 and 5 min were washed once in homogenization buffer, followed by lysis (100 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% deoxycholate, and 1% Triton X-100, supplemented with proteinase inhibitors) and TCA precipitation. 35S-incorporation levels were estimated by scintillation counting.
Constitutive AMPAR internalization assay.
The assay was performed as described previously for rat neurons (Nakamoto et al., 2007), with the following modifications: mouse WT or Fmr1 KO cells were preincubated with LY294002 (50 μm) or U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] (20 μm) for 1 h when indicated. Surface AMPARs in live neurons were labeled with a rabbit polyclonal antibody against the N terminus of the GluR1 subunit (Calbiochem) (1:5 in conditioned media) for 15 min at 37°C, 0.5% CO2. After incubation with antagonists [1 μm TTX, 10 μm NBQX (2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline), and 50 μm APV (Tocris Bioscience), as described previously (Bhattacharyya et al., 2009)] in conditioned media for 15 min at 37°C, 0.5% CO2, cells were fixed in 4% PFA and processed and analyzed as described previously (Nakamoto et al., 2007).
Analysis of dendritic protrusion density.
High-density hippocampal neurons from WT and Fmr1 KO were treated for 3 d with 10 μm LY294002 or an equal amount of vehicle (DMSO) starting at 15 DIV. Every 24 h, culture media was exchanged with fresh drug in conditioned media. After 48 h (17 DIV), cells were transfected (NeuroMag, according to the manual; OZBiociences) with GFP–Lifeact, a peptide that binds to filamentous actin (F-actin) (Riedl et al., 2008) and can be used to visualize dendrites and spines. At 16–24 h after transfection, cells were fixed and imaged with a Nikon Eclipse inverted microscope using a 60× Plan-Neofluar objective and a cooled CCD camera (Quantix; Photometrics). Images were taken as z-stacks (31 stacks, 0.2 μm per stack) and deconvolved using AutoQuant 2X (Media Cybernetics). Dendrites and spines (>40 μm distance from cell body, the analyzed region including a main dendrite of at least 50 μm length) were traced and analyzed using FilamentTracer (Imaris software; Bitplane). Total protrusion density [including spines and filopodia, classified as described previously (Harris et al., 1992)] was displayed as average number per 100 μm.
Statistics.
All statistics were performed using SSPS 17.0. Statistical tests used and n of experiments are indicated in the figure legends and text. All t tests were two tailed. All data were tested for normal distribution and homogeneity of variances with Levene's tests. Nonparametric tests were used for supplemental Figures S2, A and B, and S3H (available at www.jneurosci.org as supplemental material), for which the data were not normally distributed. In all other instances, when datasets did not meet the criterion of normal distribution and/or homogenous variances, tests were performed on square-root transformed data (see Figs. 2 A, 3 I, 7 B; supplemental Fig. S2C, available at www.jneurosci.org as supplemental material) or log-transformed data (see Fig. 2 B; supplemental Fig. S5F, available at www.jneurosci.org as supplemental material). Effects were considered significant when the p value was <0.05. Within each individual experiment, means were normalized to the mean of WT, WT control, or control, when applicable, depending on the experiment. Diagrams represent means of n normalized experiments. Error bars represent SEM of n independent experiments.
Figure 2.

Excess PI3K activity in Fmr1 KO cortical neurons can be reduced by inhibition of gp1 mGluR-mediated signaling. A, Treatment with the mGluR5 antagonist MPEP significantly reduces PI3K activity in Fmr1 KO SNS but not in WT SNS (n = 4, two-way ANOVA: genotype, p = 0.953; treatment, p = 0.425; interaction of genotype and treatment, *p = 0.006; LSD post hoc analyses: p wtctr–wtMPEP = 0.327, *p wtctr–koctr = 0.033, *p koctr–koMPEP = 0.012). B, Disruption of the mGluR5–Homer complex with a tat-fused mGluR5 C-terminal peptide but not with a mutated peptide (CT and MUT, respectively; 5 μm) decreases PI3K activity in both WT and Fmr1 KO SNS. Peptide treatment led to a significant decrease in PI3K activity in both WT and KO, but PI3K activity was still significantly increased in KO compared with WT after treatment with the mGluR5 C-terminal peptide (n = 4, two-way-ANOVA; effect of peptide, *p < 0.001; effect of genotype, *p = 0.014; interaction of peptide and genotype, p = 0.143). Moreover, there was neither a significant genotype-specific difference in PI3K activity in KO samples compared with WT after disruption of the mGluR complex, nor a significant difference in the ratios of KO versus WT PI3K activity when treated with mutated compared to C-terminal peptides (analyses shown in supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). C, PI3K activity in cultured cortical Fmr1 KO neurons is increased compared with WT, and short-hairpin-mediated knockdown of the PI3K enhancer PIKE-L (shPIKE-L) significantly reduces PI3K activity in Fmr1 KO (n = 3; two-way ANOVA: genotype, *p = 0.001; treatment, *p = 0.008; interaction of genotype and treatment, *p = 0.014; Tukey's HSD post hoc analyses: *p wtctr–koctr = 0.002, *p koctr–koPIKE = 0.007). shPIKE-L did not lead to a complete rescue of PI3K activity to WT levels (KO, 139 ± 14% of WT after PIKE knockdown). Western blots below show knockdown of PIKE-L in WT and KO neurons; tubulin was used as loading control. Additional data in supplemental Figure S2, C and D, show association of PIKE-L mRNA with FMRP in coimmunoprecipitations from brain lysates, as well as increased PIKE-L levels in Fmr1 KO SNS (available at www.jneurosci.org as supplemental material). D, E, Activation of gp1 mGluRs with DHPG (10 min, 100 μm) leads to a twofold increase in PI3K activity in WT SNS (D, n = 4, *p = 0.033, paired t test) but to decreased PI3K activity in Fmr1 KO SNS (E, n = 4, *p = 0.049, paired t test).
Figure 3.
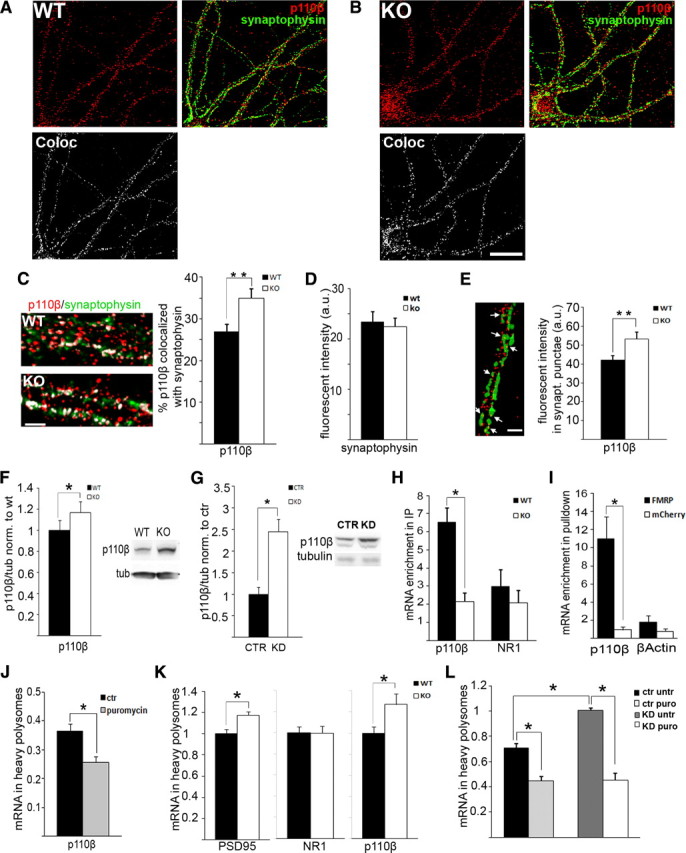
p110β protein expression and translation is regulated by FMRP. A, B, Immunocytochemical analysis of p110β expression at synapses in WT (A) and Fmr1 KO (B) neurons shows that p110β (red) colocalizes with the synaptic marker synaptophysin (green), as indicated by yellow signal; specificity of antibody is shown in supplemental Figure S3A (available at www.jneurosci.org as supplemental material). Colocalized signal is shown in white (bottom). Scale bar, 20 μm. C, Quantification of p110β signal intensities overlapping with synaptophysin reveals a significant increase of overlap in Fmr1 KO neurons compared with WT neurons (WT, 27% overlap; KO, 34.9% overlap; n = 43 dendrites each for WT and KO, 3 independent hippocampal cultures, **p = 0.008, independent t test) (additional analyses shown in supplemental Fig. S3B–D, available at www.jneurosci.org as supplemental material). Representative images of dendrites from WT and Fmr1 KO that were analyzed for colocalization are shown on the left, with colocalized signal superimposed in white. Scale bar, 3 μm. D, Total levels of synaptophysin were unchanged in Fmr1 KO compared with WT dendrites (n = 43, p = 0.73, independent t test). E, Quantification of fluorescent p110β-specific signal within three-dimensional reconstructed synaptophysin punctae reveals increased mean intensity of p110β-specific signal in synaptophysin punctae in Fmr1 KO dendrites, suggesting specific enrichment of p110β at single synapses (n = 43, **p = 0.004, independent t test). In contrast, the relative number of p110β-positive synapses was not changed in Fmr1 KO neurons (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). Example of three-dimensional reconstruction of synaptophysin punctae is shown on the left, and p110β signal within these punctae is indicated by arrows. Scale bar, 3 μm. Additional analysis is shown in supplemental Figure. S3D (available at www.jneurosci.org as supplemental material). F, Densitometric analysis of p110β-specific Western blots demonstrates increased p110β protein levels in SNS from Fmr1 KO cortices compared with WT. Signal intensities were normalized to tubulin (n = 5, *p = 0.043, paired t test); a representative western blot is shown at the right. G, siRNA-mediated reduction of FMRP expression in HEK293T cells leads to increased p110β protein levels. Signal intensities were normalized to tubulin (n = 5, *p = 0.005, paired t test). H, FMRP-specific quantitative coimmunoprecipitation from WT and Fmr1 KO brain lysates demonstrates a specific enrichment of p110β mRNA in WT immunoprecipitations, whereas NR1 mRNA is not enriched. mRNA levels were quantified by quantitative real-time PCR (n = 6, two-way ANOVA: p mRNA = 0.488, p genotype = 0.372, *p between subjects = 0.034; Tukey's HSD post hoc analyses: *p p110 β = 0.003, p NR1 = 0.884). I, Recombinant p110β 3′ UTR fused to EGFP expressed in HEK293T cells is significantly enriched in anti-flag pulldowns with coexpressed flag-tagged mCherry–FMRP but not with flag-tagged mCherry. No specific enrichment can be detected with β-actin 3′ UTR. mRNA levels in pulldowns were quantified by qRT-PCR with EGFP-specific primers and normalized to input (n = 4, two-way ANOVA: *p mRNA = 0.006, *p rec.protein = 0.001, *p between subjects = 0.01; Tukey's HSD post hoc analyses: *p p110 β = 0.001, p β -actin = 0.798). J, K, qRT-PCR quantification of mRNA levels in sucrose gradients from SNS shows that p110β mRNA in polysomal fractions is puromycin sensitive (J, n = 3, *p = 0.033, paired t test) and significantly increased in these fractions from Fmr1 KO SNS compared with WT; PSD95 and NR1 mRNAs served as positive and negative controls, respectively (K, n = 5; PSD95, *p = 0.029; NR1, p = 0.89; p110β, *p = 0.043, paired t tests; results were normalized to WT). For experimental details, see supplemental Fig. S3, E and F (available at www.jneurosci.org as supplemental material). L, Likewise p110β mRNA is shifted into actively translating polysomes after Fmr1 knockdown in HEK293T cells (n = 3, two-way ANOVA: *p genotype = 0.002, *p treatment < 0.001, *p between subjects = 0.002; Tukey's HSD post hoc analyses: *p ctruntr–ctrpuro = 0.002, *p ctruntr–KDuntr = 0.001, *p KDuntr–KDpuro < 0.001). All error bars represent SEM.
Figure 7.

A PI3K antagonist rescues increased dendritic spine density in Fmr1 KO neurons. A, Three-dimensional reconstruction of representative dendrites from WT and Fmr1 KO neurons (18 DIV) after 3 d of treatment with vehicle (ctr) or LY294002 (LY, 10 μm) illustrates that increased protrusion density in Fmr1 KO is rescued by PI3K inhibition. Scale bar, 5 μm. B, Automated quantification using FilamentTracer software (Imaris, Bitplane) shows significantly increased protrusion density in vehicle-treated Fmr1 KO, which can be restored to WT levels by LY294002 treatment, but does not change spine morphology in WT. Bar diagrams represent average spine number per 100 μm (WT control, 38.2; WT LY294002, 38.7; KO control, 46.0; KO LY294002, 33.6; n = 30, 2 independent cultures, two-way ANOVA: significant interaction between genotype and treatment, *p = 0.001; LSD post hoc tests: *p wtctr–koctr = 0.024, ***p koctr–koLY294002 < 0.0001, p wtctr–wtLY294002 = 0.996; error bars represent SEM). C, Examples of a dendrite that was analyzed with FilamentTracer (Imaris; Bitplane) illustrate accuracy of the applied method to identify protrusions. Top, Three-dimensional reconstruction of fluorescent signals from GFP–Lifeact transfected hippocampal dendrite; middle, traced and rebuilt dendrite (white) with spines (blue); bottom, overlay of rebuilt dendrite with original image. Scale bar, 10 μm.
Results
Excess PI3K activity at Fmr1 KO synapses
The molecular mechanisms underlying exaggerated receptor-mediated signaling and protein synthesis in the absence of FMRP are unknown. Some reports have revealed defects in PI3K/mammalian target of rapamycin (mTOR) signaling (Ronesi and Huber, 2008a; Sharma et al., 2010), whereas others implicate impaired ERK signaling in FMRP-deficient cells (Hou et al., 2006; Kim et al., 2008). Both pathways are crucial to regulate mGluR-induced protein synthesis in normal brain (Klann and Dever, 2004), yet it remains controversial whether one pathway is predominantly dysregulated in FXS. However, no study has directly examined or compared the activity of PI3K and ERK in FXS. Here, we quantified the activity of these two major downstream signaling molecules at synapses from Fmr1 KO mice. PI3K assays were performed with p110β, the predominant neuronal isoform of PI3K catalytic subunits, immunoprecipitated from Fmr1 KO and WT SNS. The efficiency of the precipitated p110β protein to convert phosphoinositol (PI) into phosphoinositol-3-phosphate (PI3-P) was assessed by using radiolabeled ATP to quantify PI3-P production on thin-layer chromatographs (Rong et al., 2003). We show that PI3K activity in Fmr1 KO SNS is markedly increased (threefold) compared with WT (Fig. 1 A, n = 8, *p = 0.035, paired t test) (for control experiment, see supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). In contrast, phospho-ERK1/2 immunoprecipitated from Fmr1 KO SNS displayed no difference in the efficiency to phosphorylate recombinant ELK-1 compared with WT SNS (Fig. 1 B, n = 8, p = 0.20, paired t test) (for control experiment, see supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). To confirm these initial findings, we further examined PI3K activity at synapses in cultured WT and Fmr1 KO hippocampal neurons. Accumulation of the PI3K products PI-(3,4)-bisphosphate (PIP2) and PI-(3,4,5)-triphosphate (PIP3) leads to recruitment of PH-domain-containing proteins to sites of PI3K enzymatic activity (Hawkins et al., 2006), e.g., to synaptic membranes. As an in vivo measure to assess PI3K activity at synapses, we analyzed the subcellular distribution of a recombinant PH domain of murine Akt1 fused to red fluorescent protein [RFP–PH(Akt)] in hippocampal WT and Fmr1 KO neurons. In Fmr1 KO neurons, colocalization of RFP–PH(Akt) with the synaptic marker synaptophysin was significantly increased, suggesting elevated synaptic PIP3 levels as a consequence of exaggerated PI3K activity at synapses (Fig. 1 C, n = 5 independent experiments, 6–12 cells each, ***p = 0.00002, paired t test). Total RFP-PH(Akt) levels were not changed in Fmr1 KO neurons compared to WT (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). Excess synaptic PI3K activity was further corroborated by significantly increased phosphorylation of the downstream signaling molecule Akt in SNS from Fmr1 KO mice (Fig. 1 D, n = 6, *p = 0.014, paired t test), whereas ERK phosphorylation was not altered (Fig. 1 E, n = 7, p = 0.18, paired t test). These results indicate that exaggerated gp1 mGluR signaling in FXS does not lead to excess signaling of downstream pathways in general. In contrast, the key downstream signaling molecule PI3K, but not ERK1/2, is specifically overactive at FMRP-deficient synapses.
Figure 1.
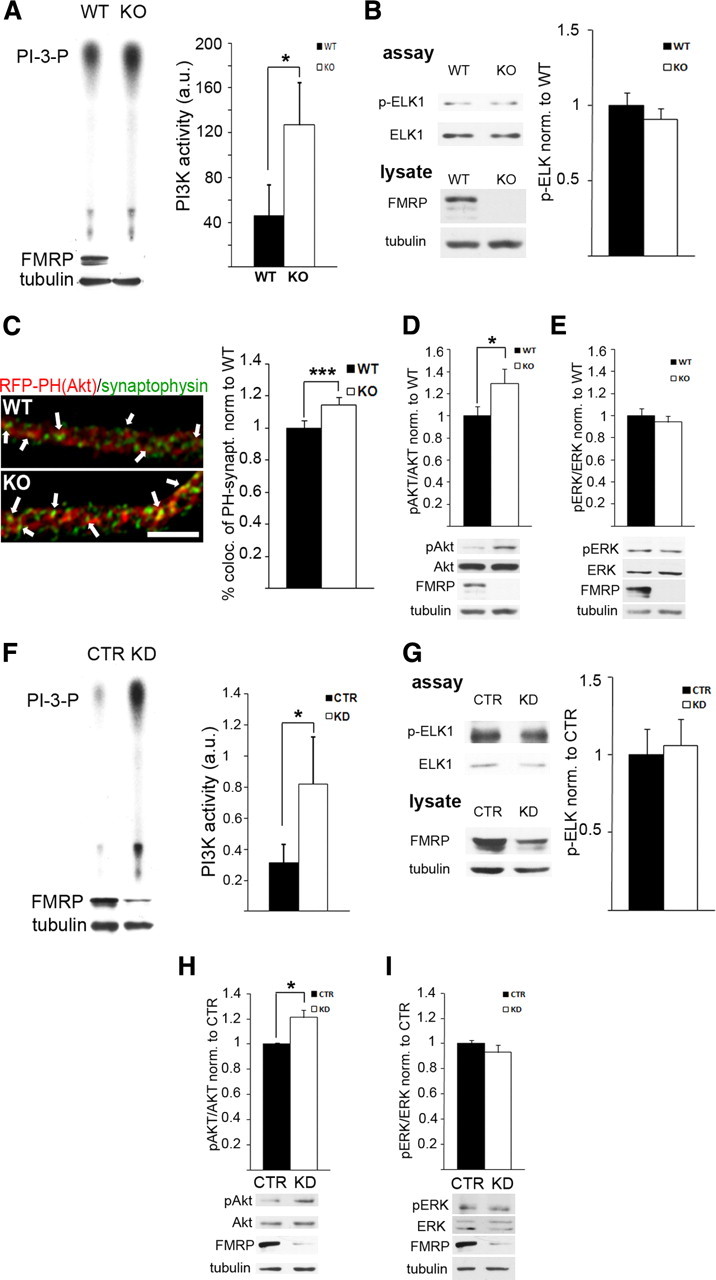
A–E, Exaggerated PI3K activity and signaling at Fmr1 KO synapses. A, PI3K activity is threefold increased in Fmr1 KO SNS compared with WT SNS, as assessed by the amount of radiolabeled PI3-P on autoradiographies from in vitro PI3K assays (n = 8, *p = 0.035, paired t test) (control experiment shown in supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). B, Quantification of phospho-ELK-1 specific Western blots of in vitro ERK1/2 kinase assays from WT and Fmr1 KO SNS using recombinant ELK-1 as a substrate shows no significant change in ERK1/2 activity in the absence of FMRP (n = 8, p = 0.20, paired t test) (control experiment shown in supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). FMRP-specific Western blot analyses of the lysate used as starting material for the assay are shown below; tubulin served as loading control. C, Quantitative analysis of recombinant RFP–PH(Akt) domain colocalized with synaptophysin in hippocampal neurons (9–10 DIV) demonstrates increased synaptic localization of RFP–PH(Akt) in Fmr1 KO neurons, suggesting elevated PI3-P3 levels at Fmr1 KO synapses, which lead to a translocation of the fluorescently labeled PH domain (scale bar, 5 μm) (n = 5 independent experiments, 6–12 cells each, ***p = 0.00002, paired t test). In contrast, total dendritic RFP–PH(Akt) levels were not different between WT and Fmr1 KO neurons (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). D, E, Phosphorylation of the PI3K downstream signaling molecule Akt (D), but not ERK1/2 (E), is significantly increased in Fmr1 KO SNS compared with WT. Phosphorylation levels were assessed by densitometric analysis of Western blots, and phospho-specific signals were normalized to total levels of the respective proteins (D, n = 6, *p = 0.014; E, n = 7, p = 0.18; paired t tests). Representative Western blots are shown below. F–I, PI3K activity and signaling is increased in FMRP-deficient HEK293T cells. In contrast to cortical SNS, HEK293T cells do not express detectable levels of mGluR1 and mGluR5 receptors (Western blot analyses shown in supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). F, G, siRNA-mediated knockdown of Fmr1 in HEK293T cells increases PI3K activity significantly (F, n = 7, *p = 0.039, paired t test), whereas in vitro ERK1/2 assays showed no significant change in activity (G, n = 7, p = 0.32, paired t test). H, I, Quantification of Akt and ERK1/2 phosphorylation after Fmr1 knockdown demonstrate significantly enhanced phospho-Akt levels (H, n = 5, *p = 0.018, paired t test) but not phospho-ERK1/2 levels (I, n = 6, p = 0.29, paired t test). Genotype or siRNA-mediated knockdown was confirmed by Western blotting with an FMRP-specific antibody and a tubulin antibody as loading control (shown below for each experiment). All error bars represent SEM. a.u., Arbitrary unit.
Excess PI3K activity in FMRP-deficient non-neuronal cells in the absence of gp1 mGluRs
To investigate whether excess PI3K activity depends on the presence of gp1 mGluRs, we performed a similar set of experiments in the embryonic kidney cell line HEK293T (Fig. 1 F–I). HEK293T cells express p110β and FMRP but neither of the gp1 mGluRs (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). FMRP expression in these cells was transiently reduced by siRNA-mediated Fmr1 knockdown. Quantification of FMRP levels by Western blot analysis showed a significant decrease of FMRP after 48 h (57.33 ± 4.57%, n = 5, *p = 0.0007, paired t test; FMRP signals were normalized to tubulin signals on the same blot; Western blots are shown for each experiment). As shown above for SNS, we also observed significantly increased PI3K enzymatic activity (Fig. 1 F, 2.6-fold, n = 7, *p = 0.039, paired t test) but not ERK1/2 enzymatic activity (Fig. 1 G, n = 7, p = 0.32, paired t test) in in vitro assays on lysates from HEK293T cells with reduced FMRP expression. Furthermore, Fmr1 knockdown led to elevated phosphorylation of endogenous Akt (Fig. 1 H, n = 5, *p = 0.018, paired t test) but not ERK1/2 (Fig. 1 I, n = 6, p = 0.29, paired t test), similar as shown for SNS (Fig. 1 D,E). Our observation that PI3K activity in FMRP-deficient cells can occur independently of gp1 mGluRs additionally suggests PI3K activity as a novel biomarker in FXS patients.
Dysregulated mGluR-mediated PI3K activity in Fmr1 KO neurons
Antagonists of gp1 mGluR signaling can rescue FXS-associated neuronal phenotypes in animal models (McBride et al., 2005; Yan et al., 2005; Nakamoto et al., 2007; de Vrij et al., 2008). To assess whether gp1 mGluR signaling can regulate excessive PI3K activity at FMRP-deficient synapses, we investigated the effects of different perturbations of mGluR signaling; each showed reduction of PI3K activity in Fmr1 KO. Antagonizing mGluR5-mediated signaling with MPEP had no significant effect on WT but significantly reduced PI3K function in Fmr1 KO (Fig. 2 A, n = 4, two-way ANOVA with least significant difference (LSD) post hoc tests: p wtctr–wtMPEP = 0.327, *p wtctr–koctr = 0.033, *p koctr–koMPEP = 0.012), indicating that inhibition of gp1 mGluRs can correct excessive PI3K activity in the absence of FMRP, which is consistent with the mGluR theory. Synaptic mGluR-dependent induction of PI3K can be regulated by different mechanisms, such as signaling through heterotrimeric G-proteins (Hawkins et al., 2006) or a neuronal complex including mGluR5, the scaffolding protein Homer, and the PI3K enhancer PIKE-L (Rong et al., 2003; Ronesi and Huber, 2008a). To investigate whether excess PI3K activity in Fmr1 KO SNS is regulated by the mGluR–Homer–PIKE signaling complex, we used two different approaches to interfere with this complex: perturbation of mGluR5–Homer interactions with a peptide comprising the C-terminal sequence of mGluR5 (Mao et al., 2005; Ronesi and Huber, 2008a) (Fig. 2 B) and short-hairpin RNA-mediated knockdown of PIKE-L in cultured cortical neurons (Fig. 2 C). Perturbation of mGluR5–Homer associations reduced PI3K activity under basal conditions independently of genotype, although PI3K activity was still increased in Fmr1 KO compared with WT SNS. Quantification of PI3K assays demonstrated a significant effect of the C-terminal peptide, which reduced PI3K activity in both WT and Fmr1 KO (Fig. 2 B). In line with our previous results (Fig. 1 A,F), a significant effect of genotype was also observed, with increased PI3K activity in Fmr1 KO SNS compared with WT regardless of the presence of the C-terminal peptide [Fig. 2 B, n = 4, two-way ANOVA, significant effects of treatment (*p < 0.001) and genotype (*p = 0.014) but no significant interaction between genotype and treatment (p = 0.143)] (additional analyses shown in supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). Of interest, quantification of shRNA-mediated PIKE reduction showed a significant interaction between genotype and treatment to reduce the excess PI3K activity observed in Fmr1 KO. In PIKE-depleted Fmr1 KO neurons, there was a significant decrease in PI3K activity (54.5 ± 9%) but only a slight decrease in PIKE-depleted WT neurons (93 ± 3%) [Fig. 2 C, n = 3, two-way ANOVA with Tukey's honestly significant difference (HSD) post hoc tests: *p wtctr–koctr = 0.002, *p koctr–koPIKE = 0.007]. However, PI3K activity is still increased in Fmr1 KO compared with WT after knockdown of PIKE (139 ± 14% of WT), suggesting an additional downstream defect leading to excess PI3K activity in Fmr1 KO.
We hypothesize that excess PI3K activity in Fmr1 KO is attributable to elevated translation of FMRP-associated mRNAs that encode PI3K subunits or modulating proteins. Of interest, PIKE was identified as an FMRP target mRNA in a high-throughput screen (J. Darnell, personal communication), and here, we show by quantitative FMRP-specific coimmunoprecipitation from brain lysates that PIKE-L mRNA specifically associates with FMRP (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). A previous study has recently reported elevated levels of PIKE-S (short isoform of PIKE) in hippocampal lysates from Fmr1 KO neurons and also suggested increased levels of both PIKE-S and PIKE-L in postsynaptic densities from Fmr1 KO (Sharma et al., 2010). In line with this previous observation, here we could show that PIKE-L protein levels are significantly enriched in SNS from Fmr1 KO (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material). Additional work is needed to assess whether possible dysregulation of PIKE-L mRNA translation in the absence of FMRP may contribute to excess PI3K activity in Fmr1 KO neurons. Nonetheless, because PI3K activity is exaggerated in FMRP-deficient non-neuronal cells, which express neither gp1 mGluRs (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material) nor PIKE-L (our own data not shown and as shown in the study by Rong et al., 2003), our results imply that excess PI3K activity in Fmr1 KO might be attributed to downstream defects within the pathway.
gp1 mGluR-mediated activation of synaptic translation was shown to be occluded in Fmr1 KO (Muddashetty et al., 2007). Because PI3K is necessary for mGluR induction of protein synthesis, we investigated whether mGluR-induced PI3K signaling was also dysregulated in the absence of FMRP and might thus underlie dysregulated translation. We analyzed PI3K activity after stimulation of gp1 mGluRs with DHPG in WT and Fmr1 KO SNS. DHPG treatment led to a twofold increase of PI3K enzymatic activity in WT SNS (Fig. 2 D, n = 4, *p = 0.033, paired t test) but failed to activate PI3K in KO SNS (Fig. 2 E). Based on our findings that PI3K activity is also increased in the absence of gp1 mGluRs, this suggests that excess synaptic PI3K activity in Fmr1 KO might not be further inducible by gp1 mGluR stimulation and thus might underlie the loss of gp1 mGluR-induced protein synthesis. This finding is consistent with a previous report showing that DHPG does not induce the phosphorylation of PDK1, which is also induced by PI3K, in hippocampal slices from Fmr1 KO mice (Ronesi and Huber, 2008a). Of interest, we observe a significant reduction of PI3K activity in Fmr1 KO SNS during DHPG stimulation (Fig. 2 E, n = 4, *p = 0.049, paired t test). The significant reduction in PI3K activity observed in Fmr1 KO SNS could be attributable to agonist-mediated mGluR5 desensitization, which was shown to decrease PI3K activity (Peavy et al., 2002). Collectively, our findings indicate that mGluR-mediated control of PI3K activity is dysregulated in the absence of FMRP.
Increased p110β protein levels at Fmr1 KO synapses
p110β is the catalytic subunit of PI3K and a putative FMRP-target mRNA (Miyashiro et al., 2003). We investigated whether p110β expression was dysregulated in Fmr1 KO, which could contribute to the excess PI3K activity. Synaptic p110β protein levels in cultured hippocampal neurons were analyzed by quantifying p110β colocalization with the synaptic marker synaptophysin (Fig. 3 A–E, specificity of the antibody was verified by siRNA-mediated knockdown of FMRP in HEK293T cells, shown in supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). In both WT and Fmr1 KO neurons, p110β showed a punctate distribution within dendrites and colocalized with synaptophysin, which is consistent with its known recruitment to active signaling complexes at synapses (Fig. 3 A,B). Quantitative analyses of distal dendrites (>50 μm from cell body) demonstrated significantly increased synaptic localization of p110β protein in Fmr1 KO neurons (Fig. 3 C, WT, 27% overlap; KO, 34.9% overlap; n = 43 dendrites each for WT and KO, 3 independent hippocampal cultures, **p = 0.008, independent t test), whereas total synaptophysin levels were unchanged (Fig. 3 D, n = 43, p = 0.73, independent t test). Furthermore, the Mander's coefficient for p110β but not synaptophysin is significantly higher in Fmr1 KO dendrites (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material), indicating that the percentage of p110β signal overlapping with synaptophysin is increased in the absence of FMRP. Using three-dimensional reconstruction, we analyzed p110β protein levels within synaptophysin punctae, here referred to as “synapses.” Although the mean intensity of p110β within synapses was increased (Fig. 3 E, n = 43, **p = 0.004, independent t test), the relative number of p110β-positive synapses was unchanged (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material), implying that there is no imbalance between synapses recruiting PI3K versus synapses not recruiting PI3K in Fmr1 KO neurons. Additional quantitative analyses of immunostainings in hippocampal neurons showed that total dendritic p110β levels are also increased in the absence of FMRP (supplemental Fig. S3D, available at www.jneurosci.org as supplemental material).
Enhanced basal p110β expression and mRNA translation in the absence of FMRP
Western blot analyses of cortical SNS corroborated the above immunocytochemical data by showing that p110β levels were significantly increased in Fmr1 KO (Fig. 3 F, n = 5, *p = 0.042, paired t test). This is in line with a previous report that showed increased p110β levels in total hippocampal lysates from Fmr1 KO mice but did not analyze synaptic levels (Sharma et al., 2010). Apart from increased synaptic levels in Fmr1 KO mice, we could also detect increased p110β protein levels in HEK293T cells after siRNA-mediated knockdown of FMRP (Fig. 3 G, n = 5, *p = 0.005, paired t test), further suggesting that gp1 mGluRs are not necessary for excess p110β expression in the absence of FMRP.
As mentioned above, a previous report suggested that p110β mRNA might be a target mRNA of FMRP, although this could not be validated using specific in vitro assays (Miyashiro et al., 2003). Here, we demonstrate that FMRP associates with p110β mRNA in vivo using FMRP-specific coimmunoprecipitation from brain lysate followed by qRT-PCR analyses (Fig. 3 H, n = 6, two-way ANOVA with Tukey's HSD post hoc analyses: *p p110 β = 0.003, p NR1 = 0.884). Furthermore, RNA pulldowns in HEK293T cells using recombinant p110β mRNA and FMRP demonstrated that the 3′ UTR of p110β is sufficient to mediate the association of p110β mRNA with FMRP in vitro (Fig. 3 I, n = 4, two-way ANOVA with Tukey's HSD post hoc analyses: *p p110 β = 0.001, p β -actin = 0.798).
The role of FMRP in p110β mRNA translation has not been studied previously. To investigate whether p110β translation was regulated by FMRP, we quantified translation levels in SNS by analyzing p110β mRNA distribution in polysomal fractions of sucrose gradients [as described previously (Muddashetty et al., 2007); additional analyses of polysomal gradients in cortical SNS shown in supplemental Fig. S3E–G, available at www.jneurosci.org as supplemental material]. The association of p110β mRNA with synaptoneurosomal polysomes is sensitive to puromycin treatment, which disrupts translocating ribosomes, indicating that it is actively translated (Fig. 3 J, n = 3, *p = 0.033, paired t test). In Fmr1 KO SNS, the association of p110β mRNA with heavy polysomes is significantly increased compared with WT (Fig. 3 K, n = 5, *p PSD95 = 0.029, p NR1 = 0.89, *p 110 β = 0.043, paired t tests), suggesting enhanced basal translation of p110β mRNA in the absence of FMRP. In contrast, total p110β mRNA levels were not altered in Fmr1 KO cortical lysates (supplemental Fig. S3H, available at www.jneurosci.org as supplemental material). Apart from these results in mouse cortical synaptoneurosomes, qRT-PCR analyses of polysomal fractions from HEK293T cells after siRNA-mediated reduction of FMRP also demonstrate that association of p110β mRNA with puromycin-sensitive polysomes is significantly elevated (Fig. 3 L, two-way ANOVA with Tukey's HSD post hoc analyses: *p ctruntr–ctrpuro = 0.002, *p ctruntr–KDuntr = 0.001, *p KDuntr–KDpuro < 0.001). Our findings in HEK293T cells support our hypothesis that gp1 mGluRs or other neuron-specific receptors might not be driving the dysregulated p110β mRNA translation and protein expression in FMRP-deficient cells. Of note, the effects on p110β protein levels and translation are larger after transient FMRP reduction in HEK293T cells than in the Fmr1 KO SNS (protein levels: 2.5-fold increase in cells vs 20% increase in SNS; polysomal RNA: 39% increase in cells vs 28% in SNS). This might be attributable to additional regulation of p110β expression at cortical synapses and/or compensatory effects in the total Fmr1 KO compared with an acute knockdown of FMRP in HEK293T cells. Nonetheless, our results suggest that excessive translation of the PI3K catalytic subunit p110β in the absence of FMRP leads to increased p110β protein levels at synapses and could therefore contribute to aberrant and dysregulated synaptic PI3K activity. Fluorescent in situ hybridization analyses in cortical and hippocampal mouse brain sections also showed that p110β mRNA can be localized to dendrites in WT and Fmr1 KO (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), consistent with our observations of FMRP-mediated translational regulation of p110β mRNA in synaptic fractions.
gp1 mGluR-dependent increase of p110β protein expression is occluded at Fmr1 KO synapses
Stimulation of gp1 mGluRs leads to activation of PI3K signaling at synapses by recruiting catalytic p110β molecules into receptor protein complexes at the membrane (Rong et al., 2003; Hawkins et al., 2006). FMRP has been shown to play a crucial role for the gp1 mGluR-induced protein synthesis (Muddashetty et al., 2007). We therefore analyzed whether, besides recruitment of existing p110β protein into receptor protein complexes, gp1 mGluR signaling could additionally lead to increased p110β mRNA translation, protein expression, and subsequently elevated p110β protein levels at synapses. Treatment of WT SNS with the gp1 mGluR agonist DHPG for 15 min led to increased p110β protein levels (Fig. 4 A, n = 6, *p = 0.008, paired t test) and induced a significant shift of p110β mRNA into puromycin-sensitive fractions (Fig. 4 B, n = 6, *p = 0.028, paired t test). In contrast, in Fmr1 KO SNS, gp1 mGluR-mediated increase in p110β protein expression (Fig. 4 C) or translation (Fig. 4 D) were occluded (C: n = 5, p = 0.613; D: n = 3, p = 0.21; paired t tests). The loss of sensitivity of p110β mRNA translation and protein expression to gp1 mGluR stimulation in the absence of FMRP parallels the loss of PI3K activation during gp1 mGluR stimulation observed in Fmr1 KO SNS (Fig. 2 E). We thus suggest that excess p110β mRNA translation, protein synthesis, and consequently activity under basal conditions in Fmr1 KO might occlude gp1 mGluR-induced PI3K activation that is necessary to stimulate protein synthesis.
Figure 4.

Dysregulated gp1 mGluR-dependent p110β expression in Fmr1 KO. A, B, DHPG treatment (15 min, 50 μm) increases p110β protein levels in WT SNS (A, normalized to control; n = 6, *p = 0.008, paired t test) and leads to a shift of p110β mRNA into puromycin-sensitive fractions (B, percentage of total mRNA in polysomal fractions; n = 6, *p = 0.028, paired t test). C, D, In Fmr1 KO SNS, DHPG treatment does not increase protein expression (C, normalized to control, n = 5, p = 0.613, paired t test) or enhance association of p110β mRNA with puromycin-sensitive polysomes (D, percentage of total mRNA in polysomal fractions, n = 3, p = 0.21, paired t test).
PI3K inhibitors rescue increased and dysregulated protein synthesis at Fmr1 KO synapses
A prominent phenotype of Fmr1 KO mice is excess and dysregulated protein synthesis (Dolen et al., 2007; Muddashetty et al., 2007), which is hypothesized to underlie many neuronal defects observed in FXS. PI3K activates mTOR and cap-dependent translation (Costa-Mattioli et al., 2009) and is therefore an important upstream regulator of protein synthesis. To examine whether excessive PI3K signaling might cause dysregulated translation in Fmr1 KO, we used metabolic radiolabeling in cortical SNS to quantify the effects of different signal transduction agonists on translation rates. We observed a significant increase of overall synaptic protein synthesis rates in Fmr1 KO SNS compared with WT (Fig. 5 A, n = 6, *p = 0.03, paired t test), which was similar to the effects observed previously in brain slices from Fmr1 KO mice (Dolen et al., 2007). Preincubation of Fmr1 KO SNS with inhibitors of either mGluR5 (MPEP) (Fig. 5 B) or mGluR1 (LY367385) (Fig. 5 C) reduced synaptic Fmr1 KO translation to WT levels, indicating that increased translation rates were modulated by gp1 mGluR-dependent signaling pathways (Fig. 5 B,C, two-way ANOVAs; B, MPEP, n = 8, *p wtuntr–kountr = 0.043, *p kountr–koMPEP = 0.002; C, LY367385, n = 5, *p wtuntr–kountr = 0.04, *p kountr-koLY367 = 0.007; Tukey's HSD post hoc tests). This is in line with our results showing that antagonizing gp1 mGluR signaling significantly reduces activity of PI3K, a crucial regulator of protein synthesis, in Fmr1 KO SNS (Fig. 2 A–C). In contrast, an NMDA receptor-specific antagonist (APV), failed to reduce augmented translation in KO (Fig. 5 D, n = 3, two-way-ANOVA: no significant interaction between genotype and treatment, p = 0.782) (also see supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material), further corroborating that excessive synaptic translation in Fmr1 KO is attributable to exaggerated signaling downstream of gp1 mGluRs. Similar to the effect of gp1 mGluR antagonists (Fig. 5 B,C), antagonizing PI3K signaling with two different drugs, LY294002 (Fig. 5 E) and wortmannin (Fig. 5 F), could specifically rescue the excessive synaptic translation in Fmr1 KO (Fig. 5 E, LY294002, n = 6, *p wtuntr–kountr = 0.001, *p kountr–koLY294 = 0.026; Fig. 5 F, wortmannin, n = 6, *p wtuntr–kountr < 0.001, *p kountr–kowort = 0.002; two-way ANOVAs with Tukey's HSD post hoc tests), whereas an inactive analog of LY294002 (LY303511) did not show any effect (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). In contrast, a specific antagonist of ERK1/2 signaling (U0126) reduced protein synthesis levels independently of genotype to the same extent [Fig. 5 G, n = 5, two-way ANOVA: significant effects of treatment (*p = 0.001) and genotype (*p = 0.001) but no significant interaction between genotype and treatment (p = 0.68)] (also see supplemental Fig. S5D, available at www.jneurosci.org as supplemental material; WT, 0.89 ± 0.05; KO, 0.88 ± 0.03), and ratios of KO to WT translation rates were the same before and after U0126 treatment (supplemental Fig. S5E, available at www.jneurosci.org as supplemental material; n = 5, control, 1.21 ± 0.04 and U0126, 1.22 ± 0.12). Together, our results suggest that excess PI3K activity causes excess basal translation in FXS and might therefore underlie the dysregulated basal and stimulus-induced protein synthesis in the absence of FMRP. Furthermore, treatment with rapamycin, which inhibits the PI3K downstream signaling molecule mTOR, significantly reduced translation in KO but not in WT, supporting the hypothesis that PI3K signaling is enhanced in the absence of FMRP (supplemental Fig. S5F, available at www.jneurosci.org as supplemental material). This result is also in line with a previous study reporting increased phosphorylation and activity of mTOR in the absence of FMRP (Sharma et al., 2010). Interestingly, this study showed that gp1 mGluR-induced mTOR phosphorylation, which is absent in Fmr1 KO, could be restored with a PI3K inhibitor. Here, we show that the PI3K antagonist wortmannin rescues the translational response of Fmr1 KO SNS to gp1 mGluR stimulation, whereas treatment with an ERK1/2 antagonist did not rescue DHPG-stimulated translation in the absence of FMRP (Fig. 5 H; wortmannin: n = 4, one-way ANOVA, *p = 0.012, Tukey's post hoc tests: *p untrctr–wortctr = 0.045, *p wortctr–wortDHPG = 0.010; U0126: n = 4, one-way ANOVA, *p = 0.001, Tukey's post hoc tests: *p untrctr–U0126ctr/DHPG = 0.006/0.001, *p untrDHPG–U0126ctr/DHPG = 0.019/0.004). In WT, inhibitors of either ERK1/2 or PI3K abolished DHPG-induced translational increase (Fig. 5 I; wortmannin: n = 4, one-way ANOVA, *p = 0.002, Tukey's post hoc tests: p untrctr–untrDHPG = 0.006, *p wortctr–wortDHPG = 0.004; U0126: n = 3, one-way ANOVA, *p < 0.001, Tukey's post hoc tests: *p untrctr–untrDHPG = 0.029, *p untrctr/DHPG–U0126DHPG = 0.022/0.001). These data further corroborate that both ERK and PI3K/mTOR pathways are crucial to activate translation in normal brain, yet impaired signaling through PI3K/mTOR underlies the dysregulated translation in FXS.
Figure 5.

PI3K antagonists rescue dysregulated basal and stimulus-induced synaptic translation rates in Fmr1 KO SNS. A, Basal translation in cortical Fmr1 KO SNS is increased ∼30% compared with WT. Translation rates were analyzed by metabolic labeling of SNS with [35S]methionine, and radioactivity incorporation during a 5 min time period was quantified (n = 6, *p = 0.03, paired t test). B, C, Pretreatment of SNS with an mGluR5 antagonist (B: MPEP, 10 μm) or an mGluR1 antagonist (C: LY367385, 10 μm) significantly decreased translation rates in Fmr1 KO but not in WT SNS (MPEP: n = 8, *p wt–ko untreated = 0.043, *p ko untreated–treated = 0.002; LY367385: n = 5, *p wt–ko untreated = 0.04, *p ko untreated–treated = 0.007; Tukey's HSD post hoc tests; two-way ANOVAs detect significant interaction between genotype and treatment; MPEP: *p < 0.001; LY367385: *p < 0.001). D, Treatment with a specific NMDAR antagonist (APV, 50 μm) affected translation similarly across genotypes (n = 3; two-way ANOVA: no significant interaction between genotype and treatment; p = 0.782) (also see supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). E, F, Treatment of SNS with two different PI3K antagonists, LY294002 (E; 50 μm) and wortmannin (F; 100 nm), significantly reduced amino acid incorporation rates in Fmr1 KO but not in WT (n = 6; E, LY294002: *p wt–ko untreated = 0.001, *p ko treated–untreated = 0.026; F, wortmannin: *p wt–ko untreated < 0.001, *p ko treated–untreated = 0.002; Tukey's HSD post hoc tests; two-way ANOVA: significant interaction between genotype and treatment; LY294002: *p < 0.001; wortmannin: *p < 0.001). LY303511, an inactive analog of LY294002, did not alter translation rates (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). G, An ERK1/2 antagonist (U0126, 20 μm) did not show a genotype-specific effect on translation rates [n = 5, two-way ANOVA: significant effects of treatment (*p = 0.001) and genotype (*p = 0.001) but no significant interaction between genotype and treatment (p = 0.68)] (also see supplemental Fig. S5D,E, available at www.jneurosci.org as supplemental material). H, DHPG-induced translational activation in Fmr1 KO SNS is occluded and can be rescued by antagonizing PI3K signaling (wortmannin) but not by ERK1/2 inhibition (U0126) (wortmannin: n = 4, one-way ANOVA, *p = 0.012, Tukey's HSD post hoc tests: *p untrctr–wortctr = 0.045, p wortctr–wortDHPG = 0.010; U0126: n = 4, one-way ANOVA, *p = 0.001, Tukey's HSD post hoc tests: *p untrctr–untrDHPG = 0.029, *p untrctr–U0126ctr/DHPG = 0.006/0.001, *p untrDHPG–U0126ctr/DHPG = 0.019/0.004). I, Inhibitors of PI3K signaling (Wort) and ERK1/2 signaling (U0126) abolish DHPG-induced (15 min, 100 μm) translational activation in WT SNS (wortmannin: n = 4, one-way ANOVA, *p = 0.002, Tukey's HSD post hoc tests: *p untrctr–untrDHPG = 0.006, *p wortctr–wortDHPG = 0.004; U0126: n = 3, one-way ANOVA, *p < 0.001, Tukey's HSD post hoc tests: *p untrctr–untrDHPG = 0.029, *p untrctr/DHPG–U0126DHPG = 0.022/0.001). Results were normalized to control. All error bars represent SEM.
PI3K inhibition corrects aberrant AMPA receptor internalization and increased protrusion density in Fmr1 KO neurons
The observation that PI3K inhibitors, but not ERK1/2 inhibitors, could rescue dysregulated protein synthesis, a fundamental phenotype in FXS believed to underlie neuronal deficits observed in animal models as well as in patients, led us to further investigate the therapeutic potential of PI3K antagonists for FXS-associated defects. We analyzed the effect of the PI3K antagonist LY294002 on two important synaptic phenotypes in Fmr1 KO neurons: (1) aberrant AMPA receptor internalization (Nakamoto et al., 2007) and (2) increased protrusion density in Fmr1 KO dendrites (Antar et al., 2006; de Vrij et al., 2008).
We have shown previously that siRNA-mediated FMRP deficiency leads to aberrant internalization of AMPA receptors in rat hippocampal neurons and that the mGluR5 antagonist MPEP rescued increased levels of internalized AMPARs (Nakamoto et al., 2007). Here, we investigated whether reduction of excess PI3K activity could also correct this phenotype. First, we showed that, in line with our observations in the rat hippocampal neuron knockdown assay, Fmr1 KO mouse hippocampal neurons similarly display excessive constitutive AMPAR internalization compared with WT neurons (Fig. 6 A,B). Treatment with the PI3K inhibitor LY294002 (50 μm, 1 h) reduced AMPAR endocytosis in Fmr1 KO neurons to WT levels (Fig. 6 A–C, n = 30, Bonferroni's t tests, *p wtctr–koctr < 0.0001, **p koctr–KO_LY294002 < 0.00001). Notably, in WT neurons, FMRP expression levels in dendrites correlated with AMPAR internalization, and antagonizing PI3K had an effect on AMPAR surface expression only in neurons with low dendritic expression of FMRP, i.e., high levels of internalized AMPAR (Fig. 6 C). As observed for the rescue of excess and dysregulated translation (see above), the effect was specific for PI3K antagonists, whereas the ERK1/2 inhibitor U0126 (20 μm, 1 h) did not rescue aberrant AMPAR internalization in Fmr1 KO neurons (Fig. 6 D,E, n = 30, Bonferroni's t test, ***p < 0.0001).
Figure 6.

A PI3K antagonist rescues increased GluR1 endocytosis in Fmr1 KO neurons. A, Representative images illustrate surface and internalized GluR1 staining in WT and Fmr1 KO primary hippocampal neurons under control conditions and after treatment with the PI3K inhibitor LY294002. Scale bar, 50 μm. B, Box-and-whisker plot of the distribution of constitutive endocytosis of AMPARs in distal dendrites shows that enhanced constitutive endocytosis of AMPARs in Fmr1 KO neurons is corrected by application with the PI3K inhibitor LY294002 (50 μm for 1 h; median: WT, 35.7; KO, 44.5; WT + LY294002, 35.3; KO + LY294002, 35.6; n = 30 each; one-way ANOVA with Bonferroni's post hoc tests: *p < 0.0001, **p < 0.00001). C, Scatter plot of correlations between FMRP signals and endocytosis of AMPARs in distal dendrites shows that AMPAR internalization in Fmr1 KO is reduced by LY294002 (see red symbols, compressed on left side). Scatter plot of signals in each distal dendrite of WT neurons show substantial variation of FMRP signals. Note however that the enhanced endocytosis of AMPARs, detected if FMRP signals are relatively low, is also corrected with LY294002 application. D, Box-and-whisker plot of the distribution of constitutive endocytosis of AMPARs in distal dendrites show that enhanced constitutive endocytosis of AMPARs in Fmr1 KO neurons is not affected by application of the ERK1/2 inhibitor U0126 (20 μm for 1 h; median: WT, 40.2; KO, 50.5; WT + U0126, 43.8; KO + U0126, 53.0; n = 30 each; one-way ANOVA with Bonferroni's post hoc test: ***p < 0.0001). E, Scatter plot of correlations between FMRP signals and endocytosis of AMPARs in distal dendrites show substantial variation of FMRP signals in WT. Note that the enhanced endocytosis of AMPARs, detected if FMRP signals are relatively low, is not affected with U0126 application.
A prominent feature of FMRP-deficient neurons in humans (Irwin et al., 2001) and mice (Comery et al., 1997) is altered dendritic spine morphology, including higher density, and immature appearance of spines, which can be corrected by genetic or pharmacologic reduction of mGluR5 signaling (Dolen et al., 2007; de Vrij et al., 2008). Using automated software (FilamentTracer in Imaris; Bitplane), we analyzed the overall protrusion density in WT and Fmr1 KO hippocampal neurons in vitro after a 3 d treatment with the PI3K inhibitor LY294002 (10 μm, 3d) (Fig. 7 A–C). To assess a potential value for a therapeutic strategy in FXS patients, a prolonged incubation with low doses of the inhibitor, as opposed to higher doses of shorter duration, was used to minimize effects on WT morphology, as well as to reduce possible side effects on neuronal function in WT and KO neurons. To visualize spines, cells were transfected with GFP–Lifeact, a small peptide that binds F-actin without altering actin morphology or dynamics (Riedl et al., 2008; Era et al., 2009; Vidali et al., 2009). We detected a significant increase in dendritic protrusions (including spines and filopodia) in Fmr1 KO neurons, and antagonizing PI3K signaling with LY294002 could rescue the elevated protrusion density observed in Fmr1 KO neurons (Fig. 7 B). In contrast, LY294002 had no effect on WT neurons (Fig. 7 B, n = 30, LSD post hoc tests, *p wtctr–koctr = 0.024, ***p koctr–koLY294 < 0.0001, p wtctr–wtLY294 = 0.996). These results indicate that exaggerated PI3K activity at synapses may underlie gp1 mGluR-dependent deficits in synapse morphology in the absence of FMRP.
Discussion
Here, we report a novel function for FMRP to control the mRNA translation, synaptic localization, and enzymatic activity of the catalytic PI3K subunit p110β. These findings have three significant implications. First, this provides an initial indication toward a molecular mechanism underlying exaggerated signaling and dysregulated protein synthesis in FXS. In the absence of FMRP, PI3K is overactive at synapses, leading to excessive protein synthesis and occlusion of agonist-induced protein synthesis. Second, these data broaden the mGluR theory of FXS, which posits that FMRP acts as a brake on mGluR signaling regulating protein synthesis. We expand this hypothesis by showing that FMRP restrains the common downstream signaling molecule PI3K, which may explain features of dysregulated translation mediated by other neurotransmitters. Finally, antagonists of PI3K, a number of which are currently being evaluated in clinical oncology trials, may thus be considered as a potential therapeutic modality for FXS.
Excessive PI3K activity and increased synaptic p110β levels in the absence of FMRP may underlie exaggerated mGluR-mediated protein synthesis
The mGluR theory of FXS proposes that many FXS-related phenotypes in mice and, by extension, human patients originate in exaggerated signaling through gp1 mGluRs (Huber et al., 2002; Bear et al., 2004). However, the underlying molecular mechanisms have not been revealed so far. Here, we show that FMRP deficiency results in excessive basal PI3K activity at synapses (Fig. 1 A,C), which we propose represents the major cause of the exaggerated mGluR-mediated protein synthesis in FXS. We hypothesize that elevated synaptic p110β protein levels in the absence of FMRP increase the amount of p110β subunits recruited into gp1 mGluR signaling complexes at synapses under basal conditions and thus contribute to excess PI3K activity resulting in exaggerated mGluR signaling and increased basal protein synthesis in FXS (Fig. 8). This hypothesis is supported by our data showing that excessive PI3K activity in the absence of FMRP is modulated but not driven or caused by overactive gp1 mGluRs. We show that excess PI3K activity in the absence of FMRP can occur independently of gp1 mGluRs (Fig. 1 F). Furthermore, our results demonstrate that perturbation of mGluR–Homer–PIKE signaling decreases PI3K activity in both WT and KO but cannot reduce the observed exaggerated PI3K activity in Fmr1 KO to WT levels (Fig. 2 B). We speculate that, during blockage of the mGluR5–Homer–PIKE pathway in Fmr1 KO, excess p110β protein can still be activated via small G-proteins, a known alternative pathway mediating gp1 mGluR-induced activation of PI3K (Hawkins et al., 2006) (Fig. 8). We speculate that this alternative pathway may be sufficient to induce LTD in Fmr1 KO during blockage of Homer signaling (Ronesi and Huber, 2008a) because of excess PI3K. However, mGluR–Homer signaling is necessary for LTD in WT because PI3K activity is limited by FMRP.
Figure 8.
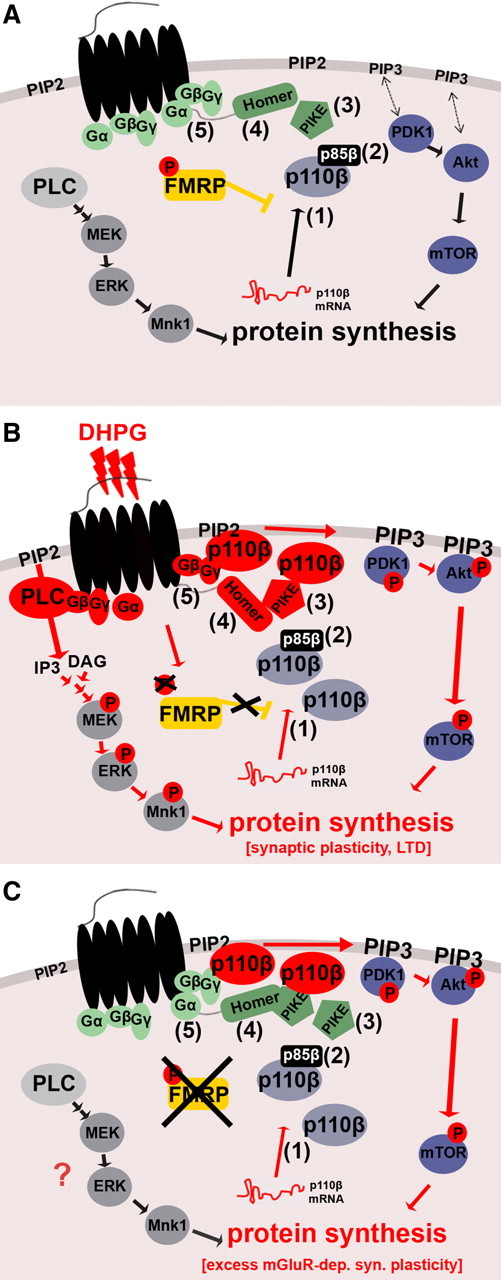
Proposed model for dysregulated mGluR signaling in FXS. A, B Regulation of the gp1 mGluR-dependent signal pathways PLC/ERK and PI3K/mTOR in WT. A, Under basal conditions, FMRP puts the break on PI3K activity. This is partially attributable to FMRP-mediated repression of p110β mRNA translation and synaptic localization of the catalytic subunit p110β (1). Additional mechanisms might include regulation of p110β-modulating subunits such as p85β (2) and PIKE (3) by FMRP. B, gp1 mGluR-mediated activation of PLC and PI3K pathways at the synapse. PLC and PI3K share the same substrate PIP2 in the membrane to produce either IP3 and DAG, or PIP3, respectively. The PLC product DAG can activate PKC, leading to induction of the mitogen-activate protein kinase kinase (MEK)/ERK pathway, whereas the PI3K product PIP3 recruits PH-containing kinases PDK1 and Akt to the membrane, thereby inducing their phosphorylation followed by activation of downstream signaling molecules including mTOR. Both pathways induce protein synthesis. During gp1 mGluR stimulation, PLC is activated by small G-proteins. PI3K was shown to be activated by at least two different mechanisms, the Homer–PIKE complex (4) and small G-proteins (5). Furthermore, gp1 mGluR stimulation leads to transient removal of FMRP-mediated translational inhibition by dephosphorylation of FMRP (Ceman et al., 2003; Narayanan et al., 2007, 2008). We hypothesize that, during this time window, synapses experience a twofold “boost” of PI3K activity composed of newly synthesized catalytic p110β subunits (1) as well as activation of preexisting and newly synthesized PI3K subunit molecules via PIKE and G-proteins (4 and 5). Together, enhanced ERK and PI3K activity lead to increased synaptic protein synthesis. C, The “molecular brake” FMRP is absent in Fmr1 KO, and FMRP-mediated inhibition of p110β translation and PI3K activity is removed constitutively. Increased p110β protein levels at synapses (1), which can be activated by basal levels of mGluR signaling (4 and 5), contribute to exaggerated PI3K signaling. Additionally, PI3K activity could be increased by dysregulation of p110β-modulating subunits (2 and 3), especially PIKE, in the absence of FMRP. DHPG-mediated transient increase in p110β protein expression is abolished and may partially account for loss of gp1 mGluR-dependent activation of PI3K signaling. Loss of this combined “brake” on PI3K signaling in FXS would elevate PI3K-dependent protein synthesis to a saturated level, which cannot be further increased by gp1 mGluR stimulation.
Finally, our analysis of synaptic p110β localization in cultured Fmr1 KO neurons demonstrates that p110β intensity within synaptophysin punctae is increased in Fmr1 KO, whereas the relative number of p110β-positive synapses was unchanged (Fig. 3 E) (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). These data indicate that, in the absence of FMRP, the population of synapses recruiting p110β into activated receptor complexes is unchanged but that, because of increased p110β protein levels in dendrites (supplemental Fig. S3D, available at www.jneurosci.org as supplemental material), more p110β subunits are translocated into these p110β-recruiting receptor complexes. This suggests that the number of synapses containing actively signaling gp1 mGluRs is not increased in FMRP-deficient neurons and is in line with the hypothesis that excess PI3K activity at FXS synapses is not caused by generally elevated mGluR activity but rather by increased synaptic p110β expression.
This study provides evidence for a molecular mechanism that could also account for other dysregulated neurotransmitter-mediated signaling pathways in FXS. We speculate that aberrant regulation of p110β provides a molecular explanation for several recent studies in Fmr1 KO mice, which reported dysregulated neuronal plasticity and signal transduction downstream of the mGluR1/5–Homer–PIKE complex (Ronesi and Huber, 2008a; Sharma et al., 2010), of D1 dopamine receptors (Wang et al., 2008), of Gq-proteins (Volk et al., 2007), and of Ras (Hu et al., 2008). PI3K regulates all of these pathways. We propose that aberrant signal transduction in FXS is caused by exaggerated p110β protein levels rather than dysregulated upstream receptors leading to PI3K activation. Although upregulated PIKE-L levels (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material) (Sharma et al., 2010) should contribute to excess PI3K activity downstream of gp1 mGluRs (Fig. 2 C), they do not explain dysregulation of other receptor pathways.
Translational regulation of p110β mRNA as a novel mechanism for gp1 mGluR-activated signaling in WT and dysregulated gp1 mGluR signaling in Fmr1 KO
Here, we show that gp1 mGluR stimulation increases mRNA translation and protein levels of the catalytic subunit p110β in WT SNS but not in Fmr1 KO SNS (Fig. 4). This reveals a previously unknown mechanism for gp1 mGluR-mediated stimulation of PI3K activity, suggesting that, in WT, gp1 mGluR signaling increases PI3K activity by at least two different mechanisms: (1) recruitment of preexisting p110β into synaptic receptor protein complexes as shown previously (Rong et al., 2003) and (2) increased synthesis of p110β protein as shown here (Fig. 4 A,B). Furthermore, it suggests that increased p110β mRNA translation contributes to the excess PI3K activity in FXS. Our work provides the first experimental indication that the loss of FMRP leads to altered functional activity of a protein encoded by its target mRNA. Apart from the FMRP target mRNA p110β (suggested by Miyashiro et al., 2003 and shown here), an inhibitory subunit of PI3K, p85β, was identified in a screen for putative FMRP targets (Brown et al., 2001). Furthermore, we show that FMRP associates with PIKE-L mRNA and that PIKE-L protein levels are increased at Fmr1 KO synapses (as shown previously by J. Darnell, personal communication, and Sharma et al., 2010). FMRP might thus regulate PI3K activity at multiple steps (Fig. 8).
PI3K versus ERK signaling in FXS
In neurons, the PI3K/mTOR pathway and the PKC/ERK pathway represent two main targets downstream of gp1 mGluRs driving the activation of protein synthesis (Banko et al., 2006). Our study provides much experimental evidence that basal ERK1/2 activity is not altered in SNS from Fmr1 KO mice, whereas PI3K activity is significantly increased. Of note, two previous studies suggested that loss of FMRP leads to dysregulated ERK1/2 phosphorylation (Hou et al., 2006; Kim et al., 2008); however, neither study directly examined and compared PI3K and ERK1/2 activity. gp1 mGluR activation of ERK1/2 is mediated by PKC, which in turn is activated by phospholipase C (PLC) (Fig. 8 B). PI3K and PLC compete for the same substrate in the membrane, PI(4,5)P2, and excessive PI3K protein at Fmr1 KO synapses could therefore tip the balance between these two pathways toward PI3K signaling. Although our results support the important role played by ERK1/2 for mGluR-stimulated protein synthesis in WT (Fig. 5 G), they indicate that it is not the ERK pathway that is primarily dysregulated in FXS. Nevertheless, this does not rule out possible indirect effects on ERK signaling that vary between different cell types or developmental status and therefore make ERK phosphorylation in lymphocytes, for example, a valid biomarker in FXS patients (Weng et al., 2008). However, we show that interfering with PI3K signaling, but not ERK1/2, rescues dysregulated synaptic translation and corrects exaggerated constitutive internalization of AMPA receptors in Fmr1 KO neurons, supporting the hypothesis that FMRP has a direct effect on PI3K activity but not on ERK1/2.
PI3K antagonists as potential treatment in FXS
A key phenotype in FMRP-deficient neurons is dysregulated synaptic protein synthesis, which results in increased basal protein synthesis in brain slices and at synapses, and loss of neurotransmitter-induced protein synthesis (Weiler et al., 2004; Dolen et al., 2007; Muddashetty et al., 2007) (Fig. 5). We hypothesize that FMRP controls protein synthesis-dependent regulation of synaptic morphology and function through regulation of PI3K signaling. We substantiate this hypothesis by showing that PI3K antagonists can reduce exaggerated basal translation at Fmr1 KO synapses and restore translational responsiveness to mGluR stimulation. Dysregulated synaptic protein synthesis was proposed to cause many, if not all, neuronal defects in FXS (Ronesi and Huber, 2008b). We therefore suggest that reducing PI3K signaling pathways might be a promising therapeutic strategy to treat FXS in humans. In support of this hypothesis, we show that PI3K antagonists can rescue two protein synthesis-dependent and physiologically relevant phenotypes in hippocampal Fmr1 KO neurons: increased number of internalized AMPA receptors (Nakamoto et al., 2007) and excessive dendritic spine density (Grossman et al., 2006). Altered spine morphology is a prominent FXS-associated phenotype, which is not only observed in all animal models for FXS but importantly also in human patients (Irwin et al., 2001). Importantly, increased spine density in Fmr1 KO can be rescued using low doses of LY294002 (10 μm, five times lower than the usual dose) in a 3 d treatment without affecting WT neurons. The absence of any effect on WT neurons after chronic treatment with a PI3K inhibitor over several days corroborates the potential therapeutic value of antagonizing PI3K. Another study detected rescue of spine morphology 4 h after treatment with a very high dose of the mGluR5 inhibitor MPEP (200 μm, 20 times more than the usually used effective dose of 10 μm) (de Vrij et al., 2008), but the therapeutic value of these findings might be questioned because, for treatment in human patients, drugs would have to be taken chronically, and lower doses are generally beneficial to minimize potential side effects and costs.
PI3K inhibitors have been intensively studied as anti-cancer and anti-inflammation drugs, including the development of isoform-specific p110β inhibitors (Crabbe et al., 2007). Thus, targeting p110β directly may be a more precise therapeutic strategy in FXS than the current focus on the upstream gp1 mGluR target.
Footnotes
This work was supported by a postdoctoral fellowship and Conquer Fragile X research grant from the National Fragile X Foundation (C.G.), National Institutes of Health Grants MH085617 (G.J.B.) and HD020521 (S.T.W.), and Fragile X Center Grant 3P30HD024064 (S.T.W., G.J.B.). We thank Dr. Ravi Muddashetty for technical assistance with polysome assays and qRT-PCRs and helpful discussions, Andrew Swanson for technical assistance with microscopes and image processing, Adam Goldberg and Mary Roth for technical assistance, Sharon Swanger for help with colocalization analyses, statistics, and critically reading this manuscript, and all members of the Bassell laboratories for helpful discussions. We are grateful to Jennifer Darnell for providing the 2F5-1 antibody.
References
- Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bear MF, Dölen G, Osterweil E, Nagarajan N. Fragile X: translation in action. Neuropsychopharmacology. 2008;33:84–87. doi: 10.1038/sj.npp.1301610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe T, Welham MJ, Ward SG. The PI3K inhibitor arsenal: choose your weapon! Trends Biochem Sci. 2007;32:450–456. doi: 10.1016/j.tibs.2007.09.001. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Bagni C. Fragile X mental retardation protein control of neuronal mRNA metabolism: Insights into mRNA stability. Mol Cell Neurosci. 2010;43:43–50. doi: 10.1016/j.mcn.2009.09.013. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. Fragile X syndrome: from molecular genetics to therapy. J Med Genet. 2009;46:577–584. doi: 10.1136/jmg.2008.064667. [DOI] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Era A, Tominaga M, Ebine K, Awai C, Saito C, Ishizaki K, Yamato KT, Kohchi T, Nakano A, Ueda T. Application of Lifeact reveals F-actin dynamics in Arabidopsis thaliana and the Liverwort, Marchantia polymorpha . Plant Cell Physiol. 2009;50:1041–1048. doi: 10.1093/pcp/pcp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C. The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol Cell Neurosci. 2007;34:343–354. doi: 10.1016/j.mcn.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Gabel LA, Won S, Kawai H, McKinney M, Tartakoff AM, Fallon JR. Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J Neurosci. 2004;24:10579–10583. doi: 10.1523/JNEUROSCI.2185-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: fragile X syndrome and beyond. J Neurosci. 2006;26:7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimentional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28:7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile x mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Kim SH, Markham JA, Weiler IJ, Greenough WT. Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci U S A. 2008;105:4429–4434. doi: 10.1073/pnas.0800257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer L, Fischer Y. Evidence that FMRP is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci. 2005;25:2741–2752. doi: 10.1523/JNEUROSCI.4360-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104:15537–155342. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, Warren ST. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem. 2008;283:18478–18482. doi: 10.1074/jbc.C800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Peavy RD, Sorensen SD, Conn PJ. Differential regulation of metabotropic glutamate receptor 5-mediated phosphoinositide hydrolysis and extracellular signal-regulated kinase responses by protein kinase C in cultured astrocytes. J Neurochem. 2002;83:110–118. doi: 10.1046/j.1471-4159.2002.01113.x. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008a;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Metabotropic glutamate receptors and fragile X mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 2008b;1:pe6. doi: 10.1126/stke.15pe6. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples gp1 mGluR to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Jang SW, Okada M, Chan CB, Feng Y, Liu Y, Luo SW, Hong Y, Rama N, Xiong WC, Mehlen P, Ye K. Netrin-1 mediates neuronal survival through PIKE-L interaction with the dependence receptor UNC5B. Nat Cell Biol. 2008;10:698–706. doi: 10.1038/ncb1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidali L, Rounds CM, Hepler PK, Bezanilla M. Lifeact-mEGFP reveals a dynamic apical F-actin network in tip growing plant cells. PLoS One. 2009;4:e5744. doi: 10.1371/journal.pone.0005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wu LJ, Kim SS, Lee FJ, Gong B, Toyoda H, Ren M, Shang YZ, Xu H, Liu F, Zhao MG, Zhuo M. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59:634–647. doi: 10.1016/j.neuron.2008.06.027. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci U S A. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Warren ST. Neuroscience: fragile dopamine. Nature. 2008;455:607–608. doi: 10.1038/455607a. [DOI] [PubMed] [Google Scholar]
- Weng N, Weiler IJ, Sumis A, Berry-Kravis E, Greenough WT. Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1253–1257. doi: 10.1002/ajmg.b.30765. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH. PIKE: a nuclear GTPase that enhances PI3Kinase activity and is regulated by protein 4.1N. Cell. 2000;103:919–930. doi: 10.1016/s0092-8674(00)00195-1. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]