

Abstract
Protein misfolding, accumulation, and aggregation characterize many aging-related diseases. Protein aggregates do not accumulate in unstressed cells primarily because of the existence of competent cellular “quality control” machinery. The endoplasmic reticulum (ER) is a major part of this quality control system. Accumulation of misfolded proteins in the ER causes ER stress and activates a signaling pathway called the unfolded protein response (UPR). The UPR limits protein load by upregulating ER chaperones such as Ig binding protein (BiP)/glucose-regulated protein 78 (GRP78) and by attenuating protein translation through eukaryotic initiation factor 2 α (eIF2α) phosphorylation. Acute sleep deprivation (6 h) in young mice leads to induction of the UPR with upregulation of BiP/GRP78 and attenuation of protein translation. We demonstrate here that aging impairs this adaptive response to sleep deprivation. Aged mice do not display an increase in BiP expression with acute sleep deprivation. In addition, there is decreased basal expression of BiP/GRP78 in aged mice. There is a decline in eIF2α phosphorylation in aged mouse cerebral cortex that is associated with higher levels of GADD34 (growth arrest and DNA damage 34) and proapoptotic proteins such as CCAAT/enhancer-binding protein-homologous protein and activated caspase-12, suggesting that young animals possess an efficient ER adaptive response that declines with aging.
Keywords: sleep deprivation, aging, ER stress, UPR, apoptotic signaling, BiP
Introduction
Age-associated declines in cellular functions are attributable in part to a progressive failure of the chaperoning systems (Macario and Conway de Macario, 2002). Protein misfolding, accumulation, and aggregation characterize many aging-related diseases. In many of these diseases, proteins or fragments of proteins convert from their normally soluble forms to insoluble fibrils or plaques that accumulate in a variety of organs, including the liver, brain, or spleen (Tan and Pepys, 1994; Kelly, 1998; Lansbury, 1999; Perutz, 1999). This group of diseases, which typically occur late in life, includes Alzheimer's disease (AD), Parkinson's disease (PD), type II diabetes, and a host of less well known but often equally serious conditions such as fatal familial insomnia (Dobson, 1999). That protein aggregates do not accumulate in unstressed cells is attributable in part to the existence of cellular “quality control” machinery. One major such quality control system is the endoplasmic reticulum (ER). It suppresses the formation of aggregates by ensuring fidelity of transcription and translation, by chaperoning nascent or unfolded proteins, and by selectively degrading improperly folded polypeptides before they can aggregate (Ellgaard et al., 1999). The ER is the site in which all secretory and integral membrane proteins are folded and posttranslationally modified in ATP-dependent chaperone-mediated processes. The ER is also the site of steroid, cholesterol, and lipid biosynthesis, and it is the major signal-transducing organelle in the cell that continuously responds to environmental cues to release calcium (Kaufman, 1999; Lin and Boyce, 2005).
The endoplasmic reticulum is a reticular membranous network that extends throughout the cytoplasm and is contiguous with the nuclear envelope. It can therefore sense and transmit signals that originate in any cellular subcompartment. Perturbations that alter ER homeostasis therefore disrupt folding and lead to the accumulation of unfolded proteins and protein aggregates, which are detrimental to cell survival. These perturbations include disturbances in calcium homeostasis, redox status, elevated secretory protein synthesis, and glucose or energy deprivation. As a consequence, the cell has evolved an adaptive coordinated response to limit accumulation of unfolded proteins in the ER. This signaling pathway is termed the ER stress response or the unfolded protein response (UPR) (for review, see Harding et al., 2002; Zhang and Kaufman, 2004; Schroder and Kaufman, 2005a,b, 2006). On a cellular level, the UPR triggers three kinds of protective cellular responses: (1) upregulation of ER chaperones such as Ig binding protein (BiP)/glucose-regulated protein 78 (GRP78); (2) attenuation of protein translation that is mediated by the serine–threonine kinase PKR-like ER kinase (PERK), which phosphorylates the eukaryotic initiation factor 2 α (eIF2α), thereby reducing translation; and (3) degradation of misfolded proteins by the proteasome by a process called ER-associated degradation (ERAD). These three responses are protective measures to alleviate ER stress; however, excessive and/or prolonged stress leads to a maladaptive response and apoptosis (Szegezdi et al., 2006).
Previous studies from our group demonstrated that sleep deprivation leads to ER stress and induction of the UPR in young mice (Naidoo et al., 2005). We have now extended the study to aged mice to assess the effect of sleep deprivation on the BiP response and UPR in these animals. Aged animals exhibit more fragmented sleep. Aging has also been shown to increase levels of abnormal proteins in cells (Berlett and Stadtman, 1997; Finkel and Holbrook, 2000), and ER chaperones have been shown to be oxidized with age (Rabek et al., 2003). We hypothesized that both the sleep fragmentation and age contribute to baseline ER stress and that there will be an age-associated impairment in protein folding in older animals. This impairment in protein folding would be exacerbated during sleep deprivation during which the UPR is induced. Oxidatively modified chaperones would be functionally less efficient in handling misfolded proteins that would lead to the UPR being turned on for a longer period of time that might result in the activation of proapoptotic factors such as CCAAT/enhancer-binding protein-homologous protein (CHOP), leading to cell injury or death. In this study, we examined each of the three cellular responses that are triggered by ER stress: we determined whether chaperones such as BiP were upregulated, we measured eIF2α phosphorylation to assess protein translation status, and we also determined the level of protein ubiquitination in young and old mice as a marker of functional efficiency of the ubiquitin–proteasome degradation pathway. Because our previous study in young (10 weeks old) mice indicated that BiP protein expression increased with 6 or more hours of sleep deprivation (Naidoo et al., 2005), we similarly performed 0, 3, 6, 9, and 12 h of sleep deprivation in the aged mice to determine whether they responded in a similar manner. Furthermore, because BiP is the key marker and master regulator of the UPR, we focused on this protein at all time points. Because the major effect of sleep deprivation on the UPR occurs at 6 h in young mice, we concentrated on this time point when examining all the other proteins. Finally, we measured the levels of proapoptotic factors CHOP, caspase-12, and growth arrest and DNA damage 34 (GADD34) in both young and aged animals subjected to sleep deprivation. We observed that the ER stress response to sleep deprivation was defective in aged mice because BiP protein levels did not increase, and neither did we observe an increase in eIF2α phosphorylation. We also found that basal expression of proapoptotic factors CHOP and activated caspase-12 were increased with aging and that they further increased with sleep deprivation. No increase in CHOP and activated caspase-12 with sleep deprivation was found in young mice.
Materials and Methods
Animal handling and behavioral assessment
Studies were performed on young 10-week-old and aged 22- to 24-month-old C57BL/6J male mice maintained on a 12 h light/dark cycle (lights on at 7:00 A.M.). Animals were kept in individual cages in a sound-attenuated room with ambient temperature at 23.5 ± 1.0°C, humidity at 40 ± 5%, and light intensity of 50–60 lux measured at the level of the mice with water and food available ad libitum. Mice were allowed to acclimate to their new environment for at least 2 weeks before starting any experiments. Animal handling and experimental procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of the University of Pennsylvania.
After acclimation, a 3 d baseline behavioral assessment was performed using infrared beam breaking activity monitors (AccuScan Instruments). Total activity was measured in 10 s epochs, i.e., 8640 epochs per 24 h. Estimates of sleep and wake were determined using an algorithm developed in our laboratory. In a previously published study in young (10 weeks old) mice, the algorithm defined sleep as 40 s or more of continuous inactivity (Pack et al., 2007). This definition of sleep/wake has an 88–94% agreement in individual mice across 8640 epochs with simultaneously EEG/EMG-defined sleep or wakefulness in young male C57BL/6J mice (Pack et al., 2007). The average sleep across the day in the mice studied was 59.4 min/2 h, and the algorithm based on sleep being considered as ≥40 s of continuous inactivity estimated this with a 1.0 min error. To estimate sleep and wake in the aged mice, in this study, by infrared beam breaks we performed a similar validation study (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We directly compared across 24 h (12 h light, 12 h dark) the EEG/EMG assessment of sleep and wake in 10 s epochs in aged (18 months old; n = 6) C57BL/6 mice to the assessment of sleep and wake by infrared beam breaks in the same mice. We performed a sensitivity analysis to assess the effects of varying the duration of continuous inactivity used to define sleep on quality of agreement between predictions of sleep based on inactivity from beam breaks and actual sleep determined from simultaneous EEG/EMG recording. We varied the duration of inactivity from 10 to 120 s in increments of 10 s. We show in supplemental Figure S1 (available at www.jneurosci.org as supplemental material) the square of average difference in estimates as a function of the duration of inactivity. The data show that a minimum is reached at 50 s. A sleep bout ends when any beam break occurs after 50 or more consecutive seconds of inactivity. Hence, we used this value of 50 s or more of continuous inactivity as our definition of sleep in the aged animals. We found that the average sleep across the day in the mice studied in this validation study was 58.7 min/2 h, and the algorithm based on sleep being considered as ≥50 s of continuous inactivity estimated this with an average 1.5 min error.
Sleep deprivation and tissue collection
Sleep deprivation was initiated at lights on (7:00 A.M.), and deprivation for 3, 6, 9, and 12 h was performed through gentle handling as described previously (Cirelli and Tononi, 1998; Naidoo et al., 2005). All the animals in the study were acclimated for at least 5 d before studies and were adapted to procedures for 3 d before the experiment. Briefly, sleep deprivation included directly observing the animal's motor activity and gently stroking the fur with an artist's brush when no activity was observed.
At each time point (10:00 A.M., 1:00 P.M., 4:00 P.M., and 7:00 P.M.), we had eight control mice (to control for circadian variations), which were left to sleep undisturbed until they were killed, and eight sleep-deprived mice. Additionally, a group of animals, for each age (n = 8), was killed at lights on (7:00 A.M.), giving us the 0 h time point. Animals were killed by cervical dislocation, their brains were rapidly dissected on wet ice, and the frontal and parietal cortex of each mouse were rapidly dissected out and flash frozen in liquid nitrogen and stored at −70°C until use. All experiments were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Tissue homogenization
The brain tissue was homogenized, on ice, in a lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mm EDTA, 1% Triton X-100, and 10% glycerol) in the presence of protease inhibitors (1 mm PMSF, 2 mg/ml pepstatin, and 4 mg/ml aprotinin). The lysate was centrifuged to remove cellular debris, and protein was determined by the Pierce micro-BCA assay.
SDS-PAGE and Western blots
Temporal expression of BiP/GRP78.
Individual sleep-deprived and matching control mouse cortex homogenates were run on SDS-PAGE gels in triplicate. For each time point, there were eight sets of gels. Commercial hamster-derived BiP/GRP78 standard (200 ng; Stressgen) was used, in two lanes, on each gel as both a loading control and an external standard. Samples (20 μg of protein) representing individual mice were run on SDS-PAGE gels (10% Tris-HCl; Bio-Rad) according to Laemmli (1970) and then transferred to nitrocellulose membranes (Bio-Rad). After transfer onto nitrocellulose, blots were incubated with rabbit polyclonal antibody against BiP/GRP78 (1:1000; Stressgen). After incubation with horseradish peroxidase-conjugated secondary antibody (anti-rabbit at 1:3000; Sigma), protein bands were detected and analyzed by enhanced chemiluminescence (Pierce Supersignal) and quantitative imaging (AlphaInnotech Fluorochem 8900). All bands were normalized to the external BiP/GRP78 standard. Densitometry was performed using the Alphaease FC software. For BiP/GRP78, we obtained data for all durations of sleep deprivation (3, 6, 9, and 12 h) in old animals and for comparison purposes data at 6 h of sleep deprivation in younger animals. These were animals specifically evaluated for this study. The choice of the 6 h time point was based on our previous study (Naidoo et al., 2005)
Other proteins.
Western blots to detect other proteins were performed as described above for BiP/GRP78. The following antibodies and dilutions were used: GADD153 (growth arrest and DNA damage 153)/CHOP (1:200; Santa Cruz Biotechnology), caspase-12 (1:2000; Stressgen), ubiquitin (1:500; Millipore Bioscience Research Reagents), GADD34 (1:200; Santa Cruz Biotechnology), phosphorylated (P) eIF2α (1:1000; Cell Signaling Technology), and P-PERK (1:1000; Cell Signaling Technology). For additional details about antibodies, see supplemental Table S1 (available at www.jneurosci.org as supplemental material). All Western blots were performed using cerebral cortex lysate from young and aged mice. For these proteins, we only studied data in both young and aged mice from the 6 h sleep deprivation time point and its diurnal control.
Immunohistochemistry
For immunohistochemistry, we compared a group of undisturbed young mice (2 months old; n = 5) with a group of undisturbed aged mice (18 months old; n = 5) that were killed at 1:00 P.M. Brain tissue was procured after pentobarbital anesthesia followed by saline and 4% paraformaldehyde transcardial perfusions. Whole brains were incubated in 4% paraformaldehyde and then cryopreserved before coronal sectioning (40 μm) from the medulla through the basal forebrain approximating murine coordinates 1.54–1.94 mm bregma (Franklin and Paxinos, 1996). Sections were stored as 1:6 or 1:3 series in 0.1% sodium azide in PBS at 4°C before use. MAP2 antibody (1:1000; Abcam) was used as a neuronal marker, and BiP presence was identified using the GRP78 (H-129; Santa Cruz Biotechnology) antibody at 1:500, whereas CHOP was detected using the GADD153/CHOP antibody at 1:500 (sc575; Santa Cruz Biotechnology). Biotinylated donkey anti-rabbit secondary antibodies diluted at 1:500 were used. Both proteins were detected using DAB.
Statistical methods
To test the effects of diurnal time and sleep deprivation (vs controls) on BiP protein expression, a two-way ANOVA was performed using the software program PROC.GLM (SAS). At each time point, eight animals were killed, and BiP protein expression was assessed by Western blots. The interaction test for condition and time assessed whether in older animals any effects of sleep deprivation differed for various time points. A t test was used to determine any differences between the young and old groups at baseline.
For other variables, including GADD34, P-PERK, and ubiquitin, a two-way ANOVA tested the effects of sleep deprivation and age (when all animals were run on the same gel). When the interaction test was significant, Tukey's-adjusted post hoc tests assessed the effect of condition within young and old animals. Finally, when young and old animals were run on separate gels, a t test was used to determine differences between sleep deprivation and control conditions separately for young and old animals.
Results
Aged mice display fragmented sleep
We determined sleep and wake in the young and aged mice in this study using infrared beam breaks and an algorithm developed in our laboratory that defines sleep as 40 s (in young mice) and 50 s (in aged mice) or more of continuous inactivity (see Materials and Methods; see supplemental data, available at www.jneurosci.org as supplemental material). We have shown that this definition of sleep/wake has an 88–94% agreement in individual mice across 8640 epochs with simultaneously EEG/EMG-defined sleep or wakefulness in male C57BL/6J mice (Pack et al., 2007). We found that aged mice displayed fragmented sleep and exhibited a greater number of sleep (p < 0.05) and wake bouts (p < 0.05) with shorter bout lengths (p < 0.05) than the young mice during lights on (Fig. 1) (supplemental Table S2, available at www.jneurosci.org as supplemental material). Similar fragmented behavior in aged mice has been described previously (Welsh et al., 1986).
Figure 1.

Sleep and wake in young and aged mice. Sleep bouts are shorter in aged mice compared with young mice during the rest phase (*p < 0.05) and longer during the active phase when compared with young mice (*p < 0.05). Shown are averaged sleep bout length (SBL) and SD during the active and rest phase.
The UPR marker BiP is increased with sleep deprivation in young mouse cerebral cortex but not in aged mouse cerebral cortex
We have shown previously that sleep deprivation of 6 h leads to the induction of the ER stress response/unfolded protein response in cerebral cortex of young mice (Naidoo et al., 2005). Our previous study demonstrated that the molecular chaperone protein BiP was upregulated at the protein level in mouse cerebral cortex with acute sleep deprivation with a concomitant decrease in protein translation. Upregulation of BiP mRNA with sleep deprivation has also been described in young rat cerebral cortex (Terao et al., 2003; Cirelli and Tononi, 2004), in Drosophila brain (Shaw et al., 2000), and birds (Jones et al., 2008). Increase in BiP protein with sleep deprivation has also been found in brain of Drosophila (Naidoo et al., 2007). In the present study, we examined the levels of BiP protein in the cerebral cortex of aged (22–24 months old) mice that had been subjected to sleep deprivation for 3, 6, 9, and 12 h. Undisturbed animals killed at the same diurnal times were used as controls. We observed no significant difference in BiP expression in the cortical lysates of sleep-deprived older animals when compared with the undisturbed controls at all time points examined (Fig. 2 A). A two-way ANOVA was used to test whether there was a difference between the control and sleep-deprived groups over time (from 3 to 12 h) in BiP expression. There was no statistically significant effect for group (F (1,60) = 1.5; p = 0.225), time (F (3,60) = 1.9; p = 0.148), or group × time interaction (F (3,60) = 0.2; p = 0.923). To ensure that we replicated our previous result that protein levels increase with sleep deprivation in young mice (Naidoo et al., 2005), we directly compared the change in protein levels of BiP in young mouse cerebral cortex in a new group of mice after 6 h of prolonged wakefulness relative to their undisturbed controls to the change in expression of BiP in aged mouse cerebral cortex subject to the same conditions. After 6 h of sleep deprivation, BiP protein levels were increased 1.6-fold in young sleep-deprived animals compared with the undisturbed diurnal controls (n = 6; p = 0.0001, t test), whereas there was no significant difference in BiP expression levels between the sleep-deprived and control samples in the aged animals (p = 0.337) at the same time point (Fig. 2 B). We know from our previous study that the handling of animals alone does not lead to an increase in BiP expression; in that study, young mice handled in an identical manner, for the first 6 h of the active lights off period, to that during sleep deprivation did not display an increase in BiP protein (Naidoo et al., 2005). We acknowledge that there is, by necessity, the potential confounder of different times of day. We also note that the circadian patterns in aged mice are different (Valentinuzzi et al., 1997).
Figure 2.
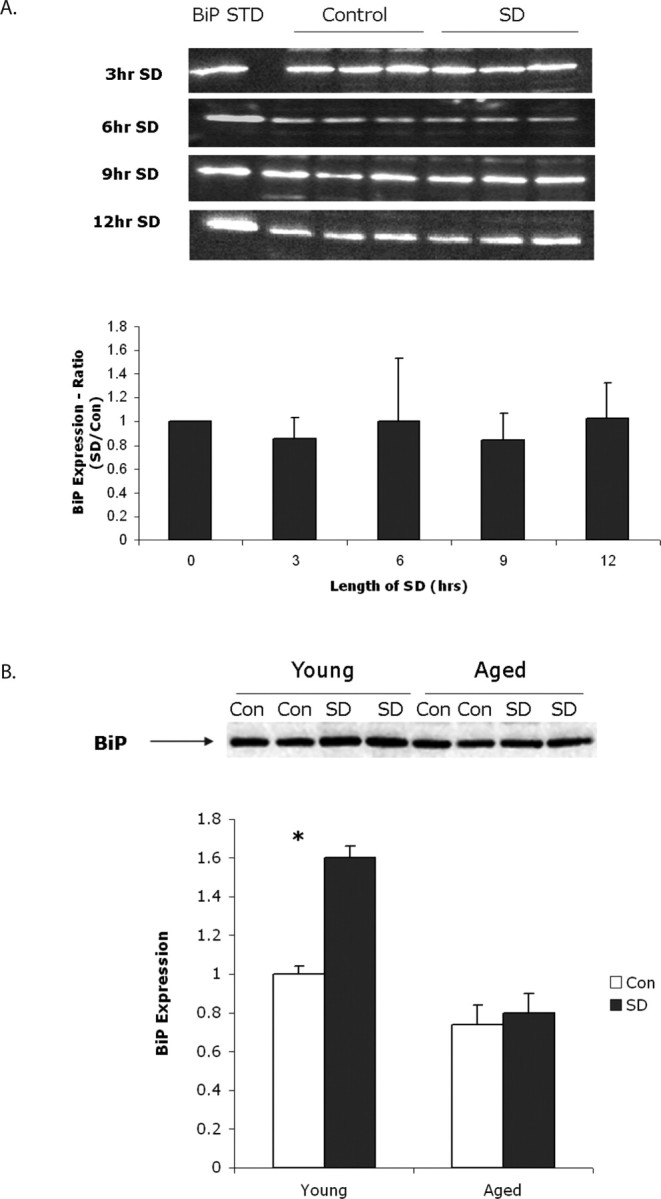
A, Top, Representative Western blots showing BiP expression in aged mouse cerebral cortex after 3, 6, 9, and 12 h of sleep deprivation (SD). Each blot shows a triplicate of a single sleep-deprived mouse lysate and its matching undisturbed control. There were eight gels run at each time point. All bands were normalized to an external commercial BiP standard (STD) run on each gel. Bottom, Densitometric quantification of BiP expression directly from chemiluminescence at 3, 6, 9, and 12 h of sleep deprivation. Averaged data with standard deviation are presented as a ratio to undisturbed controls killed at the same diurnal time. n = 8 per time point. These ratios were not significantly different than 0 h. B, Top, Representative Western blot showing BiP expression in two young and two aged mice cerebral cortices after 6 h of sleep deprivation compared with two young and two aged undisturbed mice killed at the same diurnal time. Bottom, Graph showing quantification of BiP expression from six sleep-deprived and control (Con) young and aged animals. Data are expressed as a ratio of sleep deprived to young control. Averaged data with standard deviations are shown. There is a significant increase in BiP in young animals but not in old (*p = 0.0001).
Expression of BiP is decreased in aged mouse cerebral cortex
Because BiP protein levels increased with sleep deprivation in young and not in aged animals, we wanted to ascertain basal expression of this chaperone at both ages. Studies in rat hippocampus and rat liver have indicated that this ER chaperone levels decline with age (Rabek et al., 2003; Gavilan et al., 2006). We analyzed basal levels of BiP expression in the cerebral cortex of aged undisturbed animals killed at 1:00 P.M. and compared that with BiP expression in young animals killed at the same diurnal time. As shown in Figure 3 A, we observed an average 30% decrease in BiP in the cortex of the six older animals we tested compared with young. A t test showed significantly lower BiP levels at baseline for the old animals compared with the young animals (t (16) = 5.1; p < 0.0001). Having found reductions in BiP expression by Western blots, we sought to localize BiP in the cortex. We found most of the BiP immunofluorescence in the pyramidal neurons of layers I–VI of the medial prefrontal cortex, cingulate cortex, and in the piriform cortex (Fig. 3 B). Aged mice displayed much lower expression of BiP in similar cells. In contrast, aged mice express high levels of the proapoptotic factor CHOP in these same cells (see below and Fig. 6 B).
Figure 3.
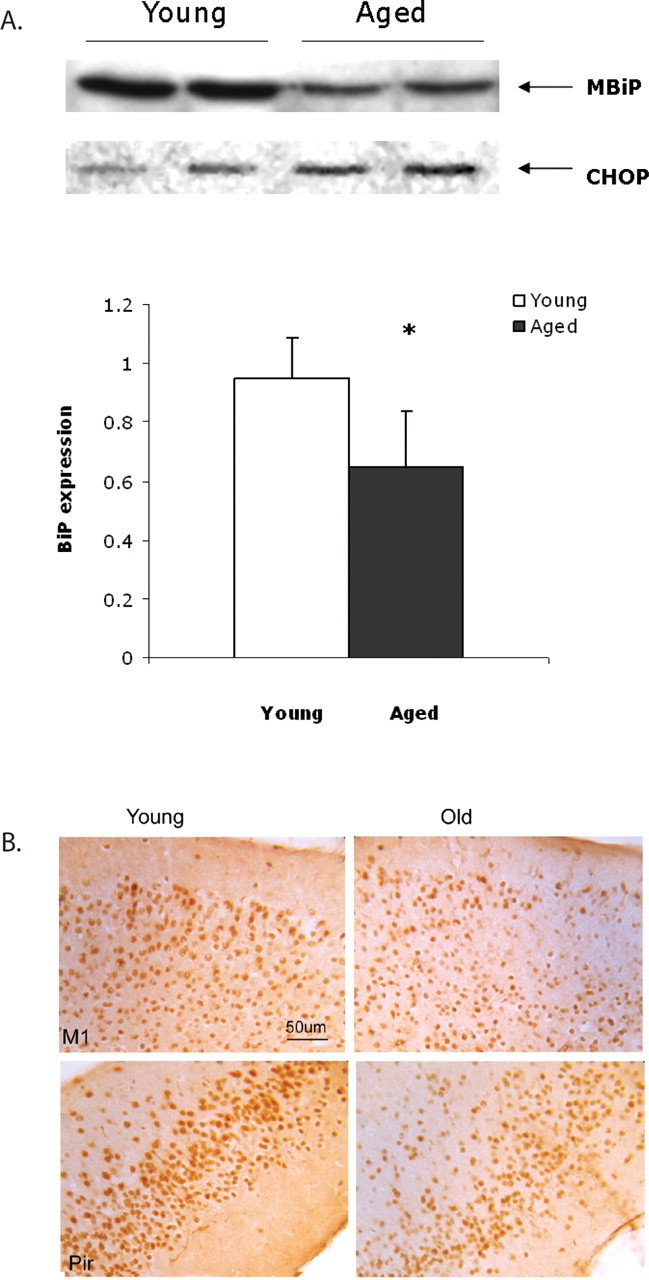
A, BiP expression is lower in aged mouse cerebral cortex. BiP expression in two young and two aged samples from the 1:00 P.M. time point are shown in a representative Western blot. CHOP, which increases with age, is shown as a loading control. The bar graph below illustrates BiP expression quantified from Western blots in young and aged tissue. Mean with SD is shown (n = 6; *p = 0.01). B, Decreased immunohistochemical labeling of BiP in with age. DAB-labeled BiP in young and aged M1 and piriform (Pir) cortical neurons. 160× magnification. Scale bar, 50 μm.
Figure 6.
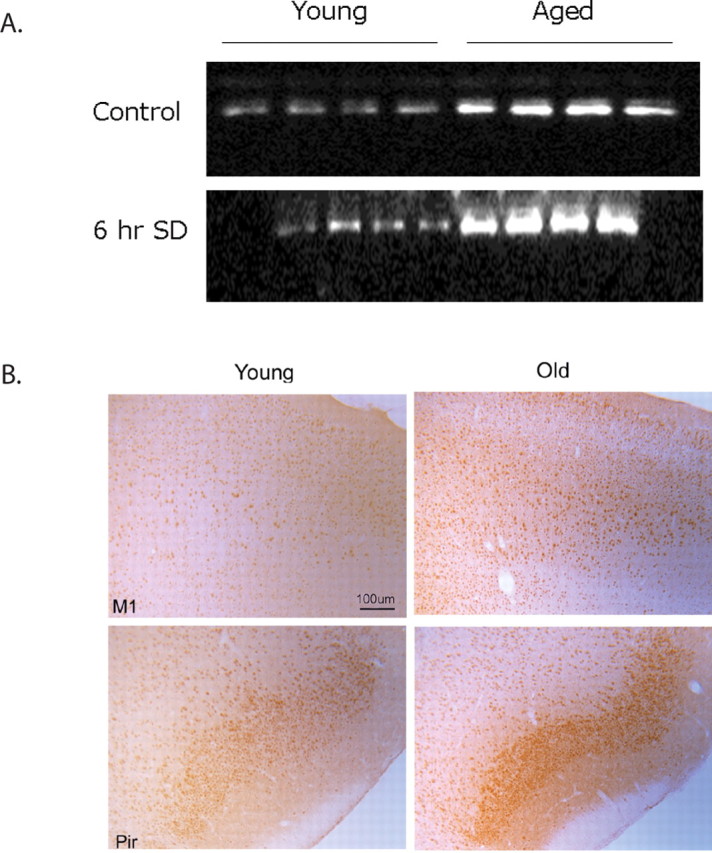
A, Western blot showing CHOP expression in young and aged control and sleep-deprived samples. CHOP levels are markedly higher in aged animals compared with young. With sleep deprivation, there was an additional increase in CHOP levels in aged animals but not in young. Each gel shows samples (20 μg of protein) from four individual young and aged animals. B, Increased expression of CHOP with age. DAB-labeled CHOP in young and aged M1 and piriform (Pir) neurons. 80× magnification. Scale bar, 100 μm (for all images).
There is no increase in markers of inhibition of protein translation, P-PERK, and P-eIF2α with sleep deprivation in aged animals, whereas young animals show this response
Attenuation of protein translation is produced by the phosphorylation of eIF2α by the serine–threonine kinase PERK when it is autophosphorylated. This is a hallmark of the UPR (Kaufman, 2002). Our previous study in young animals demonstrated that 6 h of sleep deprivation led to PERK phosphorylation, eIF2α phosphorylation, and the attenuation of protein translation (Naidoo et al., 2005). We therefore now measured PERK levels, PERK phosphorylation, and phosphorylation of eIF2α in both young and aged animals subjected to 6 h of sleep deprivation to determine whether this aspect of the UPR occurred. We were able to detect PERK expression in both control and sleep-deprived aged mouse cortex, albeit at much lower levels than in young animals. However, although we were able to detect an increase in P-PERK in young cerebral cortex with sleep deprivation, we did not see a similar increase in PERK phosphorylation in the aged animals (Fig. 4 A,B). A two-way ANOVA comparing the effects of sleep deprivation at 6 h after sleep deprivation for young and old animals showed that P-PERK levels were higher overall for animals in the sleep deprivation condition compared with controls (1.1 vs 0.7; F (1,20) = 4.7; p = 0.043) and were also higher for the young compared with the old animals (1.4 vs 0.5; F (1,20) = 24.9; p < 0.0001). There was also a significant interaction between condition and age (F (1,16) = 5.3; p = 0.032), and post hoc tests confirmed that the effect of sleep deprivation was significant for the young animals (p = 0.005) but not old animals (p = 0.912).
Figure 4.

A, PERK and eIF2α phosphorylation in young and aged cerebral mouse cortex. Western blot showing P-PERK and phospho-eIF2α expression in young and aged samples (20 μg of protein loaded per well) with and without sleep deprivation (SD). eIF2α levels shown below are used as loading controls (Con). B, Densitometric quantification of P-PERK signal directly from chemiluminescence in young and aged sleep-deprived and control samples; mean values with standard deviation shown (n = 6, *p = 0.01). C, The level of phosphorylated eIF2α is decreased in aged mouse cerebral cortex. Densitometric quantification of P-eIF2α signal directly from chemiluminescence in young and aged sleep-deprived and control samples; mean values with standard deviation are shown (n = 8; *p < 0.0001).
We were also able to detect very low levels of P-eIF2α in aged animals, much lower than in young animals (Fig. 4 A,C). The aged animals exhibit no significant increase in P-eIF2α with 6 h of sleep deprivation (Fig. 4 C). In contrast, young animals sleep deprived for 6 h displayed a robust 40% increase in phosphorylated eIF2α levels compared with control undisturbed animals (Fig. 4 B) (n = 8; p < 0.0001).
Given the low expression levels of P-eIF2α in aged mouse cerebral cortex, we decided to examine the expression of GADD34 in these samples. GADD34, a protein phosphatase 1 (PP1)-interacting protein, causes PP1 to dephosphorylate eIF2α, in the late stages of ER stress and relieve the translational block imposed by eIF2α phosphorylation (Brush et al., 2003; Szegezdi et al., 2006). Removal of the translational block by GADD34 also facilitates the synthesis of proapoptotic proteins such as CHOP (see further below). Western blot analysis of young and aged mouse cortical tissue indicate that baseline expression of GADD34 is increased in aged animals (Fig. 5); we observed 58% more GADD34 expressed in aged undisturbed control animals compared with young undisturbed control animals (n = 5; p = 0.005). Sleep-deprived aged animals displayed an additional 12% increase in GADD34 expression over the aged control animals (n = 5; p = 0.01). In contrast, there was no significant difference in GADD34 expression between young sleep-deprived animals and their matching diurnal controls.
Figure 5.

GADD34 expression is elevated in aged animals and further increased by sleep deprivation. Top, Representative Western blot showing samples (20 μg of protein) from young and aged animals after 6 h of sleep deprivation (SD) and their diurnal controls (Con). Bottom, Bar graph illustrating increased GADD34 expression in aged animals quantified from Western blots (n = 5; *p = 0.005).
Levels of proapoptotic proteins, CHOP and caspase-12, are markedly increased with age; sleep deprivation produces additional increases in old but not young animals
CHOP also known as GADD153 is a component of the ER stress-mediated apoptotic pathway. CHOP levels have been shown to increase when ER stress is not alleviated and persists (Zinszner et al., 1998); CHOP inhibits protective antiapoptotic factors such as Bcl-2 and promotes apoptotic caspase activity (McCullough et al., 2001). We measured CHOP levels in the cerebral cortex of young and aged mice. Basal levels of CHOP were markedly higher in aged animals compared with young animals. We observed eightfold more CHOP in the cerebral cortex of aged mice compared with young mice, suggesting that aged mice are subject to much more ER stress on an ongoing basis (n = 7; p = 0.00005, t test) (Fig. 6 A, top). Having established that CHOP levels are greatly increased with age, we sought to determine where in the cortex this occurred. Young mice display reduced CHOP staining in the cortex. We found increased CHOP staining in pyramidal neurons of layers I–VI of the medial prefrontal cortex (Fig. 6 B) and in the piriform cortex in aged mice. In contrast, there are reduced amounts of BiP in these same cells in the aged animals (Fig. 3 B). Young mice, as we point out above, display strong BiP immunostaining in these neurons. There appears to be an age-dependent BiP–CHOP flip-flop in regions of the cortex. This flip-flop is suggestive of cytoprotective loss.
We also measured CHOP levels in the cerebral cortex of young and aged mice that had been sleep deprived for 6 h and compared them with their respective undisturbed diurnal controls. CHOP levels increased by 40% in aged animals (p < 0.01, two-tailed t test; n = 5) that were subjected to 6 h of sleep deprivation (Fig. 6 A, bottom). Young animals did not display a significant increase in CHOP with similar sleep deprivation.
Given that CHOP levels are increased, we examined next the expression of procaspase-12 and cleaved caspase-12, components of the second main ER stress-induced apoptotic pathway (Szegezdi et al., 2003). Caspase-12 is a member of the caspase family of cysteine proteases that are key mediators of programmed cell death or apoptosis. Caspase-12 is an initiator caspase and the central player in ER-induced apoptosis (Szegezdi et al., 2006). The precursor form of all caspases is composed of a prodomain and large and small catalytic subunits. All known caspases require cleavage adjacent to aspartates to liberate one large and one small subunit, which associate into a2b2 tetramer to form the active enzyme (for review, see Nunez et al., 1998; Thornberry and Lazebnik, 1998). Once activated, caspase-12 translocates from the ER to the cytosol in which it cleaves caspase-9, which induces the cleavage of caspase-3 in a cytochrome c-independent manner and activation of the rest of the apoptotic pathway (Szegezdi et al., 2003). We compared the amount of procaspase-12 and activated cleaved caspase-12 in young and aged control and 6 h sleep-deprived samples. As shown in Figure 7, the basal level of procaspase-12 was similar in young and aged control animals. However, the ratio of cleaved caspase-12 to procaspase-12 was significantly higher in aged control animals (66.0 ± 12.6%; n = 6) compared with that in young control animals (36.0 ± 8.5%; n = 6; p = 0.001). During cleavage, caspase-12 is activated and translocates from the ER to the cytosol in which it is reported to cleave caspase-9, which in turn activates caspase-3 and the rest of the caspase cascade (Morishima et al., 2002; Szegezdi et al., 2003).
Figure 7.

Caspase-12 activation in sleep-deprived young and aged mice. A representative Western blot showing procaspase-12 and cleaved expression in young and aged animals subjected to 6 h of sleep deprivation (SD) and their matching diurnal controls (Con). Both procaspase-12 and cleaved caspase-12 was detected with a specific polyclonal antibody. Induction of caspase-12 was significantly higher in aged than in young mice (n = 6; two-tailed t test, *p = 0.01). This was further increased with sleep deprivation in old but not young animals.
We also found no additional change in the amount of cleaved caspase-12 to procaspase-12 in young animals subjected to 6 h of sleep deprivation. We do however observe a 22.5% increase in cleaved caspase-12 in sleep-deprived aged animals compared with undisturbed control animals (p = 0.016, two-tailed t test; n = 7).
Ubiquitination is increased in aged mouse cerebral cortex and is only increased in young mouse cortex with sleep deprivation
In eukaryotic cells, the ubiquitin–proteasome system (UPS) is the main pathway for eliminating misfolded proteins (Hershko and Ciechanover, 1998; McClellan et al., 2005). Misfolded or damaged proteins are tagged for UPS-mediated degradation by the covalent attachment of ubiquitin molecules that are recognized by the 26S proteasome (Hershko and Ciechanover, 1998). Ubiquitin is a small 76 amino acid protein that attaches to the lysine residues on proteins destined for degradation; a minimum of four ubiquitin molecules are required for recognition by the proteasome. We measured ubiquitin levels in tissue samples from young and aged 6 h sleep-deprived animals and their matching diurnal controls. Basal levels of ubiquitin are twofold higher in aged control cerebral cortex compared with young control cerebral cortex, indicating that there must be many more damaged or unfolded proteins in the aged tissue (Fig. 8). We also determined whether ubiquitin levels changed with 6 h of sleep deprivation. A two-way ANOVA tested whether there were differences between the control and sleep deprivation conditions, as well as looked at age differences and the interaction of condition and age. Results showed a statistically significant main effect of age (F (1,8) = 175.5; p < 0.0001), such that mean levels of ubiquitin were higher in old animals compared with young animals (2.0 vs 1.12). Although there was no significant main effect for condition (F (1,8) = 3.2; p = 0.112), there was a significant interaction of age and condition (F (1,8) = 21.6; p = 0.002). Post hoc tests showed that there was a significant difference between the control and sleep deprivation condition only for the young animals (p = 0.002) but not for the old animals (p = 0.077).
Figure 8.
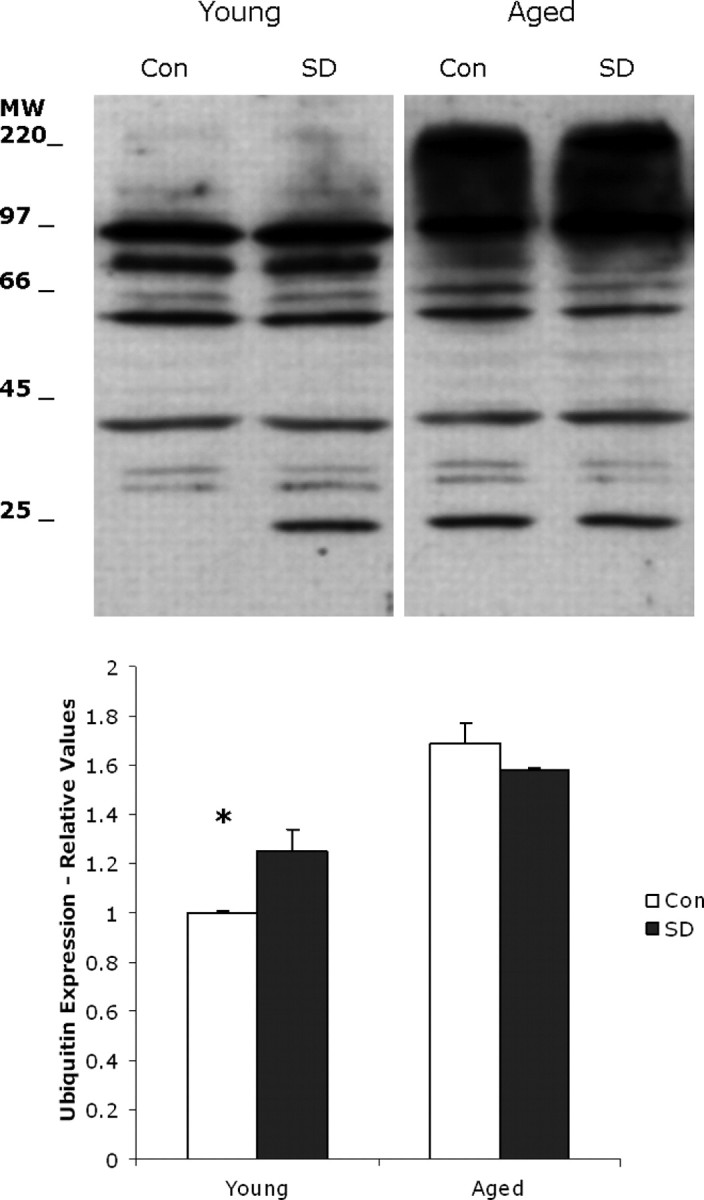
Representative profiles of ubiquitinated proteins in individual young and aged mouse cortical tissue with and without 6 h of sleep deprivation (SD). More proteins are tagged for degradation by polyubiquitinylation in aged mouse cerebral cortex than in young. Equal amounts of protein (25 μg) from young (n = 6) and aged (n = 6) mice were loaded onto SDS-PAGE gels and detected with a specific antibody to ubiquitin. Con, Control; MW, molecular weight. There is a significant increase in ubiquitination in young sleep-derived cortex (*p = 0.002).
Discussion
We have reported previously that acute sleep deprivation leads to induction of the ER stress response in young mouse cerebral cortex (Naidoo et al., 2005). The protective aspect of the ER stress response/unfolded protein response has three major components (Fig. 9 A): (1) an increased expression of ER chaperones, particularly BiP whose increased expression is considered the key indicator of ER stress; the main function of the increased levels of BiP is to bind to unfolded proteins, prevent them from aggregating, and to aid their refolding (Ma and Hendershot, 2004); (2) attenuation of protein translation produced by phosphorylation of eIF2α; and (3) increased degradation of misfolded proteins by the proteasome by a process called ERAD. We sought in this study to examine the ER stress response to acute sleep deprivation in aged animals and compared this with the young. It has been reported that ER resident proteins are more susceptible to oxidative stress with aging (Rabek et al., 2003). Thus, we hypothesized that sleep deprivation would be a greater insult to aged animals than to young animals and would result in greater cellular stress. The major findings of this study indicate that the ER stress response (UPR) to sleep deprivation is impaired in aged animals. As a result of this impairment in the initial steps of the protective response, proapoptotic factors GADD34, CHOP, and caspase-12 are increased in older animals with sleep deprivation. When cell-protective changes mediated by the UPR fail to restore folding capacity, CHOP, c-jun NH2 terminal kinase, and caspases have been implicated in mediating apoptosis in response to sustained ER stress (Fig. 9 B). This does not occur in younger animals. Thus, sleep deprivation in older animals results in more unfavorable consequences than in young animals.
Figure 9.
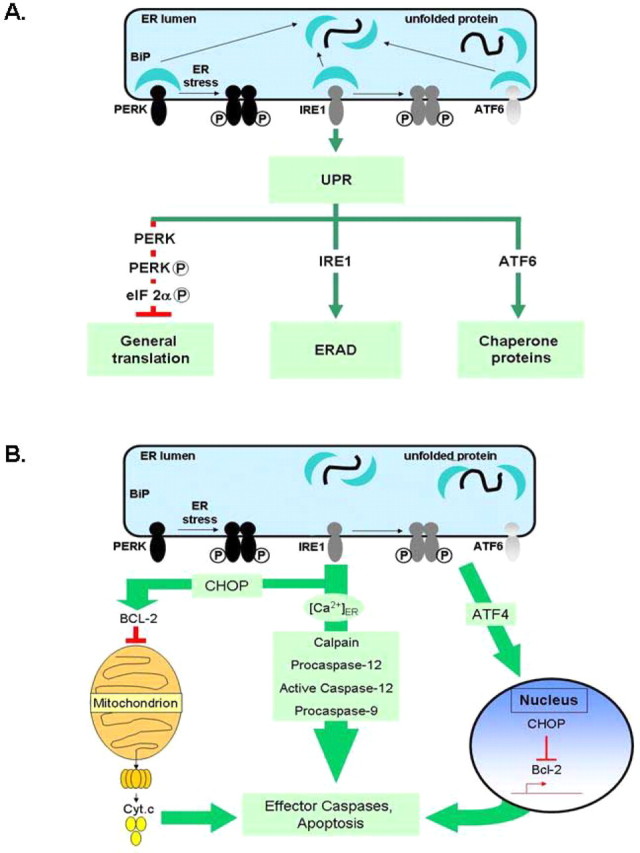
A, Simplified schematic showing the three major components of the unfolded protein response. Misfolded or unfolded proteins titrate BiP away from the three sensors of ER stress: PERK, IRE1, and ATF6. Activated PERK phosphorylates eIF2α to attenuate protein translation. Activated ATF6 leads to inductions of molecular chaperones BiP, protein disulfide isomerase (PDI), GRP94, etc. IRE1 activation leads to stimulation of protein degradation. The various ER chaperones, such as BiP and GRP94, are protective and control protein folding and components of the UPR. B, Sustained ER stress leads to proapoptotic signaling. Bcl-2 in conjunction with IP3 receptors (IP3-R) control ER calcium stores and release into the cytosol. Bcl-2 normally exerts an antiapoptotic function in the ER. CHOP inhibits Bcl-2, leading to calcium release; higher calcium levels sensitize mitochondria to other insults inducing cell death. ER specific caspases, such as the caspase-12, are thought to directly induce cell death.
BiP, the key chaperone in the ER, is not increased by sleep deprivation in aged animals. BiP expression in aged animals does not change significantly with increasing amounts of sleep deprivation, unlike young animals that express increased BiP levels with 6 or more hours of sleep deprivation. Furthermore, baseline expression of BiP in aged animals is lower than that in young animals; there is a 30% reduction in basal BiP expression in old animals compared with young. This suggests that protein folding in aged animals is compromised; there is less chaperone available at baseline and a challenge such as sleep deprivation exacerbates the problem. BiP is the first ER chaperone that a newly synthesized peptide encounters; BiP binds to hydrophobic domains and prevents misfolding as the rest of the protein is synthesized. A consequence of low levels of BiP is a decreased capacity of the ER to handle protein load, both nascent and misfolded proteins. There is also no evidence of attenuation of protein translation in aged animals in response to sleep deprivation as occurs in young mice (Naidoo et al., 2005). The phosphorylation of the translation initiation factor eIF2α by P-PERK is a key regulatory step in protein translation; it leads to the formation of stalled 43S ribosomal ternary complexes and inhibition of translation. We do not observe significant phosphorylation of eIF2α in aged animals in response to sleep deprivation, although this is seen after 6 h of deprivation in young animals (Naidoo et al., 2005). This is consistent with our observation that GADD34 is increased in response to sleep deprivation in aged animals but not in young. GADD34 is instrumental in dephosphorylating eIF2α and removing the translational block. Because eIF2α is phosphorylated by PERK, we also examined the expression of both PERK and P-PERK. Although we were able to detect PERK expression in aged tissue, we were not able to detect significant phosphorylation in response to sleep deprivation as we observed in young animals. This lack of activation of PERK is the most parsimonious explanation as to why protein translation is not inhibited.
ER-associated degradation by the ubiquitin–proteasome pathway is the third mechanism by which cells faced with ER stress and misfolded proteins reduce protein load. Our data indicating a fourfold increase in ubiquitination of proteins in aged tissue at baseline compared with young tissue indicates the presence of more misfolded proteins and a more overwhelmed ubiquitin–proteasome pathway in the aged mouse cortex. Abnormal accumulation and aggregation of cellular proteins is common to many neurodegenerative diseases. That the aged animals are unable to increase the amount of ubiquitinated proteins after sleep deprivation through ERAD further suggests impairment of the ER stress response.
It is evident that all three components of the ER stress response/UPR to sleep deprivation are compromised with aging; chaperones such as BiP are not increased, protein translation is not attenuated, and ER-associated proteasomal degradation is reduced. All these will result in an increased protein load in the ER that will further exacerbate cellular stress in the cell.
Given the inability of the aged cells to adequately respond to the ER stress presented by the challenge of sleep deprivation, it is not surprising to see that proapoptotic factors are increased by this challenge. Our data indicate that CHOP levels are elevated in aged mouse cortex as others have shown (Gavilan et al., 2006; Hussain and Ramaiah, 2007). We show that, in addition, CHOP is further increased in aged animals with sleep deprivation. Young animals in contrast display lower levels of CHOP in the cortex and show no increase in the protein with sleep deprivation. This is in agreement with previous studies directly assessing apoptosis in the cortex of young rats (Cirelli et al., 1999; Eiland et al., 2002). CHOP, which is controlled transcriptionally and translationally, is also regulated posttranslationally by phosphorylation by p38 mitogen-activated protein kinase, which increases its activity (Szegezdi et al., 2006). A role for CHOP in mediating apoptosis in response to ER stress is well established (Wang et al., 1996; Zinszner et al., 1998). It has been reported that elevated CHOP sensitizes cells to oxidative insults (Ikeyama et al., 2003) and that ectopic expression of CHOP in rat fibroblasts leads to increased levels of reactive oxygen species (McCullough et al., 2001). Targets of CHOP include Bcl-2, GADD34, ERO1a, and TRB3 (Szegezdi et al., 2003). GADD34, which we found to increase with age and sleep deprivation, promotes protein synthesis by removing the translational block. Bcl-2 and other members of the Bcl-2 family Bax and Bak confer protection against lethal ER stress and apoptosis (Szegezdi et al., 2006) as well as oxidative stress (Lee, 2001). CHOP is known to repress Bcl-2. All upstream signals, such as activation of the transcription factors described, ultimately lead to caspase activation, which results in the ordered and sequential dismantling of the cell. Our data that caspase-12 is activated in aged animals and this is further increased with sleep deprivation suggest that cellular injury and possibly cell death pathways are activated by sleep deprivation. This does not occur in younger animals in which the unfolded protein response to sleep deprivation is robust.
Misfolded proteins and the associated ER stress are emerging as common features of neurodegenerative diseases (for review, see Rao et al., 2004; Schroder and Kaufman, 2005b). Neuronal loss in both familial and sporadic forms of neurodegenerative disorders is often accompanied by aggregation of misfolded proteins (Selkoe, 2003). Accumulating evidence suggests that ER dysfunction and aberrant protein degradation play a role in dopamine neuron loss in PD (Wang and Takahashi, 2007). The ER stress response has also been implicated in AD, amyotrophic lateral sclerosis, Huntington's disease, and spinocerebellar ataxias (Rao et al., 2004; Schroder and Kaufman, 2005b; Wang and Takahashi, 2007). These diseases usually appear late in life and are associated with aging. It is well documented that patients with neurodegenerative diseases such as PD and AD experience sleep disorders (Menza and Rosen, 1995; Van Someren, 2000; Swaab et al., 2002). It is not unreasonable to speculate that the sleep disturbances place an additional burden on an already stressed protein folding and degradation system and could exacerbate protein aggregation.
Surprisingly, given our results, recovery sleep after sleep deprivation is less in older animals than young. This has been shown in humans (Bonnet, 1985; Carskadon and Dement, 1987) and in rats (Mendelson and Bergmann, 2000; Shiromani et al., 2000). Given that our results indicate that sleep deprivation in older animals is more injurious, one might have expected more, not less, recovery sleep. One interpretation of this result may be that sleep deprivation has a lesser effect in older animals. During recovery sleep after deprivation, there is progressive decline in BiP levels in Drosophila (Naidoo et al., 2007). Moreover, alteration of BiP function changes the amount of recovery sleep. Specifically, mutants with loss of function of BiP have decreased recovery sleep, whereas transgenics with increased expression of BiP have more recovery sleep (Naidoo et al., 2007). Our findings that older animals, who exhibit reduced recovery sleep after deprivation, have no BiP response to sleep deprivation is compatible with the concept that BiP itself may play a direct role in determining the magnitude of sleep recovery.
To summarize, our data indicate that the ER stress response is compromised with aging and that the additional challenge of sleep deprivation further exacerbates ER stress and leads to induction of proapoptotic signaling.
Footnotes
This work was supported by National Institute on Aging Grant AG025353 (N.N.) and Program Project Grant AG17628 (A.I.P.). We thank Ray Galante and Jie Lian for superb assistance with studies of mouse sleep validation, Dr. Sigrid Veasey for expert advice on immunohistochemistry, Jennifer Montoya for assistance with the illustrations, and Daniel Barrett for editing.
References
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bonnet MH. Effect of sleep disruption on sleep, performance, and mood. Sleep. 1985;8:11–19. doi: 10.1093/sleep/8.1.11. [DOI] [PubMed] [Google Scholar]
- Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003;23:1292–1303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carskadon MA, Dement WC. Daytime sleepiness: quantification of a behavioral state. Neurosci Biobehav Rev. 1987;11:307–317. doi: 10.1016/s0149-7634(87)80016-7. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Differences in gene expression between sleep and waking as revealed by mRNA differential display. Brain Res Mol Brain Res. 1998;56:293–305. doi: 10.1016/s0169-328x(98)00057-6. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Uncoupling proteins and sleep deprivation. Arch Ital Biol. 2004;142:541–549. [PubMed] [Google Scholar]
- Cirelli C, Shaw PJ, Rechtschaffen A, Tononi G. No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res. 1999;840:184–193. doi: 10.1016/s0006-8993(99)01768-0. [DOI] [PubMed] [Google Scholar]
- Dobson C. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- Eiland MM, Ramanathan L, Gulyani S, Gilliland M, Bergmann BM, Rechtschaffen A, Siegel JM. Increases in amino-cupric-silver staining of the supraoptic nucleus after sleep deprivation. Brain Res. 2002;945:1–8. doi: 10.1016/s0006-8993(02)02448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. New York: Academic; 1996. [Google Scholar]
- Gavilan MP, Vela J, Castano A, Ramos B, del Rio JC, Victoria J, Ruano D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27:973–982. doi: 10.1016/j.neurobiolaging.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hussain SG, Ramaiah KV. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun. 2007;355:365–370. doi: 10.1016/j.bbrc.2007.01.156. [DOI] [PubMed] [Google Scholar]
- Ikeyama S, Wang XT, Li J, Podlutsky A, Martindale JL, Kokkonen G, van Huizen R, Gorospe M, Holbrook NJ. Expression of the pro-apoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J Biol Chem. 2003;278:16726–16731. doi: 10.1074/jbc.M300677200. [DOI] [PubMed] [Google Scholar]
- Jones S, Pfister-Genskow M, Benca RM, Cirelli C. Molecular correlates of sleep and wakefulness in the brain of the white-crowned sparrow. J Neurochem. 2008;105:46–62. doi: 10.1111/j.1471-4159.2007.05089.x. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- Laemmli U. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lansbury P. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci USA. 1999;96:3342–3344. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- Lin DA, Boyce JA. IL-4 regulates MEK expression required for lysophosphatidic acid-mediated chemokine generation by human mast cells. J Immunol. 2005;175:5430–5438. doi: 10.4049/jimmunol.175.8.5430. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem. 2004;279:13792–13799. doi: 10.1074/jbc.M313724200. [DOI] [PubMed] [Google Scholar]
- Macario AJ, Conway de Macario E. Sick chaperones and ageing: a perspective. Ageing Res Rev. 2002;1:295–311. doi: 10.1016/s1568-1637(01)00005-8. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson WB, Bergmann BM. Age-dependent changes in recovery sleep after 48 hours of sleep deprivation in rats. Neurobiol Aging. 2000;21:689–693. doi: 10.1016/s0197-4580(00)00154-8. [DOI] [PubMed] [Google Scholar]
- Menza MA, Rosen RC. Sleep in Parkinson's disease. The role of depression and anxiety. Psychosomatics. 1995;36:262–266. doi: 10.1016/S0033-3182(95)71665-6. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92:1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Casiano V, Cater J, Zimmerman J, Pack AI. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep. 2007;30:557–565. doi: 10.1093/sleep/30.5.557. [DOI] [PubMed] [Google Scholar]
- Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Von Smith R, Kay T, Lian J, Svenson K, Peters LL. Novel method for high-throughput phenotyping of sleep in mice. Physiol Genomics. 2007;28:232–238. doi: 10.1152/physiolgenomics.00139.2006. [DOI] [PubMed] [Google Scholar]
- Perutz MF. Glutamine repeats and neurodegenerative diseases: molecular aspects. Trends Biochem Sci. 1999;24:58–63. doi: 10.1016/s0968-0004(98)01350-4. [DOI] [PubMed] [Google Scholar]
- Rabek JP, Boylston WH, III, Papaconstantinou J. Carbonylation of ER chaperone proteins in aged mouse liver. Biochem Biophys Res Commun. 2003;305:566–572. doi: 10.1016/s0006-291x(03)00826-x. [DOI] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005a;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005b;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [Erratum (2004) 428:445] [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster . Science. 2000;287:1834–1837. doi: 10.1126/science.287.5459.1834. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Lu J, Wagner D, Thakkar J, Greco MA, Basheer R, Thakkar M. Compensatory sleep response to 12 h wakefulness in young and old rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R125–R133. doi: 10.1152/ajpregu.2000.278.1.R125. [DOI] [PubMed] [Google Scholar]
- Swaab DF, Dubelaar EJ, Hofman MA, Scherder EJ, van Someren EJ, Verwer RW. Brain aging and Alzheimer's disease; use it or lose it. Prog Brain Res. 2002;138:343–373. doi: 10.1016/S0079-6123(02)38086-5. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann NY Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Reports. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Pepys M. Amyloidosis. Histopathology. 1994;25:403–414. doi: 10.1111/j.1365-2559.1994.tb00001.x. [DOI] [PubMed] [Google Scholar]
- Terao A, Steininger TL, Hyder K, Apte-Deshpande A, Ding J, Rishipathak D, Davis RW, Heller HC, Kilduff TS. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience. 2003;116:187–200. doi: 10.1016/s0306-4522(02)00695-4. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Valentinuzzi VS, Scarbrough K, Takahashi JS, Turek FW. Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. Am J Physiol. 1997;273:R1957–R1964. doi: 10.1152/ajpregu.1997.273.6.R1957. [DOI] [PubMed] [Google Scholar]
- Van Someren EJ. Circadian rhythms and sleep in human aging. Chronobiol Int. 2000;17:233–243. doi: 10.1081/cbi-100101046. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson's disease. Antioxid Redox Signal. 2007;9:553–561. doi: 10.1089/ars.2006.1524. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Richardson GS, Dement WC. Effect of age on the circadian pattern of sleep and wakefulness in the mouse. J Gerontol. 1986;41:579–586. doi: 10.1093/geronj/41.5.579. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]