

Abstract
Transforming growth factor-β (TGF-β) is thought to play a role in the pathobiological progression of ovarian cancer because this peptide hormone is overexpressed in cancer tissue, plasma, and peritoneal fluid. In the current study, we investigated the role of the TGF-β/Smad3 pathway in ovarian cancer metastasis by regulation of an epithelial-to-mesenchymal transition. When cancer cells were cultured on plastic, TGF-β1, TGF-β2, and TGF-β3 induced pro–matrix metalloproteinase (MMP) secretion, loss of cell-cell junctions, down-regulation of E-cadherin, up-regulation of N-cadherin, and acquisition of a fibroblastoid phenotype, consistent with an epithelial-to-mesenchymal transition. Furthermore, Smad3 small interfering RNA transfection inhibited TGF-β–mediated changes to a fibroblastic morphology, but not MMP secretion. When cancer cells were cultured on a three-dimensional collagen matrix, TGF-β1, TGF-β2, and TGF-β3 stimulated both pro-MMP and active MMP secretion and invasion. Smad3 small interfering RNA transfection of cells cultured on a collagen matrix abrogated TGF-β–stimulated invasion and MMP secretion. Analysis of Smad3 nuclear expression in microarrays of serous benign tumors, borderline tumors, and cystadenocarcinoma revealed that Smad3 expression could be used to distinguish benign and borderline tumors from carcinoma (P = 0.006). Higher Smad3 expression also correlated with poor survival (P = 0.031). Furthermore, a direct relationship exists between Smad3 nuclear expression and expression of the mesenchymal marker N-cadherin in cancer patients (P = 0.0057). Collectively, these results implicate an important role for the TGF-β/Smad3 pathway in mediating ovarian oncogenesis by enhancing metastatic potential.
Introduction
More than 80% of ovarian cancers are thought to originate from the single layer of epithelia surrounding the ovary, called the ovarian surface epithelia (OSE). OSE cells are distinct from other epithelia in that they possess a relatively uncommitted phenotype, expressing both epithelial and mesenchymal characteristics (1). The coelemic epithelium gives rise to OSE cells and cells that form the Müllerian duct, which subsequently differentiate into the endocervix, endometrium, and Fallopian tube (2). Ovarian cancer is unique from other cancer models, because cancer cells acquire more specialized epithelial markers, such as E-cadherin, when compared with their hypothesized tissue of origin, the OSE (3). Ovarian cancer is thought to arise from neoplastic changes that result in the pathologic differentiation of OSE into the established histologic subtypes, which include serous (fallopian tube–like), endometrioid (endometrium-like), and mucinous (endocervical-like) adenocarcinoma among others. Although OSE have a more uncommitted phenotype than malignant epithelia, we hypothesize that cancer cells undergo an epithelial-to-mesenchymal transition (EMT) to disseminate within the i.p. cavity or metastasize to a distant site.
EMTs occur as a part of developmental and physiologic programs that orchestrate events, such as neural crest formation, palatal fusion, and wound healing (4). Cancer cells in advanced stages become aggressive and invasive due to the pathologic initiation of an EMT that often recapitulates developmental signaling cascades. EMTs involve coordination of a complex genetic program that results in the loss of epithelial markers (e.g., E-cadherin, occludin) and the acquisition of mesenchymal markers (e.g., N-cadherin, vimentin). Cancer cells that undergo an EMT change from a compact, cuboidal (epithelial) shape to a spindle-like, fibroblastic (mesenchymal) morphology. The transition causes cells to lose cell-cell contacts and cell polarity, become motile, and secrete proteases that degrade extracellular matrix (5). Transforming growth factor-β (TGF-β) is thought to contribute to oncogenesis by inducing an EMT that promotes tumor metastasis (6).
The TGF-β superfamily participates in a diverse array of reproductive, differentiation, and developmental programs (7). Family members are subdivided into the TGF-β/activin/nodal branch and the bone morphogenetic protein/growth and differentiation factor branch because of the specific receptor-regulated Smads they activate (8). TGF-β1, TGF-β2, and TGF-β3 exert biological effects by binding to type I (TβRI) and type II (TβRII) receptors. Upon ligand binding to TβRII, TβRI is activated and phosphorylates the receptor-regulated Smads, Smad2 and Smad3. Phosphorylated receptor-regulated Smads then bind to the co-Smad, Smad4, and translocate to the nucleus to modulate gene expression. TGF-β also initiates Smad-independent pathways, including those mediated by the mitogen-activated protein kinase family members (TAK1, extracellular signal-regulated kinase, p38, c-Jun-NH2-kinase) and phosphatidylinositol 3-kinase (9).
TGF-β can function as a tumor suppressor by inhibiting cell proliferation, but can also promote metastasis in various cancer models (10, 11). Although TGF-β plays an important role in ovarian physiology, much remains to be elucidated about how this hormone contributes to ovarian cancer progression, particularly in the regulation of an EMT. TGF-β1 elevates matrix metalloproteinase (MMP) secretion and invasion through Matrigel in ovarian cancer cells (12, 13). Another study reports that TGF-β1 induces an EMT in two ovarian adenocarcinoma cell lines, derived from a rare variant of a Müllerian mixed tumor (14).
Several clinical studies suggest that TGF-β is involved in ovarian tumor progression. TGF-β1, TGF-β2, and TGF-β3 are overexpressed in 44%, 66%, and 66% of malignant ovarian tumors, respectively (15). Notably, TGF-β1 expression is less frequent than TGF-β2 and TGF-β3, underscoring the relevance of studying the TGF-β2 and TGF-β3 isoforms. Furthermore, TGF-β1 and latent TGF-β1-binding protein mRNAs are up-regulated in ovarian carcinoma tissue relative to benign tissue (16), and TGF-β1 levels are elevated in the plasma and peritoneal fluid of ovarian cancer patients (17). Although the role of TGF-β1 in oncogenesis has been studied extensively, the function of TGF-β2 and TGF-β3 in ovarian cancer and other cancers remains relatively unexplored.
In the current study, we investigated the potential of TGF-β1, TGF-β2, and TGF-β3 to initiate an EMT in ovarian cancer cells. All three isoforms were able to efficiently induce an EMT in ovarian cancer cells, disrupting cell-cell junctions and stimulating MMP secretion. Furthermore, Smad3 is required for the EMT mediated by all three TGF-β family members, and down-regulation of Smad3 expression inhibits the loss of cell-cell contacts and transition to a fibroblastic morphology. Within the context of a three-dimensional collagen matrix, Smad3 is required for TGF-β–regulated secretion of pro-MMP and active MMP and invasion. Consistent with the in vitro findings, analysis of serous cystadenocarcinoma revealed that higher Smad3 nuclear expression correlates with poor outcome. Taken together, these results suggest that the TGF-β/Smad3 pathway plays an important role in ovarian tumor progression by promoting the dissemination of cancer cells.
Results
TGF-β1, TGF-β2, and TGF-β3 Induce an EMT and MMP Secretion in Ovarian Cancer Cells
The role of TGF-β, particularly that of TGF-β2 and TGF-β3, in regulating EMTs in ovarian cancer remains relatively unexplored. Therefore, we treated the human cancer cell line, OVCA429, with TGF-β1, TGF-β2, and TGF-β3 to determine if any of these family members can initiate an EMT, causing cells to lose cell-cell contacts and to change from a compact, epithelial morphology to a spindle-shaped fibroblastic morphology. Notably, all three ligands efficiently induced a fibroblastic morphology in OVCA429 cells after 72 h (Fig. 1A, brightfield images).
FIGURE 1.

TGF-β1, TGF-β2, and TGF-β3 mediate an EMT and MMP secretion in ovarian cancer cells. OVCA429 cells were serum-starved for 24 h and then treated with vehicle (Veh), 10 ng/mL TGF-β1, TGF-β2, TGF-β3, or activin A for 72 h. A. The TGF-β ligands stimulate an EMT in cancer cells. Top, brightfield images of cells showing that TGF-β initiates the loss of cell-cell junctions and the change from an epithelial to a fibroblastic morphology (bar, 20 μm); bottom, OVCA429 cells immunostained with anti–E-cadherin, anti-occludin, anti–N-cadherin, or anti-vimentin (FITC, white arrowheads) and counterstained with 4′,6-diamidino-2-phenylindole (blue) to show nuclei (bar, 50 μm). B. Anti–E-cadherin and anti–N-cadherin immunoblots of quiescent cells treated with vehicle, activin A, TGF-β1, TGF-β2, or TGF-β3. The same blot was stripped and reprobed with anti–β-actin as a loading control. C. TGF-β1, TGF-β2, and TGF-β3 trigger pro–MMP-2 and pro–MMP-9 secretion in cancer cells, but not in the normal cell line IOSE80. Conditioned media from quiescent OVCA429 and IOSE80 cells treated with vehicle, TGF-β1, TGF-β2, or TGF-β3 were subjected to gelatin zymography to analyze MMP secretion.
We further investigated the effects of TGF-β1, TGF-β2, and TGF-β3 on the epithelial markers, E-cadherin and occludin, and the mesenchymal markers, vimentin and N-cadherin, by indirect immunofluorescent staining. All three ligands elicited a fibroblastic, undifferentiated phenotype in OVCA429 cells, disrupting adherens junctions and tight junctions, as shown by E-cadherin (Fig. 1A, i-iv) and occludin immunofluorescence (Fig. 1A, v-viii), respectively. E-cadherin staining was present at adherens junctions in vehicle-treated cells (Fig. 1A, i, white arrowhead), but was markedly decreased in TGF-β1–treated, TGF-β2–treated, and TGF-β3–treated cells (Fig. 1A, ii-iv). Similarly, occludin staining was present at tight junctions in vehicle-treated cells (Fig. 1A, v, white arrowhead), but was severely diminished in TGF-β–treated cells (Fig. 1A, vi-viii). Furthermore, treatment with all three ligands increased the immunostaining of the mesenchymal marker vimentin, an intermediate filament protein (Fig. 1A, x-xii, white arrowheads), and N-cadherin (Fig. 1A, xiv-xvi, white arrowheads) when compared with vehicle-treated cells (Fig. 1A, ix and xiii). Consistent with these observations, TGF-β1, TGF-β2, and TGF-β3 up-regulated N-cadherin and down-regulated E-cadherin protein expression (Fig. 1B). Cells were treated with activin A, for 72 h, as a negative control, because activin A did not mediate an EMT in OVCA429 cells (data not shown). Treatment with activin A did not affect E-cadherin or N-cadherin expression (Fig. 1B). Because the TGF-β ligands caused ovarian cancer cells to undergo dramatic changes in cell morphology suggestive of an EMT, the effect of these ligands on MMP secretion was also evaluated. Conditioned media was collected from cells treated with TGF-β and subjected to gelatin zymography. TGF-β treatment had no effect on MMP-2 and MMP-9 secretion in the cell line, IOSE80 (normal human OSE cells transfected with SV40 large T antigen to allow for longer passage times in vitro; Fig. 1C, top), but augmented both pro–MMP-2 and pro–MMP-9 secretion in OVCA429 carcinoma cells (Fig. 1C, bottom).
Collectively, these results suggest that all three TGF-β ligands exert prometastatic effects on ovarian cancer cells by inducing an undifferentiated cell morphology and MMP secretion.
Smad3 Regulates TGF-β–Mediated Changes in Cell Morphology, but not MMP Secretion in Cells Cultured on Plastic
To determine the relevant Smad pathway activated down-stream of TGF-β signaling, the phosphorylation of Smad2 and Smad3 was investigated. TGF-β1, TGF-β2, and TGF-β3 stimulated phosphorylation of Smad3 (Fig. 2A), but only low level or no phosphorylation of Smad2 was detected (Fig. 2B). Lysates from the mouse gonadotrope cell line LβT2, treated with activin A, were used as a positive control for the phosphorylated Smad2 immunoblot (Fig. 2B). The effects of the TGF-β ligands on Smad3 nucleocytoplasmic shuttling were also assessed using a Smad3-GFP construct, because Smad3-GFP has been shown to behave similarly to endogenous, wild-type Smad3 (18). Treatment of OVCA429 cells with TGF-β ligands for 30 minutes resulted in predominantly nuclear localization of Smad3-GFP (Fig. 2C) in contrast to the diffuse cytoplasmic and weak nuclear Smad3-GFP localization in vehicle-treated cells (Fig. 2C). Taken together, these results suggest that Smad3 is activated as a result of TGF-β stimulation.
FIGURE 2.

TGF-β1, TGF-β2, and TGF-β3 stimulate Smad3 phosphorylation and nuclear localization. OVCA429 cells were made quiescent and then treated with vehicle, 10 ng/mL TGF-β1, TGF-β2, or TGF-β3. A. Anti–phosphorylated Smad3 immunoblot. OVCA429 cells were treated with TGF-β ligands for 0, 10, 20, 30, 45, and 60 min. The same blot was stripped and reprobed with anti-Smad3 as a loading control. B. Anti–phosphorylated Smad2 immunoblot. OVCA429 cells were treated with TGF-β ligands for 0, 10, 20, 30, 45, and 60 min. The same blot was reprobed with anti-Smad2 as a loading control. Lysates from LβT2 cells treated with activin A were used as a control for anti–phosphorylated Smad2 immunoreactivity. C. Fluorescent micrograph of cells transfected with Smad3-GFP for 24 h before treatment with vehicle, TGF-β1, TGF-β2, or TGF-β3 for 30 min. Bar, 50 μm.
We next analyzed the effects of inhibition of Smad3 expression on TGF-β–coordinated changes in cell morphology and MMP secretion. To perform this analysis, cells were transfected with Smad3-specific small interfering RNA (siRNA) to repress Smad3 expression. Cells were also transfected with nontargeting siRNA as a negative control. Smad3 siRNA transfection dramatically decreased Smad3 expression (Fig. 3A, lanes 5-8), whereas control siRNA treatment displayed minimal effect on Smad3 protein expression (Fig. 3A, lanes 1-4). Transfection of control siRNA did not dramatically alter TGF-β–induced loss of cell-cell junctions as evidenced by the diminution of E-cadherin staining in TGF-β–treated cells when compared with the robust E-cadherin staining in vehicle-treated cells (Fig. 3B, arrowhead; control siRNA). However, attenuation of Smad3 expression abrogated TGF-β–regulated cell scattering and loss of cell-cell junctions, as indicated by the presence of anti–E-cadherin immunofluorescence in both vehicle-treated and TGF-β–treated cells (Fig. 3B, top and bottom, arrowheads; Smad3 siRNA). In addition, TGF-β–stimulated pro–MMP-2 and pro–MMP-9 secretion were equivalent in both Smad3 and control siRNA-transfected cells (Fig. 3C, compare lanes 2-4 and lanes 6-8).
FIGURE 3.

Smad3 is required for TGF-β–mediated EMT, but not MMP secretion in ovarian cancer cells cultured on plastic. Cells were cultured on plastic dishes and transfected with 50 nmol/L Smad3 siRNA or control (nontargeting) siRNA and then treated with vehicle, 10 ng/mL TGF-β1, TGF-β2, or TGF-β3 for 72 h. A. Anti-Smad3 immunoblot showing down-regulation of Smad3 protein in Smad3-siRNA–transfected cells. The same blot was also reprobed with anti–β-actin as a loading control. B. Brightfield images of quiescent cells transfected with control or Smad3 siRNAs and then treated with vehicle and TGF-β ligands (top); Anti–E-cadherin immunofluorescence (white arrowheads) showing that Smad3-siRNA–transfected cells do not undergo an EMT in response to TGF-β1, TGF-β2, or TGF-β3, in contrast to control-siRNA–transfected cells (bottom); bar, 20 μm. C. Gelatin zymography of conditioned media from cells transfected with 50 nmol/L Smad3 or control (nontargeting) siRNA showing effects on MMP secretion.
These data indicate that Smad3 is required for TGF-β–mediated loss of cell-cell adhesion and cell shape changes, but does not substantially alter TGF-β–triggered MMP secretion.
Inhibition of Smad3 Expression Ablates TGF-β–Stimulated Cellular Invasion and MMP Secretion in Ovarian Cancer Cells Cultured on a Collagen Matrix
Because Smad3 is required for TGF-β–regulated of loss of cell-cell contacts and changes in cell morphology that would promote ovarian cancer dissemination, we next determined the requirement for Smad3 in cellular invasion through a three-dimensional collagen matrix. Invasion through a type I collagen matrix was evaluated, because ovarian cancer cells preferentially adhere to this collagen type (19). OVCA429 cells were transfected with nontargeting, control siRNA or Smad3 siRNA and then allowed to invade through the three-dimensional matrix in the presence of TGF-β ligands. When cells were transfected with control siRNA, treatment with TGF-β1, TGF-β2, or TGF-β3 enhanced invasion 2.1-fold, 1.7-fold, and 1.8-fold (Fig. 4A, columns 2-4), respectively, compared with vehicle treatment (Fig. 4A, column 1). However, Smad3 siRNA transfection dampened basal invasive potential (Fig. 4A, column 5) by 44.7% and repressed TGF-β1–triggered, TGF-β2–triggered, and TGF-β3–triggered invasion (Fig. 4A, columns 6-8) by 68.1%, 53.8%, and 62.8%, respectively, when compared with the corresponding treatments in control siRNA-transfected cells (Fig. 4A, columns 1-4). Most importantly, Smad3 siRNA transfection abrogated TGF-β1–induced, TGF-β2–induced, and TGF-β3–induced cellular invasion when compared with vehicle treatment (Fig. 4A, compare column 5 with columns 6-8).
FIGURE 4.
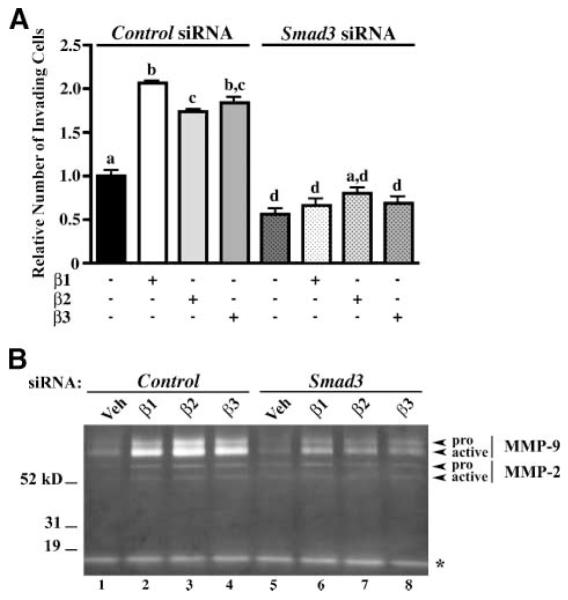
TGF-β–induced invasion through a three-dimensional collagen matrix and (pro and active) MMP secretion is Smad3 dependent. To assay invasive potential, OVCA429 cells were transfected with 50 nmol/L control (nontargeting) siRNA or Smad3 siRNA for 24 h and then cultured on a three-dimensional type I collagen matrix in a modified Boyden chamber setup. Cells were allowed to invade for 72 h in the presence of 10 ng/mL TGF-β1, TGF-β2, or TGF-β3. As a negative control, cells were treated with vehicle. A. Quantification of cellular invasion through a type I collagen matrix. Columns, means relative to control siRNA-transfected cells treated with vehicle; bars, SE (n = 3). Bars labeled with different letters are statistically significant as analyzed by repeated measures ANOVA, followed by Tukey’s multiple comparison test (P < 0.05). B. Gelatin zymography of conditioned media collected from cells subjected to invasion assay through a three-dimensional type I collagen matrix. Zymogram shows secreted pro–MMP-2, active MMP-2, pro–MMP-9, active MMP-9, and an unidentified gelatinase (*).
Gelatin zymography of conditioned media from invading cells reveals that, within the context of a three-dimensional matrix, TGF-β1, TGF-β2, and TGF-β3 augmented the production of both pro- and active MMP-2 and MMP-9 (Fig. 4B, compare lane 1 with lanes 2-4) although MMP-9 expression is preferentially enhanced over that of MMP-2 (recall that TGF-β ligands stimulate pro-MMP and not active MMP expression when cultured on plastic). Cells treated with vehicle expressed low levels of pro- and active MMP-2 and MMP-9 (Fig. 4B, lanes 1 and 5). An unidentified, low–molecular weight gelatinase was also detected, but TGF-β had no effect on the levels of this gelatinase (Fig. 4B). Furthermore, Smad3 siRNA transfection dramatically diminishes TGF-β1–stimulated, TGF-β2–stimulated, and TGF-β3–stimulated expression of both the pro-MMP and active forms of MMPs (Fig. 4B, compare lanes 6-8 with lanes 2-4).
Together, these results suggest that Smad3 is required for TGF-β to promote pro-MMP and active MMP expression and invasion within a three-dimensional type I collagen matrix.
Smad3 and N-Cadherin Expression in Benign Serous Cystadenoma, Borderline Tumor, and Cystadenocarcinoma Microarrays
To evaluate the relevance of Smad3 to the pathologic status of OSE, we analyzed Smad3 nuclear expression in ovarian epithelia from three microarrays consisting of benign serous cystadenoma, borderline tumor, and cystadenocarcinoma tissues. The expression of N-cadherin was also analyzed, because all three TGF-β ligands up-regulated the expression of this mesenchymal marker. For both protein markers, immunostaining was classified as 0 (no staining), 1 (weak to moderate staining), or 2 (strong staining). The scores for all tissue cores were averaged for each patient within a microarray. Only patients with at least three tissue cores displaying epithelia that could be scored for immunostaining were included in our data analyses. The average scores for benign, borderline, and carcinoma patients were then divided into three categories: (a) no expression (average score = 0), (b) low expression (average score < 1.5), and (c) high expression (average score ≥ 1.5).
Smad3 expression was present in epithelia from all tissue categories, but there was a statistically significant difference in average nuclear Smad3 score among benign serous cystadenoma, borderline tumor, and cystadenocarcinoma (Mantel-Haenszel 03C72 test, P = 0.0065; Table 1). Low nuclear Smad3 expression was detected in 20% of benign, 20% of borderline, and 51.3% of cystadenocarcinoma patients (Table 1). High nuclear Smad3 expression was detected in 80% of benign, 80% of borderline, and 48.7% of carcinoma patients (Table 1). Pairwise comparison showed that average nuclear Smad3 scores were statistically different between benign and cystadenocarcinoma tissues and between borderline and cystadenocarcinoma tissues, but not between benign and borderline tumors (P < 0.05). More specifically, benign and borderline tumors were more likely to exhibit high Smad3 nuclear expression (average score ≥ 1.5) than cystadenocarcinoma, exhibiting odds ratios of 4.2 (95% confidence interval, 1.2-14.9; P = 0.0257) and 4.2 (95% confidence interval, 1.4-12.6; P = 0.0099), respectively (Table 1).
Table 1.
Nuclear Smad3 and N-Cadherin Expression in Benign Cystadenoma, Borderline Tumor, and Cystadenocarcinoma Tissue Microarrays
| Frequency of Average Scores |
P | Total No. Patients | |||
|---|---|---|---|---|---|
| 0 | <1.5 | ≥1.5 | |||
| Smad3 | 0.031* | ||||
| Benign | 0 | 4 | 16 | 1.0 (benign versus borderline) | 20 |
| Borderline | 0 | 6 | 24 | 0.0099 (borderline versus carcinoma) | 30 |
| Cystadenocarcinoma | 0 | 20 | 19 | 0.0257 (benign versus carcinoma) | 39 |
| N-Cadherin | 0.996* | ||||
| Benign | 2 | 12 | 5 | 0.0669 (benign versus borderline) | 19 |
| Borderline | 5 | 24 | 1 | 0.773 (borderline versus carcinoma) | 30 |
| Cystadenocarcinoma | 14 | 22 | 4 | 0.0319 (benign versus carcinoma) | 40 |
NOTE: Pairwise contrast (logistic regression) between tissue categories are comparing (a) Smad3 average scores of <1.5 versus average scores of ≥1.5 and (b) N-cadherin average scores of 0 versus average scores of ≥1.5.
Cox proportional hazards regression analysis using Smad3 or N-cadherin as a continuous variable.
The relationship between Smad3 nuclear expression and survival in serous cystadenocarcinoma patients was also evaluated. All cystadenocarcinoma tissues exhibited Smad3 nuclear staining. Representative brightfield images of Smad3 nuclear expression in tissue core punches that scored 1 and 2 from the carcinoma microarray are shown in Fig. 5A. Using Smad3 as a continuous variable in a proportional hazards model, higher Smad3 nuclear expression correlated with shorter survival (P = 0.031; Table 1). Comparison of survival times (in a dichotomized analysis) between patients exhibiting low Smad3 nuclear expression (average score < 1.5) versus high nuclear Smad3 expression (average score ≥ 1.5) revealed a marginal correlation between survival and Smad3 expression (P = 0.0513; Fig. 5B). Patients exhibiting low Smad3 nuclear expression had a median survival time that was 10.3 months longer than that of patients exhibiting high nuclear Smad3 expression (Fig. 5B).
FIGURE 5.

Smad3 and N-cadherin expression in serous cystadenocarcinoma and relationship to survival. A. Brightfield images (20×) showing immunostaining of Smad3 or N-cadherin assigned a score of 1 or 2 in serous cystadenocarcinoma tissue cores counterstained with hematoxylin; bar, 100 μm. B. Survival curves for serous cystadenocarcinoma patients based on average scores for Smad3 (nuclear) and N-cadherin expression. For Smad3, survival curves were plotted for patients exhibiting low (average score < 1.5) and high (average score ≥ 1.5) nuclear expression (P = 0.0513). For N-cadherin, survival curves were plotted for patients with no (average score = 0) expression, low expression (average score < 1.5), and high expression (average score > 1.5; P = 0.9295). Scores for all tissue cores from each patient were averaged and then subjected to Kaplan-Meier estimate and log-rank test.
Because TGF-β1, TGF-β2, and TGF-β3 treatment increased N-cadherin protein levels (Fig. 1B), N-cadherin expression in benign serous cystadenoma, borderline tumors, and cystadenocarcinoma was also analyzed. Representative brightfield images of N-cadherin immunostaining in serous cystadenocarcinoma core punches that received a score of 1 and 2 are shown in Fig. 5A. No N-cadherin expression (average score = 0) was detected in 10.5% of benign, 16.7% of borderline, and 35% of serous cystadenocarcinoma patients (Table 1). Low N-cadherin expression (average score < 1.5) was observed in 63.2% of benign, 80% of borderline, and 55% of carcinoma patients (Table 1). High N-cadherin expression (average score ≥ 1.5) was detected in 26.3% of benign, 3.3% of borderline, and 10% of carcinoma patients (Table 1). Pairwise analysis of the three tissue categories revealed that serous cystadenocarcinoma was 8.75 times (95% confidence interval, 1.21-63.43; P = 0.0319) more likely to show no N-cadherin expression than high N-cadherin expression when compared with benign tumors (Table 1).
The relationship between N-cadherin expression and survival was also evaluated. Using N-cadherin as a continuous variable in a proportional hazards model, we did not find a correlation between N-cadherin expression and outcome (P = 0.996; Table 1). Survival for patients exhibiting no expression (average score = 0), low expression (average score < 1.5), and high N-cadherin expression (average score ≥ 1.5) did not differ in a statistically significant manner (P = 0.9295; Fig. 5B). However, there was a correlation between N-cadherin expression and Smad3 nuclear expression (P < 0.01). Cystadenocarcinoma tissues that displayed no N-cadherin expression were more likely to exhibit low Smad3 nuclear expression, and cystadenocarcinoma tissues that displayed N-cadherin expression were more likely to exhibit high Smad3 nuclear expression (odds ratio, 10.39; 95% confidence interval, 1.88-57.42; P = 0.0057; Table 2). For carcinoma patients that showed no N-cadherin expression, 84.6% exhibited low Smad3 nuclear expression and 15.4% exhibited high Smad3 nuclear expression (Table 2). For carcinoma patients that were positive for N-cadherin expression, 34.6% exhibited low Smad3 nuclear expression and 65.4% exhibited high Smad3 nuclear expression (Table 2).
Table 2.
Relationship Between Smad3 (Nuclear) and N-Cadherin Expression in Serous Cystadenocarcinoma
| Frequency of N-Cadherin and Smad3 Average Scores | |||
|---|---|---|---|
| N-Cadherin | Smad3 |
Total | |
| <1.5 | ≥1.5 | ||
| =0 | 11 | 2 | 13 |
| >0 | 9 | 17 | 26 |
| Total | 20 | 19 | 39 |
NOTE: Patients with N-cadherin average scores of 0 or >0 were subdivided according to low (average score < 1.5) and high (average score ≥ 1.5) Smad3 nuclear expression (P = 0.0057). The total number of patients that fall into each category for N-cadherin and Smad3 expression was also tabulated.
In summary, these results show that Smad3 can be used to distinguish ovarian epithelia of different pathologic status. Higher Smad3 nuclear expression also correlated with poor survival, supporting a role for Smad3 in ovarian oncogenesis. Finally, there was a correlation between N-cadherin expression levels and Smad3 nuclear expression levels in cancer epithelia, which is consistent with our in vitro experimental findings.
Discussion
Ovarian cancer is the most lethal gynecologic malignancy in the Western world and is often diagnosed during late stages when metastasis has occurred (20). Five-year survival rates increase dramatically from 20% to 30% to over 90% if cancer is detected during early stages when disease is confined to the ovary (20-22). Therefore, understanding the mechanisms that govern ovarian cancer metastasis is a crucial step in controlling and treating this disease.
In the current study, we evaluated the role of TGF-β1, TGF-β2, and TGF-β3 in mediating an EMT to enhance ovarian cancer metastatic potential. All three TGF-β family members were capable of inducing an EMT, MMP secretion, and invasion. Furthermore, these TGF-β ligands signaled through Smad3 to stimulate changes in cell morphology and regulate MMP secretion within the context of a three-dimensional collagen matrix. Consistent with these in vitro observations is the correlation between (a) higher Smad3 expression and higher N-cadherin expression in cystadenocarcinomas and (b) higher Smad3 expression and poor outcome in cancer patients.
Because all three TGF-β ligands elicited similar responses in ovarian cancer cells, it is predicted that elevated levels of TGF-β1, TGF-β2, and TGF-β3 observed in cancer tissues (15) would promote dedifferentiation of cancer cells and enhance i.p. dissemination. Our analysis of Smad3 expression in human ovarian tissues revealed that serous cystadenocarcinoma was more likely to display lower levels of Smad3 nuclear expression than benign tumors. In addition, N-cadherin expression was higher in benign epithelia than in cancer epithelia, which was more likely to exhibit no N-cadherin staining. These observations raise the possibility that down-regulation of Smad3 and loss of the mesenchymal marker N-cadherin is a consequence of the pathologic differentiation of the putative precursor OSE to become fallopian tube–like, serous carcinoma (1). In fact, normal OSE have a more uncommitted phenotype than malignant epithelia, and ovarian cancer cells acquire expression of the epithelial marker E-cadherin after neoplastic transformation. Expression of E-cadherin in SV40-immortalized OSE cells induced a mesenchymal-to-epithelial transition, which resulted in the formation of simple epithelia (in three-dimensional culture) that secreted CA125, a protein produced by metaplastic and neoplastic OSE (23). Normal OSE seldom express E-cadherin (24-26), but do express N-cadherin (27, 28), which may play a role in the mesenchymal conversion of OSE that occurs when cultured in vitro. A final point to consider is that carcinoma cells expressing high levels of N-cadherin and nuclear Smad3 may be the result of elevated TGF-β production in these cells. Likewise, low TGF-β production may result in lower nuclear Smad3 and N-cadherin expression.
Analysis of ovarian carcinoma revealed higher Smad3 expression correlated with poor survival, suggesting that Smad3 plays a key role in ovarian tumorigenesis. One possible explanation for this observation is that carcinoma displaying high nuclear Smad3 expression may possess greater metastatic potential than carcinoma expressing low Smad3 nuclear expression. These results are consistent with a recent study that analyzed Smad3 expression in a microarray consisting of 244 specimens from serous epithelial ovarian tumors. This study reported that Smad3 expression could be used to differentiate between grade 0 and grade 1 or grade 0 and grade 2 serous tumors. Furthermore, high Smad3 expression correlated with patients having survival times of <18 months (29). This particular study scored only two tissue core punches, whereas we scored three tissue core punches for each patient. We chose to score three tissue core punches, because one disadvantage of the tissue microarray is that the small sample of tissue derived from a single core punch may not fairly represent the pathologic status of the entire tumor. Rosen et al. (30) did a study to validate the use of tissue microarrays for serous ovarian carcinoma and concluded that three tissue cores could correctly represent the expression of the markers, p53, estrogen receptor, and Ki-67 in whole tissue section with 98% probability. Also, we scored both Smad3 cytoplasmic staining and nuclear staining and did statistical analyses on whether (a) Smad3 cytoplasmic staining, (b) Smad3 nuclear staining, or (c) the ratio of Smad3 nuclear to cytoplasmic staining could stratify disease or predict survival (data not shown). Only Smad3 nuclear staining could be used to stratify disease and predict survival.
There is an apparent contradiction between our findings that carcinoma tended to display lower Smad3 expression and higher Smad3 expression correlated with poor prognosis. We reconcile these findings with the hypothesis that Smad3 is down-regulated as a consequence of the pathologic differentiation to become serous adenocarcinoma, but carcinomas which subsequently undergo changes resulting in up-regulation of Smad3 expression are more likely to spread beyond the ovary and result in poor survival.
To our knowledge, a role for Smad3 in the regulation of EMTs, MMP secretion, and invasion in ovarian cancer has not been reported in the literature, but a role for Smad3 as both a tumor suppressor and tumor promoter for various cancer models has been described. Gastric tumors express low to undetectable levels of Smad3, and cancer cells that express Smad3 show a decrease in tumorigenicity in vivo (31). Also, Smad3 null mice develop colorectal adenocarcinomas that metastasize to the lymph nodes (32). Intriguing studies done on Smad2 and Smad3 heterozygous mice, using a chemically induced skin carcinogenesis model, revealed that the Smads might play opposing roles during oncogenesis. Smad3(+/−) mice developed significantly less tumors compared with wild-type mice (33). However, Smad2(+/−) mice developed more undifferentiated tumors than wild-type mice. These observations show that Smad3 plays a tumor-promoting role, whereas Smad2 plays a tumor-suppressing role. Results from our current study suggest that Smad3 may act as a tumor promoter during late-stage ovarian oncogenesis by initiating changes to an undifferentiated phenotype and enhancing metastatic potential. Furthermore, because OSE acquire more specialized epithelial characteristics upon transformation to become the various ovarian cancer histologic subtypes, it will be important to determine if Smad3 can act as a tumor suppressor in early disease by promoting the mesenchymal phenotype.
In vitro data from the current study suggests that Smad3 regulation of MMP secretion is dependent upon the surrounding microenvironment. Smad3 is required for TGF-β–initiated morphologic changes, but not MMP secretion when cells are grown on cell culture plastic. However, Smad3 can positively mediate TGF-β–induced pro-MMP and active MMP secretion when cancer cells are cultured on a three-dimensional collagen matrix. This suggests that Smad3 regulation of MMP secretion and/or activation may require integrin/collagen interactions. Changes in integrin/extracellular matrix interactions can alter adhesion, cell shape, and protease expression, secretion, and activity in multiple cell types (34-38). In addition, mechanotransduction by Smad3 has been shown in a mouse model for incisional wound closure (39). Future studies should address the role of Smad3 in mechanotransduction that modulates changes in gene expression to alter ovarian cancer cell differentiation state and metastatic potential. It will also be important to determine if Smad3 modulates N-cadherin expression, which, consequently, contributes to the initiation of metastatic signaling cascades.
In summary, our results implicate a role for the TGF-β/Smad3 pathway in ovarian tumor development and metastasis. These findings are consistent with other studies that report a role for Smad3 in EMTs for other pathologies, including fibrosis and cancer. Elucidation of Smad-mediated pathways that regulate the epithelial cell plasticity required in the transformation of OSE to carcinoma and in the dissemination of cancer cells will lead to the identification of important therapeutic targets crucial to treating the disease.
Materials and Methods
Cell Culture, Treatments, and Transfections
The OVCA429 late-stage serous adenocarcinoma cell line, a generous gift from Dr. M. Sharon Stack (Northwestern University), was cultured as previously described (40). For all experiments, cells were cultured to 80% confluency and serum-starved for 24 h before treatment with 10 ng/mL human TGF-β1, TGF-β2, TGF-β3 (R&D Systems) or activin A, purified in our laboratory (41) for 72 h. For transient transfections, cells were plated in complete media and cultured overnight before transfecting with siGENOME SMARTPOOL siRNAs (Dharmacon) or Smad3-GFP using LipofectAMINE 2000 Reagent (Invitrogen) according to the manufacturer’s instructions. After 24 h, cells were cultured in serum-free media and treated with TGF-β or activin.
Immunofluorescence and Imaging Techniques
Cells were cultured on glass coverslips, serum-starved for 24 h, and then treated with vehicle, TGF-β1, TGF-β2, or TGF-β3. For occludin immunolocalization, cells were fixed in 4% paraformaldehyde in 20 mmol/L HEPES (pH 7.4) and 150 mmol/L NaCl for 20 min, permeablized in 0.1% Triton X-100/PBS for 10 min, blocked with 1% bovine serum albumin/PBS for 1 h, and then incubated at room temperature for 1 h with anti-occludin (Invitrogen) at 1:100 in blocking solution. Coverslips were then rinsed in PBS, incubated with donkey anti-mouse Cy3 or donkey anti-mouse FITC secondary antibody (Jackson Immunoreseach) and mounted in Vectashield media (Vector Laboratories). For anti–E-cadherin (Invitrogen) and anti-vimentin (Sigma-Aldrich) indirect immunofluorescence, cells were fixed in methanol for 2 min and then processed as described for anti-occludin. Images were acquired on a Nikon Eclipse TE2000-U microscope using the SPOT digital camera and Metamorph 5.0 software.
Gelatin Zymography
To analyze pro–MMP-2 processing, cells were cultured to 80% confluency in 12-well plates (Corning), grown overnight, and then treated as described above. Conditioned media was subjected to SDS-PAGE, under nonreducing conditions, using 9% polyacrylamide gels containing 0.1% gelatin. Gels were washed twice in 2.5% Triton X-100 for 30 min, incubated in 20 mmol/L glycine, 10 mmol/L CaCl2, and 1 μmol/L ZnCl2 (pH 8.3) at 37°C for 24 h, and then stained in Coomassie Blue to detect areas of gelatinolytic activity.
Immunoblotting
Whole-cell lysates were prepared by lysing cells in 50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1% TritonX-100, and 0.1% SDS supplemented with phosphatase (Sigma-Aldrich) and protease (Roche Diagnostics) inhibitor cocktail. Lysates were separated on 4% to 12% polyacrylamide gradient gels (Invitrogen) and then transferred to nitrocellulose. Blots were blocked for 1 h in 5% milk/0.1% Tween 20 in TBS (TBS-T) and then incubated with primary antibody in 3% bovine serum albumin/TBS–T overnight at 4°C. Blots were then washed thrice for 15 min in TBS-T, incubated in goat anti-mouse or goat anti-rabbit antibodies conjugated to horseradish peroxidase in 5% milk/TBS-T for 1 h, washed thrice for 15 min in TBS-T and then twice for 10 min in TBS, and developed with ECL Plus Reagent (Amersham). Primary antibodies used were as follows: anti–E-cadherin, anti–N-cadherin, anti-Smad2, and anti-Smad3 were from Invitrogen; anti–β-actin was from Sigma-Aldrich; anti–phosphorylated Smad2 and anti–phosphorylated Smad3 were from Cell Signaling Technology.
Invasion Assay
Cells were transfected with either nontargeting or Smad3 siRNA as described above, and then 2 × 105 cells were cultured in inserts with 8-μm pores (Corning) overlaid with 20 μg of a type I collagen matrix (Sigma-Aldrich). Cells were allowed to invade for 72 h in the presence of serum-free media containing vehicle, 10 ng/mL TGF-β1, TGF-β2, or TGF-β3. Noninvading cells were removed with a cotton swab, and cells that invaded through the matrix were fixed and stained with Dif-Quik (Becton Dickinson) according to the manufacturer’s instructions. For each insert, 10 different fields of cells were counted to quantify the number of invading cells. Data were presented as means ± SE from three independent experiments. Statistical analysis was done using repeated measures ANOVA, followed by Tukey’s multiple comparison test (Prism 4 software).
Immunohistochemistry
Immunohistochemistry was carried out as previously described (42). Briefly, slides were deparaffinized and rehydrated in a graded series of ethanol. Antigen retrieval was done in a 10 mmol/L sodium citrate buffer (pH 6.0) and permeablized in TBS-T. Endogenous peroxidase activity was quenched in a 3% H2O2 solution before blocking with the Avidin-Biotin kit (Vector Laboratories). Slides were incubated for 1 h at room temperature in blocking solution and then incubated, overnight at 4°C, with primary antibody in blocking solution. Slides were then washed in TBS-T and incubated with 2.5 μg/mL biotin-labeled secondary antibody (Vector Laboratories) for 30 min at room temperature. Slides were washed again in TBS-T and incubated in ABC reagent (Vector Laboratories) for 30 min. Horseradish peroxidase was detected with the diaminobenzidine reagent kit (Vector Laboratories) and counterstained with Harris-modified hematoxylin (Sigma-Aldrich). Anti–N-cadherin (Invitrogen) and anti-Smad3 (Invitrogen) were used at 1:100 dilutions.
Ovarian Tissue Microarrays and Statistical Analyses
Human tissues were used in experiments after approval by an institutional review board. Tissue microarrays were created from archival, paraffin-embedded, human ovarian tissues from Evanston Northwestern Healthcare and classified into three categories: benign serous cystadenoma (n = 44), borderline serous tumor (n = 36), and malignant serous cystadenocarcinoma (n = 40). Core tissue punches (0.6 μm) were used to assemble the microarray, with three core punches for each patient in the borderline and carcinoma category and five core punches for each patient in the benign category. The mean ages for patients included in the serous benign, borderline, and carcinoma microarrays were 58.0 ± 13.7 y (range, 27-86 y), 51.6 ± 15.7 y (range, 27-85 y), and 62.4 ± 11.8 y (range, 37-82 y), respectively. Tumors were analyzed and staged by a pathologist according to the criteria of the Federation Internationale des Gynaecologistes et Obstetristes. The borderline microarray consisted of 2 stage I, 13 stage IA, 4 stage IB, 9 stage IC, 1 stage IIB, 3 stage IIC, 3 stage IIIA, and 1 stage IIIB. The carcinoma microarray consisted of 1 stage IA, 1 stage IIC, 3 stage III, 1 stage IIIB, 31 stage IIIC, and 2 stage IV patients. For all protein markers analyzed by immunohistochemistry, positive staining was defined as staining present in >25% of epithelia within a tissue core. Only those patients who had at least three tissue cores, which contained epithelia that could be scored for immunostaining, were used for data analysis. Because the number of tissue cores that could be scored varied from slide to slide for each microarray, the total number of patients included in data analysis for a specific immunohistochemistry marker could be less than the total number of patients included in each microarray. Protein expression was scored on a scale: 0 (no staining), 1 (weak to moderate staining), and 2 (strong staining). immunohistochemistry results were evaluated and scored by two pathologists, and the scores for each patient’s tissue cores were averaged, so that each patient was assigned one average score for each marker. Statistical analyses were done using SAS 9.1 and Prism 4 software. For comparison of average scores among the three microarrays, the Mantel-Haenszel 03C72 test was done. Survival curves were generated for serous cystadenocarcinoma patients using the Kaplan-Meier estimate and log-rank test. Cox proportional hazards regression analyses using Smad3 or N-cadherin as a continuous variable were also done.
Acknowledgments
Grant support: NIH grant RO1 HD044464 (T.K. Woodruff).
References
- 1.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–88. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez M, Dubeau L. Ovarian tumor development: insights from ovarian embryogenesis. Eur J Gynaecol Oncol. 2001;22:175–83. [PubMed] [Google Scholar]
- 3.Van Niekerk CC, Ramaekers FC, Hanselaar AG, Aldeweireldt J, Poels LG. Changes in expression of differentiation markers between normal ovarian cells and derived tumors. Am J Pathol. 1993;142:157–77. [PMC free article] [PubMed] [Google Scholar]
- 4.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–65. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 7.Peng C. The TGF-β superfamily and its roles in the human ovary and placenta. J Obstet Gynaecol Can. 2003;25:834–44. doi: 10.1016/s1701-2163(16)30674-0. [DOI] [PubMed] [Google Scholar]
- 8.Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-β superfamily signalling. Genes Cells. 2002;7:1191–204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- 9.ten Dijke P, Hill CS. New insights into TGF-β-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 11.Tang B, Vu M, Booker T, et al. TGF-β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin SW, Lee MT, Ke FC, et al. TGFβ1 stimulates the secretion of matrix metalloproteinase 2 (MMP2) and the invasive behavior in human ovarian cancer cells, which is suppressed by MMP inhibitor BB3103. Clin Exp Metastasis. 2000;18:493–9. doi: 10.1023/a:1011888126865. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez GC, Haisley C, Hurteau J, et al. Regulation of invasion of epithelial ovarian cancer by transforming growth factor-β. Gynecol Oncol. 2001;80:245–53. doi: 10.1006/gyno.2000.6042. [DOI] [PubMed] [Google Scholar]
- 14.Kitagawa K, Murata A, Matsuura N, et al. Epithelial-mesenchymal transformation of a newly established cell line from ovarian adenosarcoma by transforming growth factor-β1. Int J Cancer. 1996;66:91–7. doi: 10.1002/(SICI)1097-0215(19960328)66:1<91::AID-IJC16>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.Bartlett JM, Langdon SP, Scott WN, et al. Transforming growth factor-β isoform expression in human ovarian tumours. Eur J Cancer. 1997;33:2397–403. doi: 10.1016/s0959-8049(97)00304-3. [DOI] [PubMed] [Google Scholar]
- 16.Higashi T, Sasagawa T, Inoue M, et al. Overexpression of latent transforming growth factor-β 1 (TGF-β 1) binding protein 1 (LTBP-1) in association with TGF-β 1 in ovarian carcinoma. Jpn J Cancer Res. 2001;92:506–15. doi: 10.1111/j.1349-7006.2001.tb01123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santin AD, Bellone S, Ravaggi A, et al. Increased levels of interleukin-10 and transforming growth factor-β in the plasma and ascitic fluid of patients with advanced ovarian cancer. BJOG. 2001;108:804–8. doi: 10.1111/j.1471-0528.2001.00206.x. [DOI] [PubMed] [Google Scholar]
- 18.Nicolas FJ, De Bosscher K, Schmierer B, Hill CS. Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci. 2004;117:4113–25. doi: 10.1242/jcs.01289. [DOI] [PubMed] [Google Scholar]
- 19.Fishman DA, Kearns A, Chilukuri K, et al. Metastatic dissemination of human ovarian epithelial carcinoma is promoted by α2β1-integrin-mediated interaction with type I collagen. Invasion Metastasis. 1998;18:15–26. doi: 10.1159/000024495. [DOI] [PubMed] [Google Scholar]
- 20.Cvetkovic D. Early events in ovarian oncogenesis. Reprod Biol Endocrinol. 2003;1:68. doi: 10.1186/1477-7827-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bast RC., Jr Status of tumor markers in ovarian cancer screening. J Clin Oncol. 2003;21:200–5. doi: 10.1200/JCO.2003.01.068. [DOI] [PubMed] [Google Scholar]
- 22.Society AC Cancer facts and figures. 2003.
- 23.Auersperg N, Pan J, Grove BD, et al. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci U S A. 1999;96:6249–54. doi: 10.1073/pnas.96.11.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maines-Bandiera SL, Auersperg N. Increased E-cadherin expression in ovarian surface epithelium: an early step in metaplasia and dysplasia? Int J Gynecol Pathol. 1997;16:250–5. doi: 10.1097/00004347-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Sundfeldt K, Piotkewitz Y, Ivarsson K, et al. E-cadherin expression in human epithelial ovarian cancer and normal ovary. Int J Cancer. 1997;74:275–80. doi: 10.1002/(sici)1097-0215(19970620)74:3<275::aid-ijc7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 26.Davies BR, Worsley SD, Ponder BA. Expression of E-cadherin, α-catenin, and β-catenin in normal ovarian surface epithelium and epithelial ovarian cancers. Histopathology. 1998;32:69–80. doi: 10.1046/j.1365-2559.1998.00341.x. [DOI] [PubMed] [Google Scholar]
- 27.Peralta Soler A, Knudsen KA, Tecson-Miguel A, et al. Expression of E-cadherin and N-cadherin in surface epithelial-stromal tumors of the ovary distinguishes mucinous from serous and endometrioid tumors. Hum Pathol. 1997;28:734–9. doi: 10.1016/s0046-8177(97)90184-2. [DOI] [PubMed] [Google Scholar]
- 28.Wong AS, Maines-Bandiera SL, Rosen B, et al. Constitutive and conditional cadherin expression in cultured human ovarian surface epithelium: influence of family history of ovarian cancer. Int J Cancer. 1999;81:180–8. doi: 10.1002/(sici)1097-0215(19990412)81:2<180::aid-ijc3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 29.Ouellet V, Guyot MC, Le Page C, et al. Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int J Cancer. 2006;119:599–607. doi: 10.1002/ijc.21902. [DOI] [PubMed] [Google Scholar]
- 30.Rosen DG, Huang X, Deavers MT, et al. Validation of tissue microarray technology in ovarian carcinoma. Mod Pathol. 2004;17:790–7. doi: 10.1038/modpathol.3800120. [DOI] [PubMed] [Google Scholar]
- 31.Han SU, Kim HT, Seong do H, et al. Loss of the Smad3 expression increases susceptibility to tumorigenicity in human gastric cancer. Oncogene. 2004;23:1333–41. doi: 10.1038/sj.onc.1207259. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–14. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 33.Tannehill-Gregg SH, Kusewitt DF, Rosol TJ, Weinstein M. The roles of Smad2 and Smad3 in the development of chemically induced skin tumors in mice. Vet Pathol. 2004;41:278–82. doi: 10.1354/vp.41-3-278. [DOI] [PubMed] [Google Scholar]
- 34.Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J Cell Biol. 1994;124:401–4. doi: 10.1083/jcb.124.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomasek JJ, Halliday NL, Updike DL, et al. Gelatinase A activation is regulated by the organization of the polymerized actin cytoskeleton. J Biol Chem. 1997;272:7482–7. doi: 10.1074/jbc.272.11.7482. [DOI] [PubMed] [Google Scholar]
- 36.Halliday NL, Tomasek JJ. Mechanical properties of the extracellular matrix influence fibronectin fibril assembly in vitro. Exp Cell Res. 1995;217:109–17. doi: 10.1006/excr.1995.1069. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh S, Brown R, Jones JC, Ellerbroek SM, Stack MS. Urinary-type plasminogen activator (uPA) expression and uPA receptor localization are regulated by a 3β 1 integrin in oral keratinocytes. J Biol Chem. 2000;275:23869–76. doi: 10.1074/jbc.M000935200. [DOI] [PubMed] [Google Scholar]
- 38.Ellerbroek SM, Wu YI, Overall CM, Stack MS. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J Biol Chem. 2001;276:24833–42. doi: 10.1074/jbc.M005631200. [DOI] [PubMed] [Google Scholar]
- 39.Arany PR, Flanders KC, Kobayashi T, et al. Smad3 deficiency alters key structural elements of the extracellular matrix and mechanotransduction of wound closure. Proc Natl Acad Sci U S A. 2006;103:9250–5. doi: 10.1073/pnas.0602473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lau KM, Mok SC, Ho SM. Expression of human estrogen receptor-α and -β, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A. 1999;96:5722–7. doi: 10.1073/pnas.96.10.5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pangas SA, Woodruff TK. Production and purification of recombinant human inhibin and activin. J Endocrinol. 2002;172:199–210. doi: 10.1677/joe.0.1720199. [DOI] [PubMed] [Google Scholar]
- 42.Pangas SA, Rademaker AW, Fishman DA, Woodruff TK. Localization of the activin signal transduction components in normal human ovarian follicles: implications for autocrine and paracrine signaling in the ovary. J Clin Endocrinol Metab. 2002;87:2644–57. doi: 10.1210/jcem.87.6.8519. [DOI] [PubMed] [Google Scholar]