

Abstract
During inflammatory immune responses, the innate cytokine IL-12 promotes CD4+ T helper-1 (Th1) development through the activation of the second messenger STAT4 and the subsequent expression of T-bet. In addition, type I interferon (IFN--α/β), secreted primarily during viral and intracellular bacterial infections, can promote STAT4 activation in human CD4+ T cells. However, the role of IFN--α/β in regulating Th1 development is controversial, and previous studies have suggested a species-specific pathway leading to Th1 development in human but not mouse CD4+ T cells. In this study, we found that although both IFN-α and IL-12 can promote STAT4 activation, IFN-α failed to promote Th1 commitment in human CD4+ T cells. The difference between these innate signaling pathways lies with the ability of IL-12 to promote sustained STAT4 tyrosine phosphorylation which correlated with stable T-bet expression in committed Th1 cells. IFN-α did not promote Th1 development in human CD4+ T cells due to attenuated STAT4 phosphorylation, which was insufficient to induce stable expression of T-bet. Further, the defect in IFN-α-driven Th1 development was corrected by ectopic expression of T-bet within primary naïve human CD4+ T cells. These results indicate that IL-12 remains unique in its ability to drive Th1 development in human CD4+ T cells, and that IFN-α lacks this activity due to its inability to promote sustained T-bet expression.
Keywords: Human, T cells, cytokines
Introduction
During infections, innate cytokines direct the development of CD4+ T cells toward effector T helper subsets that secrete distinct cytokines. This process is initiated by pathogen-associated molecular patterns (PAMPs) that are recognized by specific Toll-like receptors (TLRs) expressed within professional antigen-presenting cell (APC) subsets (1, 2). In cases of bacterial infections, PAMPs such as peptidoglycan and LPS are sensed by TLRs 2 and 4 that are expressed predominantly by monocyte-derived dendritic cells (mDC) (3). As a consequence, mDCs secrete high levels of IL-12, which directly promotes CD4+ Th1 development (4, 5). In contrast, virus-derived PAMPs such as double-stranded and single-stranded RNA signal through TLRs 3 and 7, respectively, both of which are potent inducers of type I interferon (IFN-α/β) (1, 6). Thus, IL-12 and IFN-α/β represent two critical innate cytokines that provide instructive cues for adaptive T cell responses (7).
A role for IL-12 in promoting Th1 development has been well established (5). IL-12 signaling promotes the phosphorylation of signal-transducer and activator of transcription-4 (STAT4) (8, 9) that is required for Th1 commitment (10). Further, IL-12-dependent STAT4 activation is conserved across species, and disruptive mutations in this signaling pathway significantly impair Th1 responses in both mice (10, 11) and humans (12). Likewise, early reports demonstrated a potential role for IFN-α/β in promoting Th1 development in human peripheral blood mononuclear cells (PBMCs) (13, 14). These initial studies were followed by findings that IFN-α, like IL-12, could promote STAT4 activation in human lymphocytes (15–17). These early reports concluded that IFN-α/β could directly regulate Th1 commitment in CD4+ T cells through the activation of STAT4 (18).
However, unlike IL-12, the role of IFN-α/β in promoting Th1 development has been met with considerable controversy. Initial reports demonstrating IFN-α/β-dependent STAT4 activation and Th1 development were performed with human PBMCs or with human T and NK cell lines (13–15). In parallel studies with murine T cells, IFN-α/β was insufficient to promote Th1 development (17, 19) or to activate STAT4 when compared to human CD4+ T cells (16, 17), indicating a potential species-specific role for IFN-α/β in regulating Th1 development. Further, human STAT2 was implicated in facilitating STAT4 recruitment to the human IFN-α/β receptor (IFNAR) complex in a species-specific manner (16, 20). These findings seemed to explain the presumed ability of IFN-α/β to promote STAT4 activation and Th1 development in human, but not murine CD4+ T cells. However, CD4+ T cells from STAT2 knock-in mice that expressed a humanized Stat2 gene failed to exhibit either STAT4 phosphorylation or Th1 commitment in response to IFN-α/β activation (21). These results suggested that although human STAT2 was required for STAT4 activation in human cells, it was not sufficient to restore this pathway when expressed in the context of the murine IFNAR.
Recent studies have called this species-specific pathway into question (22, 23). Biron and colleagues demonstrated that IFN-α/β could promote STAT4 phosphorylation in murine T cells, although this effect was more pronounced in CD8+ than in CD4+ T cells (22). A further examination comparing various subtypes of IFN-α demonstrated that although mIFN-αA indeed could induce weak STAT4 phosphorylation, it was insufficient to drive IFN-γ production and Th1 development in murine CD4+ T cells (24). In a related study, Hilkens and colleagues demonstrated that even in human T cells, IFN-α/β-driven STAT4 activation was attenuated compared to the effects of IL-12 (25). This study suggested that IL-12 was more efficient than IFN-α/β at promoting Th1 development in human CD4+ T cells. One explanation was that IL-12 stimulation was able to maintain STAT4 phosphorylation over a longer period of time compared to IFN-α/β. We have recently confirmed this observation in murine CD4+ T cells (26), however a molecular explanation for this difference in signaling between IFN-α and IL-12 has not been examined. At the core of this issue is whether IL-12 and IFN-α/β share redundant roles in T helper commitment, and whether there truly is a species-specific pathway that operates in human but not murine T cells.
In this study, we have addressed this central issue by comparing the ability of IL-12 and IFN-α/β to direct the commitment of primary human CD4+ T cells toward the Th1 fate. We found that IFN-α does not promote Th1 development from naïve human CD4+ T cells, and we have linked this developmental defect to the inability of IFN-α to induce sustained T-bet expression. Further, we demonstrate that in primary naïve human CD4+ T cells, T-bet expression is sufficient to drive Th1 commitment in a cytokine-independent manner. Thus, in contrast to previous reports, IFN-α/β does not promote Th1 development in the absence of IL-12 due to the lack of sustained T-bet expression, and this effect is consistent across species.
Materials and Methods
Human subjects
100–120 ml of peripheral blood was obtained from healthy adults by venipuncture. Informed consent was obtained from each donor according to guidelines established by the Internal Review Board (UT Southwestern Medical Center).
Mice
DO11.10 TCR transgenic mice were housed in specific pathogen free conditions in accordance with guidelines established by the Institutional Animal Care and Use Committee (UT Southwestern Medical Center).
Cytokines, antibodies, and reagents
Recombinant human IL-12, IFN-αA, IFN-γ, and anti-human IL-4 were purchased from R&D Systems (Minneapolis, MN). Recombinant human IL-18 was purchased from Biosource (Camarillo, CA). Recombinant human IL-2 was a generous gift from M. Bennett (UT Southwestern Med. Ctr.). Neutralizing anti-human IFNAR2 was purchased from Calbiochem (La Jolla, CA). The phycoerythrin (PE)- and tricolor (TC)-conjugated anti-human CD4, anti-human IFN-γ, anti-murine-CD4, and anti-murine-IFN-γ were purchased from Caltag Laboratories (Burlingame, CA). FITC-conjugated anti-human CD45RA antibody and PE-conjugated anti-human IL-12Rβ2 antibodies were purchased from BD Pharmingen (San Diego, CA). Polyclonal rabbit α-human STAT4, α-human STAT1, and α-human T-bet antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-STAT4 was purchased from Zymed (San Francisco, CA), and anti-phospho-STAT1 was purchased from Upstate (Lake Placid, NY). Horseradish peroxidase (HRP)-conjugated goat α-rabbit immunoglobulin antiserum was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Purification of hPBMCs and naïve CD4+ T cells from peripheral blood
Human peripheral blood mononuclear cells (hPBMCs) were isolated by ficol density centrifugation using Lymphocyte Separation Media (Cellgro) and cultured in complete Iscove’s Modified Dubelcco’s Medium (HyClone, Logan, UT) supplemented with 10% FBS (Valley Biomedical, Inc., Winchester, VA) (cIMDM). hPBMCs utilized for purification of naïve cells were stained with anti-human CD45RA-FITC and either anti-human CD4-PE or anti-human CD4-TC. Cells were sorted on a MoFlo cytometer (Dako Cytomation, Fort Collins, CO) to >93% purity. Both hPBMCs and naïve CD4+ T cells were activated for 3 days at 1–5 × 106 cells/ml in cIMDM containing 50–100 U/ml rhIL-2 using either 1 μg/ml PHA (Calbiochem, La Jolla, CA) or culture plates coated with 5μg/ml anti-CD3 (OKT3) and 5 μg/ml anti-CD28 (clone 37.51). Cytokines and neutralizing anti-cytokine antibodies were added at the following concentrations as indicated in the figures: rhIL-12, 10 ng/ml; rhIFN-αA, 1000 U/ml; rhIFN-γ, 10 ng/ml; anti-human IL-12 (20C2), 5 μg/ml; anti-human IFNAR2, 2–5 μg/ml; anti-human IFN-γ (4SB3), 5 μg/ml; anti-human IL-4, 5 μg/ml. On day 3, cells were split into cIMDM containing 50 U/ml rhIL-2 and rested to day 7. Restimulation was performed using either 0.8 μg/ml PMA (A.G. Scientific, Inc. San Diego, CA) + 1 μM ionomycin (Sigma-Aldrich St. Louis, MO) or plates coated with 5 μg/ml anti-CD3 (OKT3).
Intracellular cytokine and phospho-STAT4 staining
For intracellular cytokine staining, resting Th cells were restimulated with 0.8 μg/ml PMA plus 1 μM ionomycin for 4 hours. Brefeldin A (1 μg/ml, Epicentre, Madison, WI) was added during the last 2 hours of stimulation. For Th1 effector function assays, cells were stimulated with either PMA + ionomycin, 10 ng/ml rhIL-12, 1000 U/ml IFN-αA, 50 ng/ml IL-18, or a combination of IL-12/IL-18 or IFN-αA/IL-18. After stimulation, cells were harvested and intracellular cytokine staining was performed as previously described (21).
Intracellular tyrosine phosphorylated STAT4 was detected by intracellular staining as described (27). Freshly isolated PBMCs were activated with cytokines as indicated in the text and were then incubated in 5% formalin/PBS followed by fixation in cold 100% methanol. Cells were washed extensively in staining buffer (PBS, 0.5% BSA, and 1 mM NaVO4) followed by permeabilization in staining buffer containing 0.1% saponin. Cells were stained with 0.5 μg of anti-STAT4 (SC-486, Santa Cruz) or with anti-P-tyr STAT4 (Zymed, San Francisco, CA). A goat-anti-mouse Ig-biotin (Jackson Immunoresearch, West Grove, PA) was used for secondary detection followed by staining with streptavidin-PE-Cy5. Cells were also stained with anti-hCD4-PE and anti-hCD45RA-FITC. Cells were analyzed on a FACScan (Becton Dickinson), and the data were processed through CellQuest software.
Analysis of cytoplasmic and nuclear STAT4
Human PBMCs activated with IL-12 or IFN-α for various times were stained for CD4, CD45RA and P-tyr-STAT4 as described above and counterstained with 20 ng/ml propidium iodide. Alternatively, the biotinylated P-tyr-STAT4 was stained with streptavidin FITC and the nucleus counterstained with 5 μM DRAQ5™ (Axxora, San Diego, CA). These cells were also stained with anti-hCD4-PE. Image files of 5000 to 10,000 events were collected for each sample using the ImageStream® imaging flow cytometer (Amnis Corp., Seattle, WA) and analyzed using IDEAS® software (Amnis). In-focus single cells were identified by gating on propidium iodide or DRAQ5 positive events with high nuclear aspect ratios (minor to major axis ratio, a measure of circularity) and high nuclear contrast (as measured by the Gradient Max feature). CD4+CD45RA+ lymphocytes (low SSC/low area cells) were then gated. Nuclear localization of P-tyr-STAT4 within these cells was measured using the Similarity score, which quantifies the correlation of pixel values of the nuclear and P-tyr-STAT4 images on a per-cell basis (28). If the transcription factor is nuclear, the two images will be similar and have large positive values. If it is cytoplasmic, the two images will be anti-similar and have large negative values. Events with positive values greater than 1 had visually apparent nuclear distributions of transcription factor, and were gated to quantify the percentage of cells within the CD4+CD45RA+ lymphocyte population with nuclear-localized P-Y-STAT.
Quantitative real-time polymerase chain reaction (qPCR) analysis
Total RNA was extracted utilizing the RNeasy Mini Kit (QIAgen, Valencia, CA) according to the manufacturer’s instructions followed by reverse transcription. The resultant cDNA was subjected to qPCR analysis with SYBR Green Master Mix (Stratagene, Cedar Creek, TX) on a ABI7300 real-time thermocycler (Applied Biosystems, Foster City, CA). Human GAPDH was used as a reference. Relative changes in mRNA expression were calculated by the 2−ΔΔCT method (29), and all treatment groups were referenced to the unstimulated controls. The primers used to detect mRNA transcripts are as follows: hIFN-γ 5′-TGATTACAAGGCTTTATCTCAGGG, 5′-GGCAGTAACTGGATAGTATCAC TTCAC; hIL-12Rβ2 5′-ACCTTCCCACCCATGATGGC, 5′-GAAAACAGA AAGGGAGATGTGCTG; hT-bet 5′-CGTCCAACAATGTGACCCAG, 5′-GCAGTCACGGCAATGAACTG; hGAPDH 5′-ACATCGCTCAGACACCATGG, 5′-CATGTAGTTGAGGTCAATGAAGGG.
Cytometric Bead Array and ELISA analyses of human cytokines
Naïve human CD4+ T cells were activated for 7 days as described above, and were then restimulated at 5 × 105 cells/ml for 24 hours on anti-CD3-coated plates. Supernatants were analyzed for IL-4, IL-5, and IFN-γ cytokine concentrations by either cytometric bead arrays (BD Pharmingen) or ELISAs (eBioscience, San Diego, CA) according to the manufacturer’s protocols.
Immunoprecipitation and immunoblotting
Human PBMCs were stimulated and expanded for two consecutive weeks with PHA under Th1 conditions (10 ng/ml rhIL-12). On day 14, cells were washed in cIMDM and rested at 1 × 107 cells/ml for 30 minutes at 37°C, 5% CO2. Cells were stimulated with either 10 ng/ml rhIL-12 or 1000 U/ml rhIFN-αA for the indicated periods of time at 37°C, 5% CO2. Lysis was performed for 1 hour at 4°C in RIPA buffer containing proteinase inhibitors. Samples were pre-cleared using rabbit pre-immune serum, and immunoprecipitation of human STAT4 was performed using 3 μg per sample rabbit anti-human STAT4 bound to Protein G Sepharose beads (Amersham Biosciences, Uppsala, Sweden). Immunoprecipitates were resolved by 7% SDS-PAGE and transferred to PVDF membranes (Bio-Rad Laboratories, Hercules, CA). Western blotting was performed using rabbit anti-phosphorylated STAT4 with an HRP-conjugated goat anti-rabbit secondary antibody. Detection was performed using ECL Detection Reagents followed by exposure to Hyperfilm ECL (Amersham Biosciences). The membranes were then stripped and reprobed with anti-human STAT4 polyclonal antiserum followed by HRP-conjugated goat anti-rabbit immunoglobulin.
Retrovirus cDNA expression constructs
The full-length murine IFNAR2 cDNA was amplified from a cDNA clone (Clone ID: 4187603, Invitrogen) with primers: 5′-AAAAAGATCTAGCTGAGCAGGATGCG-TTCACGG and 5′-AAAAAGATCTCTTTGGAGTCATCTCATGATGTAGCC. The PCR product was digested with BglII and cloned into the BamHI site of GFPRV. A full-length human T-bet cDNA clone (Accession #BC039739) was purchased from ATCC (Manassa, VA). The open reading of this cDNA clone was amplified by PCR with primers: 5′-AAAACTCGAGCCCGGATGGGCATCGTGGAG and 5′-AAAACTCGAGCTGCTCAGTTGGGAAAATAGTTATAAAACTGTCC. The PCR product was digested with XhoI and ligated into the XhoI site within the retrovirus vector GFPRV (30), and all constructs were confirmed by DNA sequencing.
Retroviral transduction of primary naïve human CD4+ T cells
Retroviral supernatants were generated by transfection of the Phoenix-amphotropic cell line as described (31). Naïve CD4+/CD45RA+ human T cells were purified by cell sorting as described above. Purified cells were activated with plate-bound anti-CD3/anti-CD28 in complete medium with 600 U/ml hIL-2 for 24 hours. The cells were subjected to two rounds of retroviral transduction by spinning the cells at 1000 x g for 90 min in the presence of retroviral supernatants containing 5 μg/ml polybrene. 24 hours after the first round of retroviral transduction, cytokines or anti-cytokine antibodies were added to the cultures as indicated in the text and figure legends. Cells were expanded on day 7 by restimulating the cells on anti-CD3 coated plates. The cells were rested to day 14 prior to intracellular cytokine analysis.
Retroviral transduction was performed in murine CD4+ T cells as previously described (31). Briefly, infectious retrovirus supernatants were generated in the Phoenix-ecotropic packaging cell line transfected with retrovirus plasmids. DO11.10 splenic T cells were transduced with retrovirus supernatants supplemented with 2 μg/ml protamine sulfate and 10 ng/ml IL-12 to generate polarized Th1 cells. Transduced T cells were purified on day 7 after activation by flow cytometric sorting based on GFP expression. Purified cells were expanded by restimulation with OVA peptide and irradiated BALB/c splenocytes.
Results
IFN-α does not promote Th1 development in the absence of IL-12
We began this study by first assessing the ability of IFN-α to promote Th1 development within highly purified naïve CD4+ T cells. For these experiments, human CD4+/CD45RA+ cells were sorted to >95% purity from peripheral blood obtained from healthy adult donors. Sorted cells were then immediately activated on plate-bound anti-CD3/anti-CD28-coated plates in the presence of either purified cytokines or neutralizing anti-cytokine monoclonal antibodies. Seven days following primary activation, resting cells were restimulated with either PMA + ionomycin (Fig. 1A and D) or plate-bound anti-CD3 (Fig. 1B and C) and assessed for cytokine protein or mRNA expression. As shown in Fig. 1A, IL-12 was capable of enhancing IFN-γ expression, and this effect was more pronounced when IL-4 was neutralized during primary stimulation (Fig. 1A, condition 3). Further, IFN-α did not inhibit the ability of IL-12 to induce IFN-γ expression either in the absence or presence of neutralizing anti-IL-4 antibody (Fig. 1A, condition 4). Stimulation with IFN-γ alone did not promote Th1 development (Fig. 1A, condition 5), as expected and as previously demonstrated (19, 32). However, in contrast to previous reports (17, 22, 23), IFN-α was also ineffective at promoting Th1 development in the absence of IL-12, and the percentage of cells capable of secreting IFN-γ was less in IFN-α-treated cells compared to neutralizing conditions (Fig. 1A, compare conditions 1 and 2). Further, cells primed with IFN-α did not induce IFN-γ mRNA in response to anti-CD3 stimulation (Fig. 1B, condition 3) indicating a general defect in Th1 commitment.
Figure 1. IL-12, but not IFN-α, promotes Th1 development in highly purified naïve human CD4+ T cells.

Purified human CD4+/CD45RA+ T cells were activated with plate-bound anti-CD3 and anti-CD28 in the presence of IL-2, anti-IL-4, and the indicated cytokines and neutralizing antibodies, where “+” indicates addition of cytokine and “−” indicates addition of neutralizing anti-cytokine antibody. On day 3, cells were diluted into fresh medium containing IL-2 and rested to day 7. A, Parallel cultures were stimulated in the absence (open bars) or presence (closed bars) of neutralizing anti-hIL-4 antibody. Cells were restimulated for 4 hours in the presence of PMA + ionomycin. Intracellular cytokine staining was performed with antibodies specific for hCD4 and hIFN-γ. Data were gated on live cell populations and expressed as a percentage of CD4+/IFN-γ+ cells. B, Total RNA was isolated from cells that were resting (open bars) or restimulated (closed bars) with plate-bound anti-CD3 for 2 hours. Analysis of IFN-γ transcript levels was performed by qPCR, and transcript levels were normalized to GAPDH. Data were normalized relative to non-stimulated (Control) cells activated under neutralizing conditions. C, Cells were rested (open bars) or restimulated for 24 hours with plate-bound anti-CD3 (closed bars). Cell culture supernatants were analyzed for the presence of IFN-γ (upper panel), IL-4 (middle panel), and IL-5 (lower panel) by cytometric bead array. D, Purified CD4+/CD45RA+ T cells from 3 separate donors (D1, D2, and D3) were stimulated as in (C) under neutralizing conditions (“Control”) or with IL-12 + anti-IFNAR2 (“IL-12”) or anti-IL-12 + IFN-αA (“IFN-αA”). On day 7, cells were restimulated and stained for CD4 and intracellular IFN-γ as described above. Data were gated on live cell populations.
We considered the possibility that IFN-α was promoting Th2 development. To address this, purified naïve human CD4+ T cells were activated under polarizing conditions as described above, and IFN-γ, IL-4 and IL-5 secretion was measured by cytometric bead arrays (Fig. 1C). IL-12 stimulation enhanced IFN-γ secretion (Fig. 1C, upper panel, compare conditions 1 and 2) and did not promote IL-4 or IL-5 secretion compared to neutralizing conditions (Fig. 1C, middle and lower panels, compare conditions 1 and 2). As expected, stimulation of cells in the presence of IL-4 inhibited IFN-γ secretion while enhancing IL-4 and IL-5 secretion (Fig. 1C, condition 3). Although IFN-α did not inhibit the ability of IL-12 to promote IFN-γ secretion (Fig. 1C, upper panel, compare conditions 2 and 5), in the absence of IL-12 or IL-4, IFN-α did not promote the secretion of either IFN-γ or IL-4 and IL-5 (Fig. 1C, condition 4). Thus, in the absence of IL-12 or IL-4, IFN-α did not promote either Th1 or Th2 development.
Each experiment described above was repeated at least 3 times. We observed heterogeneity in the overall percentages of cells that were capable of secreting IFN-γ from one donor to the next. This level of heterogeneity in IFN-γ expression is depicted in Fig. 1D. Here, naïve CD4+/CD45RA+ cells were purified from 3 separate healthy human donors and activated under neutralizing conditions (anti-IL-12 + anti-IFNAR2, “Control”), or with IL-12 + anti-IFNAR2 (“IL-12”) or IFN-αA + anti-IL-12 (“IFN-αA”). The percentage of IFN-γ-secreting CD4+ T cells that developed in the absence of IL-12 or IFN-α varied significantly from each of the 3 donors. However, in all cases, IL-12 stimulation increased while IFN-α decreased the percentage of cells capable of secreting IFN-γ compared to cells activated under neutralizing conditions.
Early reports demonstrating a role for IFN-α in Th1 development used mixed populations of peripheral or cord blood lymphocytes (13, 14). Thus, it was possible that IFN-α was acting indirectly on other populations of cells to promote Th1 development in CD4+ T cells. Therefore, we addressed this issue by attempting to reproduce those early studies. For these experiments, non-fractionated human PBMCs were activated with plate-bound anti-CD3 + anti-CD28 in the presence of cytokines or neutralizing anti-cytokine antibodies as indicated in Fig. 2A. Cells were expanded in culture for 7 days followed by restimulation with PMA and ionomycin in order to assess IFN-γ expression by intracellular cytokine staining. IL-12 was sufficient to increase the percentage of CD4+ T cells capable of secreting IFN-γ, as expected (Fig. 2A, condition 3), and the absence or presence of neutralizing anti-IL-4 antibody did not significantly enhance this effect. However, differentiation of human PBMCs in the presence of IFN-α alone was not sufficient to expand IFN-γ-secreting CD4+ T cells compared to neutralizing conditions (Fig. 2A, compare conditions 1 and 2). Although IFN-α failed to enhance Th1 development, it did not inhibit the ability of IL-12 to expand a population of IFN-γ-secreting CD4+ T cells (Fig. 2A, condition 4).
Figure 2. IL-12, but not IFN-α, promotes Th1 development in human PBMC cultures.

A, Human PBMCs were stimulated with plate-bound anti-CD3 + anti-CD28 in the presence of IL-2 and the indicated cytokines and/or neutralizing antibodies, where “+” indicates addition of cytokine, and “−” indicates addition of neutralizing anti-cytokine antibody. Parallel cultures were stimulated in the absence (open bars) or presence (closed bars) of neutralizing anti-hIL-4 antibody. B, Human PBMCs were activated with plate-bound anti-CD3 and anti-CD28 in the presence of IL-2 and the indicated cytokines and neutralizing antibodies, where “+” indicates addition of cytokine, “−” indicates addition of neutralizing anti-cytokine antibody, and “o” indicates that the cytokine was not manipulated. On day 3, cells were diluted 1:8 into fresh medium containing IL-2 and rested to day 7. Cells were restimulated for 4 hours in the presence of PMA + ionomycin. Intracellular cytokine staining was performed with antibodies specific for hCD4 and hIFN-γ. Data were gated on live cell populations and expressed as a percentage of CD4+/IFN-γ+ cells.
The differences observed between our data and previous reports could have resulted from our strict control of endogenous IL-12 levels with the use of neutralizing anti-IL-12 antibodies with cells cultured in the presence of IFN-α. In some cases, previous studies did not neutralize IL-12 during the primary activation (13, 14). To address this, human PBMCs were activated in the presence of IL-12 or IFN-α in the absence or presence of neutralizing antibodies for 7 days (Fig. 4B). IFN-γ expression from CD4+ T cells was assessed as described above. Even in the absence of neutralizing antibodies against IL-12, IFN-α alone was still unable to induce Th1 development in CD4+ T cells (Fig 2B, compare conditions 6 and 7).
Figure 4. IL-12 and IFN-α differentially regulate the kinetics of STAT4 tyrosine phosphorylation in human CD4+ T cells.
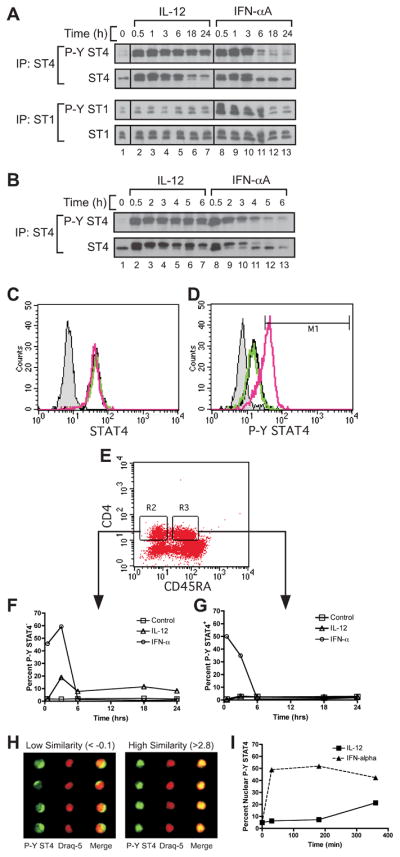
A and B, Human PBMCs were activated and expanded for two consecutive weeks with PHA in the presence of IL-2 and IL-12. On day 14, cells were rested in fresh medium for 30 minutes and stimulated with either IL-12 or IFN-αA for the indicated periods of time. Immunoprecipitation was performed using polyclonal antisera for human STAT4 or STAT1, and Western blotting was performed for phosphorylated STAT4 or STAT1, as indicated. Membranes were then stripped and re-probed for total STAT4 or STAT1. A, Time course of STAT4 and STAT1 phosphorylation in human Th1 PBMCs stimulated with IL-12 (lanes 2–7) or IFN-αA (lanes 8–13) for 0–24 hours. B, Time course of STAT4 phosphorylation in human Th1 PBMCs stimulated with IL-12 (lanes 2–7) or IFN-αA (lanes 8–13) for 0–6 hours. C through G, Freshly isolated PBMCs were activated with medium alone, IL-12, or IFN-α at various time points up to 24 hours. Cells were stained for CD4, CD45RA, and intracellular STAT4 and phosphotyrosine STAT4 (P-Y STAT4) as described in the Materials and Methods. C and D, Total STAT4 (C) and P-Y STAT4 (D) were measured from cells activated for 30 min by flow cytometry. Data are gated on live, CD4+, and CD45RA+ cells. Black line, unstimulated; green line, IL-12 stimulated; red line, IFN-α stimulated; gray shaded, non-immune rabbit Ig control. E, Gating scheme is shown for the analysis of CD4+, CD45RA- (R2) and CD45RA+ (R3) cells. F and G, CD45RA- (F) and CD45RA+ (G) gated cells are represented as a percentage of cells that display increased P-Y STAT4 staining over a 24 hour period. Open square, unstimulated; open triangle, IL-12 stimulated; open circle, IFN-α stimulated. H and I, Human PBMCs were activated and stained as above and analyzed on the ImageStream® imaging flow cytometer. Single cell images were gated on the live, CD4+, and CD45RA+ populations. H, Cells displaying positive staining for P-Y STAT4 were categorized for either low similarity (left panel) or high similarity (right panel) of co-localization of P-Y STAT4 within the nucleus based on nuclear dye staining (Draq-5). I, Cells displaying elevated P-Y STAT4 staining were quantified for co-localization of P-Y STAT4 within the nucleus of cells treated with IL-12 (closed square) or IFN-α (closed triangle).
We considered the possibility that subtypes of IFN-α/beta; other that IFN-αA may differentially regulate Th1 commitment. To address this, naïve human CD4+ T cells were activated in parallel cultures with IFN-αB2, IFN-αD, IFN-αA/D, and IFN-β. Consistent with results obtained with IFN-αA (Figs. 1 and 2), the other IFN-α subtypes (data not shown), IFN-β (Fig. 3A) failed to promote Th1 development. In each case, these cultures were activated with 1000 U/ml of IFN-α/β. Previous reports demonstrating a role for IFN-α/β in murine Th1 development used IFN-α at concentrations ranging from 50,000 – 100,000 U/ml (22, 23). Thus, it was possible that extremely high concentrations of IFN-α/β were required to promote Th1 development. However, we titrated IFN-αA in primary stimulation cultures and found that IFN-αA at concentrations up to 100,000 U/ml was still incapable of driving Th1 development in human CD4+ T cells (Fig. 3B).
Figure 3. Type I interferon does not promote Th1 commitment.
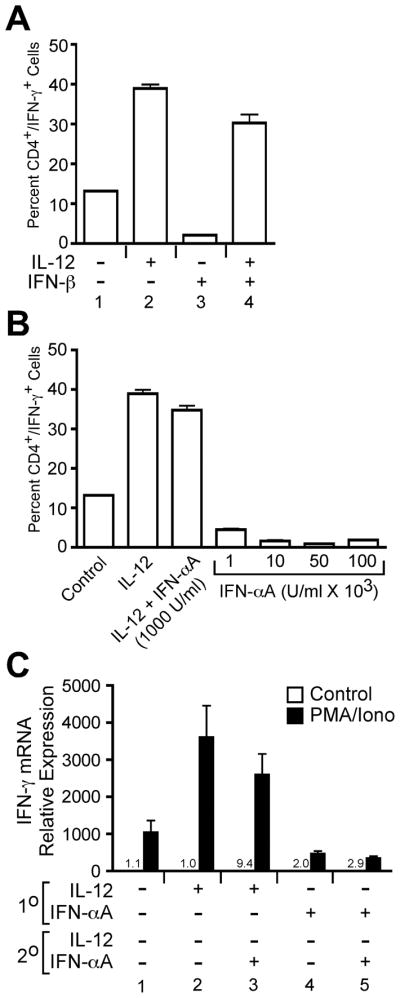
A, Purified naïve CD4+ T cells were stimulated with plate bound anti-CD3/anti-CD28 in the presence of IL-12 or IFN-β (1000 U/ml) as indicated in the figure. On day 7, cells were restimulated with PMA + ionomycin, and IFN-γ expression was measured by intracellular staining. B, Purified naïve CD4+ T cells were stimulated with IFN-α at concentrations indicated in the figure, and IFN-γ was measured by intracellular staining as describe above. C, Purified naïve CD4+ T cells were activated as described above for 7 days (1° conditions) where “+” indicates addition of cytokine and “−” indicates addition of neutralizing anti-cytokine antibody. These cells were then restimulated with plate-bound anti-CD3 under cytokine conditions indicated in the figure for an addition 7 days (2° conditions). Cells were then restimulated with either medium alone, or PMA + ionomycin for 1 hour. Total RNA was isolated from these cells, and IFN-γ mRNA induction was measured by qPCR. All data are referenced to the unstimulated control in condition 1.
Studies of in vitro-activated murine T cells suggest that repeated stimulation under Th1 polarizing conditions tends to reinforce Th1 commitment as measured by increased IFN-γ secretion and the inability of these cells to be redirected to the Th2 phenotype (33). Based on these observations, we wished to determine if repeated IFN-α stimulation could eventually lead to Th1 commitment. Naïve human CD4+/CD45RA+ T cells were purified and activated for 1 week with plate-bound anti-CD3 + anti-CD28 in the presence of cytokines or neutralizing anti-cytokine antibodies as indicated in Fig. 3C (1° conditions). These cells were then restimulated for a second week with either additional cytokines or neutralizing anti-cytokine antibodies (2° conditions). IFN-γ mRNA was quantified from cells that were restimulated with either medium or with PMA + ionomycin. Cells that were stimulated for the first week with IL-12 displayed 3–4-fold induction of IFN-γ mRNA compared to cells that were differentiated under neutralizing conditions during the primary and secondary stimulations (Fig. 3C, compare conditions 1 and 2). However, cells that received IFN-α during the 1°stimulation alone (Fig. 3C, condition 4) or during both the 1° and 2° stimulations (Fig. 3C, condition 5) failed to induce IFN-γ mRNA compared to cells polarized under neutralizing conditions (Fig. 3C, condition 1). Although IFN-α failed to promote Th1 development in the absence of IL-12, it did not significantly inhibit IFN-γ expression from cells differentiated in the presence of IL-12 during the 1° stimulation (Fig. 3C, compare conditions 2 and 3, p=0.17). Collectively, these data demonstrate that IFN-α is not sufficient to promote Th1 development in human CD4+ T cells.
Cytokine-driven STAT4 phosphorylation and acute IFN-γ expression
IL-12 drives Th1 development in CD4+ T cells by efficiently promoting STAT4 tyrosine phosphorylation (9, 34). In addition, IL-12, in synergy with IL-18, induces sustained secretion of IFN-γ from Th1 cells that, in mice, has been shown to be dependent upon STAT4 (35–37). Thus, IL-12-dependent STAT4 activation plays significant roles in both Th1 development and effector function. Although both IL-12 and IFN-α/β can promote STAT4 phosphorylation in human CD4+ T cells (38, 39), several reports have suggested that either the magnitude or kinetics of IFN-α-induced STAT4 phosphorylation is decreased compared to IL-12 signaling (25, 38). Based on these observations, we wished to determine which aspects of Th1 development and effector function could be directly regulated by IFN-α through STAT4 activation.
First, the kinetics of IL-12- and IFN-α-dependent STAT4 tyrosine phosphorylation were assessed in human peripheral blood mononuclear cells (PBMC) by immunoblotting (Fig. 4A and B). IL-12 stimulation of Th1-polarized PBMCs promoted STAT4 phosphorylation that was sustained up to 24 hours post-stimulation (Fig. 4A, upper panel, lanes 2–7). In contrast, IFN-αA-dependent STAT4 phosphorylation was attenuated (Fig. 4A, upper panel, lanes 8–13), peaking at 0.5–2 hours and extinguished by 6 hours (Fig. 4B, lanes 8–13). In addition, IFN-α-dependent STAT1 phosphorylation was sustained up to 6 hours post-stimulation (Fig. 4A, lower panel, lanes 8–13), indicating that the rate at which STAT4 and STAT1 are dephosphorylated may be regulated through different mechanisms.
We wished to determine whether the kinetics of IFN-α-dependent STAT4 tyrosine phosphorylation differed between fully polarized Th1 cells and uncommitted naïve CD4+ T cell progenitors. To address this, we assessed the kinetics of STAT4 tyrosine phosphorylation in naïve CD4+/CD45RA+ T cells within freshly isolated human PBMCs (Fig. 4C through G). For these experiments, both total STAT4 protein and tyrosine phosphorylated (P-Y) STAT4 was measured by intracellular staining from cells activated with either IL-12 or IFN-α between 0–24 hours. The overall expression of STAT4 protein in CD4+/CD45RA+ cells was not altered by cytokine treatment (Fig. 4C) and remained constant throughout the 24 hour time course (data not shown). In naïve CD4+/CD45RA+ T cells, IL-12 did not increase the percentage of cells displaying elevated P-Y STAT4 compared to unstimulated cells (Fig. 4D), presumably due to the lack of IL-12Rβ2 expression (32). However, IFN-α stimulation promoted STAT4 phosphorylation in naïve CD4+ T cells (Fig. 4D). Further, consistent with our observations in Th1 cells (Fig. 4A and B), we found that IFN-α-driven STAT4 phosphorylation was rapidly induced by 30 min and then extinguished by 6 hours post-stimulation in naïve CD4+ T cells (Fig. 4E and G). Further, the kinetics of IFN-α-dependent STAT4 phosphorylation was similar in the CD45RA+ and CD45RA−populations (Fig. 4F and G). We also observed that IL-12 promoted STAT4 phosphorylation in approximately 15–20% of the CD45RA− cells (Fig. 4F) indicating that these cells were responsive to IL-12. Further, IL-12-dependent STAT4 phosphorylation was maintained in this population through the entire 24 hour time course. Taken together, these data indicate that unlike IL-12, IFN-α does not promote sustained STAT4 tyrosine phosphorylation in naïve human CD4+ T cells.
It was possible that the reason IFN-α failed to promote Th1 development was due to lack of sustained STAT4 phosphorylation. However, it was also possible that phosphorylated STAT4 was not efficiently translocated to the nucleus in response to IFN-α. This possibility was addressed by visualizing the relative nuclear accumulation of phosphorylated STAT4 within naïve human CD4+ T cells (Fig. 4H and I). We utilized a novel technology that combines multiparametric flow cytometry with single cell microscopy. For these experiments, freshly isolated human PBMCs were stimulated with either IL-12 or IFN-α followed by intracellular staining for CD4, CD45RA and P-Y STAT4 as described above. Cells were imaged with the use of the ImageStream® flow cytometer. Single cell images were processed through the live, CD4+ and CD45RA+ gates. Cells that exhibited an increase in phosphorylated STAT4 signal were divided into populations that displayed either P-Y STAT4 cytoplasmic accumulation (Fig. 4H, left panel) or nuclear accumulation (Fig. 4H, right panel). As shown in Fig. 4I, approximately 50% of the CD4+/CD45RA+/P-Y STAT4+ cells displayed accumulation of P-Y STAT4 in the nuclei in response to IFN-α. Although the level of P-Y STAT4 was significantly diminished by 6 hours (Fig. 4G), the residual amount of P-Y STAT4 within those cells was still retained within the nucleus (Fig. 4I). Thus, these data suggest that although IFN-α-induced STAT4 phosphorylation was attenuated compared to IL-12, STAT4 nuclear translocation remained intact in naïve human CD4+ T cells.
We wished to correlate the duration of IFN-α-dependent STAT4 phosphorylation with Th1 effector function. IFN-γ secretion by Th1 cells is regulated independently by both antigen activation and by innate cytokines (36, 40, 41). In the case of innate cytokines, IL-12 synergizes with IL-18 to promote IFN-γ secretion from fully polarized Th1 cells in a STAT4-dependent manner. Although IFN-α + IL-18 stimulation was reported to induce IFN-γ secretion from human T cells (39), we suspected that IFN-α and IL-12 differentially regulated acute IFN-γ secretion due to the altered kinetics of STAT4 activation observed above. To test this possibility, human CD4+ T cells were activated under Th1-inducing conditions (IL-12 + α-IL-4) in order to generate a population of cells capable of responding to IL-12 and secreting IFN-γ upon restimulation with innate cytokines. Human Th1 cells re-stimulated for 4 hours with either IL-12 + IL-18 or IFN-αA + IL-18 secreted IFN-γ to equivalent levels (Fig. 5A and B). Stimulation with each cytokine alone failed to promote IFN-γ secretion (Fig. 5B) as previously reported (36, 38). However, if cells were stimulated for 24 hours, the level of IFN-γ secreted into the culture supernatants was significantly lower in response to IFN-α + IL-18 as compared to IL-12 + IL-18 (Fig. 5C). These data correlate with the attenuated activation of STAT4 in response to IFN-α observed above.
Figure 5. Cytokine-dependent IFN-γ secretion from fully polarized human Th1 cells.

Purified human CD4+/CD45RA+ T cells were activated with plate-bound anti-CD3 and anti-CD28 in the presence of IL-2, anti-IL-4, anti-IFNAR2, and IL-12 for 3 days (Th1-inducing conditions). Cells were diluted into fresh medium and rested to day 7. A and B, Cells were restimulated for 4 hours in the presence of IL-12, IFN-αA, IL-18, or a combination of these cytokines. PMA + ionomycin was used as a positive control. Intracellular cytokine staining was performed with antibodies specific for hCD4 and hIFN-γ. Data were gated on live cell populations. A, Representative dot plots showing unstimulated cells and cells stimulated in the presence of IL-12 + IL-18 or IFN-αA + IL-18. B, Graphical representation of the proportion of CD4+ IFN-γ+ cells. C, Cells were restimulated for 24 hrs in the presence of the indicated cytokines or with plate-bound anti-CD3 antibody as described above. Cell culture supernatants were harvested and analyzed for the presence of IFN-γ protein by ELISA.
Ectopic IFNAR2 expression enhances IFN-α-dependent STAT4 phosphorylation and acute IFN-γ secretion
Several mechanisms could account for the differential activation of STAT4 by IL-12 versus IFN-α. First, IFN-α signaling may induce the expression of negative regulators of JAK/STAT signaling such as SOCS proteins or Ubp43. However, we found that SOCS mRNA induction was equivalent in cells activated with either IL-12 or IFN-a (data not shown). In addition to SOCS proteins, Ubp43 has recently been shown to specifically inhibit JAK kinase activation by the IFNAR (42), and Ubp43-deficient mice display enhanced anti-viral and anti-bacterial responses to IFN-α signaling (43, 44). We examined STAT4 activation and IFN-γ secretion in Ubp43-deficient T cells but detected no differences in these parameters compared to strain-matched wild type control T cells (data not shown).
Previous studies have suggested that acute IFN-α/β signaling negatively regulates expression of the human IFNAR1 subunit, thereby extinguishing downstream signaling events (25). However, Berenson et. al. found that the murine IFNAR1 subunit was stably expressed on murine CD4+ T cells following IFN-α activation (26). This recent study concluded that like in human T cells, STAT4 tyrosine phosphorylation was attenuated in response to IFN-α compared to IL-12. Further, we recently demonstrated that the IFNAR2 subunit plays an important role in STAT4 activation (45). Here, we found that the STAT4 N-domain interacts with the cytoplasmic domain of the IFNAR2 subunit. Based on these collective observations, we wished to determine if ectopic overexpression of either the IFNAR1 or IFNAR2 subunit could restore IFN-α-dependent STAT4 activation in murine T cells. To test this, we expressed the full length murine IFNAR1 and IFNAR2 subunits in murine Th1 cells and tested both the kinetics of STAT4 phosphorylation and acute IFN-γ secretion in response to IFN-α. Retroviral expression of the IFNAR1 did not alter either the kinetics of STAT4 phosphorylation or the secretion of IFN-γ in response to IFN-α + IL-18 stimulation (data not shown). However, as shown in Fig. 6A, we found that expression of the IFNAR2 subunit increased both the magnitude and duration of IFN-α-dependent STAT4 tyrosine phosphorylation. In contrast, IL-12-induced STAT4 activation was not altered by expression of the IFNAR2 subunit. The increased duration of STAT4 activation correlated well with enhanced induction of IFN-γ in response to IFN-α + IL-18 stimulation (Fig 6B). Here, purified transduced cells were activated for 24 hours with either IL-12 + IL-18 or with IFN-α + IL-18 in order to assess IFN-γ secretion. IL-12 + IL-18 activated cells displayed ~60% GFP+ cells, and this effect was independent of the expression of the IFNAR2 subunit. However, we found that expression of the IFNAR2 subunit increased the IFN-γ+ population by 10 fold in response to IFN-α + IL-18 activation (Fig. 6B and C).
Figure 6. Ectopic IFNAR2 expression promotes sustained STAT4 phosphorylation and IFN-γ secretion in response to IFN-α.

(A) Spleen and lymph node cells from DO11.10 mice were activated with OVA peptide under Th1-inducing conditions and transduced with retrovirus vectors expressing GFP alone (GFPRV) or the full-length mIFNAR2 subunit. Cells were sorted on day 7 based on GFP expression and restimulated with irradiated BALB/c splenocytes and OVA peptide. Following expansion for an additional 7 days, resting cells were activated with either IL-12 or IFN-α for the times indicated in the figure. Cells were then stained and analyzed for intracellular tyrosine-phosphorylated STAT4 as described in Fig. 5. (B and C) Day 14 transduced Th1 cells were activated with either IL-12 + IL-18, IFN-α + IL-18, or with the individual cytokines indicated in the figure for 24 hours. Brefelden A was added during the last 4 hours of stimulation. Cells were then stained for mCD4 and IFN-γ and analyzed by flow cytometry. Data were gated on live cells and GFP expression.
Differential induction of T-bet expression by IL-12 and IFN-α
The differential kinetics of STAT4 phosphorylation observed in response to IL-12 and IFN-α could result in differences in commitment of naïve human CD4+ T cells to the Th1 phenotype. One aspect of Th1 commitment is the acquisition of IL-12 responsiveness, mediated through induction of the IL-12Rβ2 subunit (46, 47). As both IL-12 and IFN-α have been reported to induce IL-12Rβ2 expression (32), we examined whether IL-12 and IFN-α differed in their induction of IL-12Rβ2 in developing human CD4+ T cells. For these experiments, naïve human CD4+/CD45RA+ cells were purified from human peripheral blood and activated with plate-bound anti-CD3 + anti-CD28 with the cytokines or neutralizing anti-cytokine antibodies indicated in Fig. 7. When analyzed at 48 hours post-stimulation, both IL-12 and IFN-α were able to promote expression of IL-12Rβ2 cell surface protein (Fig. 7A) and mRNA transcripts (Fig. 7B, upper panel) to similar levels. In addition, IL-12 + IFN-α acted synergistically to promote enhanced IL-12Rβ2 expression at 48 hours (Fig. 7A, and Fig. 7B, upper panel). However, IFN-α failed to maintain IL-12Rβ2 expression in cells activated for 7 days (Fig. 7B).
Figure 7. IFN-α does not promote stable T-bet expression in human CD4+ T cells.

Purified human CD4+/CD45RA+ T cells were activated with plate-bound anti-CD3 + anti-CD28, IL-2, and anti-IL-4, and with either anti-IFNAR2 and anti-IL-12 (“Ctl”), anti-IFNAR2 and IL-12 (“IL-12”), IFN-αA and anti-IL-12 (“IFN-aA”), or with IFN-αA and IL-12 (“IL-12 + IFN-αA”). A, After 72 hours, cells were stained for surface expression of IL-12Rβ2: filled histogram, neutralizing antibodies alone; dashed line, IL-12 + anti-IFNAR2; dotted line, IFN-αA + αIL-12; solid line, IL-12 + IFN-αA. B, Total RNA was isolated from cells harvested 48 hours or 7 days after activation. Analysis of IL-12Rβ2 and T-bet transcript levels was performed by quantitative real-time PCR (qPCR) using the primers listed in Materials and Methods. Transcript levels for each condition were normalized to GAPDH, and the data were further normalized relative to cells activated under neutralizing conditions. C, Whole-cell lysates were prepared from day 7 activated cells and assessed for expression of T-bet protein by Western blotting (upper panel). Blots were stripped and re-probed for Lamin protein expression (lower panel).
In mice, T-bet is known to be involved in the regulation of IL-12Rβ2 (46, 48) and also acts downstream of IL-12 signaling to promote Th1 development (49–51). In addition, previous studies have demonstrated that IFN-α induces expression of both IL-12β2(32) and T-bet(52) in human CD4+ T cells. Thus, it has been assumed that IL-12 and IFN-α play redundant roles in Th1 commitment in human T cells through the induction of T-bet. However, given that IFN-α failed to induce Th1 development (Figs. 1–3), we re-examined the ability of IL-12 and IFN-α to regulate T-bet expression during early phases of Th1 commitment and in fully polarized Th1 cells. Similar to the regulation of IL-12Rβ2, both IL-12 and IFN-α independently induced expression of T-bet at 48 hours, and IL-12 + IFN-α co-stimulation further enhanced T-bet expression (Fig. 7B, lower panel). However, T-bet expression was not maintained in response to IFN-α alone in fully polarized cells analyzed on day 7 (Fig. 7B, lower panel). The low expression of T-bet mRNA in response to IFN-α correlated well with decreased T-bet protein accumulation in these cells (Fig. 7C).
Based on these observations, we considered the possibility that IFN-α failed to promote Th1 development due to the lack of stable T-bet expression within polarized Th cells. If this hypothesis is correct, then Th1 development should be restored by ectopic T-bet expression regardless of initial polarizing conditions. To test this hypothesis, we expressed T-bet by retroviral transduction of primary naïve human CD4+ T cells (Fig. 8). For these experiments, purified CD4+/CD45RA+ T cells were activated with plate-bound anti-CD3 + anti-CD28 and transduced with retrovirus constructs expressing GFP only (GFPRV) or human T-bet and GFP (hT-bet-GFP). Upon retroviral transduction, polarizing cytokine conditions were imposed with the addition of cytokines or neutralizing anti-cytokine antibodies as indicated in Fig. 8. These cells were expanded on day 7 with plate-bound anti-CD3, and IFN-γ expression was assessed on day 14 by intracellular cytokine staining. We were able to achieve approximately 15–20% transduction efficiency as measured by GFP expression, and the non-GFP expressing cells served as an additional internal negative control. As shown in Fig. 8A, cells transduced with the hT-bet-GFP construct were gated on GFP− and GFP+ populations. Within the GFP- population, IL-12 increased the frequency of IFN-γ+ cells, whereas IFN-α did not promote IFN-γ expression, as expected. However, expression of T-bet within the GFP+ population increased the percentage of cells capable of secreting IFN-γ, and this effect was independent of initial cytokine conditions (Fig. 8A, compare left and right panels). Expression of GFP alone from the GFPRV vector had little effect on IFN-γ expression (Fig. 8B). Further, T-bet was sufficient to promote IFN-γ expression even when cells were polarized under Th2-inducing conditions (Fig. 8A, bottom panels, and Fig. 8B, condition 8). Thus, these data place T-bet down-stream of STAT4 and suggest that T-bet is sufficient to mediate Th1 development in human CD4+ T cells independent of innate cytokine priming. These data also suggest that IFN-α fails to mediate Th1 development due to lack of sustained T-bet expression.
Figure 8. Ectopic T-bet expression promotes Th1 development independent of IL-12 or IFN-α in naïve human CD4+ T cells.

Purified naïve human CD4+ T cells were transduced with retrovirus constructs expressing GFP only (GFPRV) or with co-expression of human T-bet (hT-bet-GFP). During retroviral transduction, separate groups of cells were simultaneously activated in the presence or absence of cytokines or anti-cytokine antibodies as indicated in the figure. Cells were expanded on day 7 by restimulation on anti-CD3-coated plates. On day 14, resting cells were restimulated with PMA + ionomycin and analyzed for IFN-γ expression by intracellular cytokine staining. A, hT-bet-GFP-transduced cells were gated on live and either GFP negative (GFP-, left panels) or positive (GFP+, right panels) populations. The percentages of CD4+ and either IFN-γ- or IFN-γ+ populations are indicated within their respective quadrants. B, Triplicate cultures were analyzed for IFN-γ expression by intracellular cytokine staining. The percentage of CD4+/IFN-γ+ cells transduced with either the control GFPRV or hT-bet-GFP vectors are compared between the GFP- (open bars) and GFP+ (closed bars) populations. The error bars represent the standard deviation of the mean. These experiments were performed three times with similar results.
Discussion
Type I interferon is one of the first lines of defense against many viral and some bacterial infections. In recent years, the importance of type I interferon during innate responses has been highlighted by the discovery of IFN-α-secreting plasmacytoid dendritic cells (pDCs) (53, 54) as well as Toll-like receptor signaling pathways which promote IFN-α/β secretion from innate cells (1, 2). Thus, IFN-α/β represents an innate cytokine with the potential to shape adaptive immune responses. Further, in vitro virus infection of human pDC (CD4+/CD11c−lin−) promotes their differentiation into high IFN-α/β-producing cells (IPCs) (55), suggesting a role for these cells in regulating adaptive responses. However, in the absence of other innate signals, the ability of IFN-α/β to promote Th1 development is controversial. The role of IFN-α/β in promoting type I responses has been suggested by studies of LCMV infections in mice (22). Here, Biron and colleagues correlated the induction of IFN-γ from splenocytes with LCMV infection that was IFN-α/β- and STAT4-dependent, but IL-12-independent. Collectively, these studies have concluded that like IL-12, IFN-α/β can directly promote Th1 development through the activation of STAT4 in naïve CD4+ T cells. However, more recent studies raise the possibility that the effects of IFN-α/β observed in vivo might be more pronounced in CD8+ than in CD4+ T cells (21, 24, 25).
In this study, we found that in the absence of IL-12, IFN-α was insufficient to promote Th1 development. This defect was correlated with attenuated STAT4 tyrosine phosphorylation and the lack of stable expression of the Th1-specific transcription factor T-bet. Whether STAT4 directly regulates the T-bet promoter in human CD4+ T cells is unclear at this point. However, we found that ectopic expression of T-bet within primary naïve human CD4+ T cells circumvented the requirement for innate cytokines in Th1 commitment.
Although we found that IL-12 was sufficient to promote Th1 development, human CD4+ T cells failed to commit to the Th1 fate in response to IFN-α/β. In some cases, we also observed that cells cultured with IL-12 + IFN-α expressed less IFN-γ that cells differentiated with IL-12 alone. However, this effect was not consistent from one donor to the next. Nonetheless, we consistently observed that IFN-α alone failed to promote Th1 development, and in some cases suppressed IFN-γ expression compared to cells developing under neutralizing conditions. Given these results, how can our observations be reconciled with previous reports suggesting that IFN-α/β regulates type I responses in vivo? Clearly, there are many examples of virus infections that promote IFN-α/β secretion and generate populations of T cells capable of secreting IFN-γ (22, 56–59). In such cases, adaptive antiviral responses rely heavily on the activation and expansion of CD8+ cells that commit to IFN-γ expression independent of either IL-12 or STAT4 (37). Thus, it is possible that the type I responses observed in vivo do not originate from CD4+ cells, but rely on other cell types such as CD8+ and NK cells. Indeed, type I responses to certain viruses such as LCMV are diminished in mice deficient in STAT4 and IFN-α/β receptor signaling (22). However, these defects were observed predominantly in the CD8+ T cell compartment.
Alternatively, it is possible that IFN-α, in combination with other innate cytokines, could promote CD4+ Th1 development in vivo. Our data indicate only that IFN-α is insufficient to promote Th1 development. IFN-α may be necessary in vivo, in the context of other innate signals, to drive Th1 development when IL-12 is limiting. For example, IL-18 and IFN-α were demonstrated to be required for productive IFN-γ responses from CD8+ T cells during LCMV infections (60). These data suggest that IFN-α/β collaborates with IL-18 to promote IFN-γ secretion from CD8+ T cells in vivo. However, the sufficiency of IFN-α/β to drive CD4+ Th1 development in vivo has not been demonstrated. Taken together, our results suggest that IFN-α/β may act in synergy with IL-12 to positively regulate early stages of Th1 commitment, but in the absence of IL-12, IFN-α/β is insufficient to promote full commitment of human CD4+ T cells to the Th1 lineage.
Acknowledgments
We thank Dr. Dong-Er Zhang for sharing spleen tissues from Ubp43-deficient mice. The authors gratefully acknowledge Drs. L. Hooper and J. Niederkorn for critically reviewing the manuscript. We thank Dr. K. Murphy, L. Berenson, L. Matthews and members of Dr. Hooper’s laboratory for helpful discussions.
ABBREVIATIONS
- STAT
signal transducer and activator of transcription
- IFNAR
IFN-α/β receptor
Footnotes
This study was supported by a grant from the NIH/NIAID (AI56222) awarded to JDF. HJR was supported by a pre-doctoral fellowship from the NIH/NIAID (AI68622), and AMD was supported by a training grant from the NIH/GM (GM00820317).
References
- 1.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 3.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ, O’Garra A. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101–109. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 7.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 12.Verhagen CE, de Boer T, Smits HH, Verreck FA, Wierenga EA, Kurimoto M, Lammas DA, Kumararatne DS, Sanal O, Kroon FP, van Dissel JT, Sinigaglia F, Ottenhoff TH. Residual type 1 immunity in patients genetically deficient for interleukin 12 receptor beta1 (IL-12Rbeta1): evidence for an IL-12Rbeta1-independent pathway of IL-12 responsiveness in human T cells. J Exp Med. 2000;192:517–528. doi: 10.1084/jem.192.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parronchi P, De Carli M, Manetti R, Simonelli C, Sampognaro S, Piccinni MP, Macchia D, Maggi E, Del Prete G, Romagnani S. IL-4 and IFN (alpha and gamma) exert opposite regulatory effects on the development of cytolytic potential by Th1 or Th2 human T cell clones. J Immunol. 1992;149:2977–2983. [PubMed] [Google Scholar]
- 14.Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993;178:1655–1663. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho SS, Bacon CM, Sudarshan C, Rees RC, Finbloom D, Pine R, O’Shea JJ. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–4789. [PubMed] [Google Scholar]
- 16.Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J Biol Chem. 2000;275:2693–2697. doi: 10.1074/jbc.275.4.2693. [DOI] [PubMed] [Google Scholar]
- 17.Rogge L, D’Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, Adorini L, Sinigaglia F. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol. 1998;161:6567–6574. [PubMed] [Google Scholar]
- 18.Sinigaglia F, D’Ambrosio D, Rogge L. Type I interferons and the Th1/Th2 paradigm. Dev Comp Immunol. 1999;23:657–663. doi: 10.1016/s0145-305x(99)00039-7. [DOI] [PubMed] [Google Scholar]
- 19.Wenner CA, Guler ML, Macatonia SE, O’Garra A, Murphy KM. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 20.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–69. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 21.Persky ME, Murphy KM, Farrar JD. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297:2063–2066. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]
- 23.Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol. 2002;169:1665–1668. doi: 10.4049/jimmunol.169.4.1665. [DOI] [PubMed] [Google Scholar]
- 24.Berenson LS, Farrar JD, Murphy TL, Murphy KM. Frontline: absence of functional STAT4 activation despite detectable tyrosine phosphorylation induced by murine IFN-alpha. Eur J Immunol. 2004;34:2365–2374. doi: 10.1002/eji.200324829. [DOI] [PubMed] [Google Scholar]
- 25.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172:61–69. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 26.Berenson LS, Gavrieli M, Farrar JD, Murphy TL, Murphy KM. Distinct Characteristics of Murine STAT4 Activation in Response to IL-12 and IFN-{alpha} J Immunol. 2006;177:5195–5203. doi: 10.4049/jimmunol.177.8.5195. [DOI] [PubMed] [Google Scholar]
- 27.Krutzik PO, Clutter MR, Nolan GP. Coordinate analysis of murine immune cell surface markers and intracellular phosphoproteins by flow cytometry. J Immunol. 2005;175:2357–2365. doi: 10.4049/jimmunol.175.4.2357. [DOI] [PubMed] [Google Scholar]
- 28.George TC, Fanning SL, Fitzgeral-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, Morrissey PJ, Lynch DH. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods. 2006;311:117–129. doi: 10.1016/j.jim.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Ranganath S, Ouyang W, Bhattarcharya D, Sha WC, Grupe A, Peltz G, Murphy KM. GATA-3-dependent enhancer activity in IL-4 gene regulation. J Immunol. 1998;161:3822–3826. [PubMed] [Google Scholar]
- 31.Farrar JD, Ouyang W, Lohning M, Assenmacher M, Radbruch A, Kanagawa O, Murphy KM. An instructive component in T helper cell type 2 (Th2) development mediated by GATA-3. J Exp Med. 2001;193:643–650. doi: 10.1084/jem.193.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogge L, Barberis-Maino L, Biffi M, Passini N, Presky DH, Gubler U, Sinigaglia F. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;185:825–831. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy E, Shibuya K, Hosken N, Openshaw P, Maino V, Davis K, Murphy K, O’Garra A. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J Exp Med. 1996;183:901–913. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobson NG, Szabo SJ, Guler ML, Gorham JD, Murphy KM. Regulation of interleukin-12 signalling during T helper phenotype development. Adv Exp Med Biol. 1996;409:61–73. doi: 10.1007/978-1-4615-5855-2_9. [DOI] [PubMed] [Google Scholar]
- 35.Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, Hartley SB, Menon S, Kastelein R, Bazan F, O’Garra A. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7:571–581. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 37.Carter LL, Murphy KM. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J Exp Med. 1999;189:1355–1360. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, Sareneva T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol. 2001;31:2236–2245. [PubMed] [Google Scholar]
- 39.Sareneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J Immunol. 1998;160:6032–6038. [PubMed] [Google Scholar]
- 40.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol. 2001;2:157–164. doi: 10.1038/84264. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Tran E, Zhao Y, Huang Y, Flavell R, Lu B. Gadd45 beta and Gadd45 gamma are critical for regulating autoimmunity. J Exp Med. 2005;202:1341–1347. doi: 10.1084/jem.20051359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. Embo J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim KI, Malakhova OA, Hoebe K, Yan M, Beutler B, Zhang DE. Enhanced antibacterial potential in UBP43-deficient mice against Salmonella typhimurium infection by up-regulating type I IFN signaling. J Immunol. 2005;175:847–854. doi: 10.4049/jimmunol.175.2.847. [DOI] [PubMed] [Google Scholar]
- 44.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med. 2004;10:1374–1378. doi: 10.1038/nm1133. [DOI] [PubMed] [Google Scholar]
- 45.Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/beta receptor-2 subunit is mediated by the STAT4 N-domain. Molecular immunology. 2007;44:1864–1872. doi: 10.1016/j.molimm.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 47.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O’Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 50.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 51.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 52.Hibbert L, Pflanz S, De Waal Malefyt R, Kastelein RA. IL-27 and IFN-alpha signal via Stat1 and Stat3 and induce T-Bet and IL-12Rbeta2 in naive T cells. J Interferon Cytokine Res. 2003;23:513–522. doi: 10.1089/10799900360708632. [DOI] [PubMed] [Google Scholar]
- 53.Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells--virus experts of innate immunity. Semin Immunol. 2005;17:253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 54.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 55.Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med. 2000;192:219–226. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roman E, Miller E, Harmsen A, Wiley J, Von Andrian UH, Huston G, Swain SL. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med. 2002;196:957–968. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cousens LP, Peterson R, Hsu S, Dorner A, Altman JD, Ahmed R, Biron CA. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J Exp Med. 1999;189:1315–1328. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101:3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 59.Divekar AA, Zaiss DM, Lee FE, Liu D, Topham DJ, Sijts AJ, Mosmann TR. Protein vaccines induce uncommitted IL-2-secreting human and mouse CD4 T cells, whereas infections induce more IFN-gamma-secreting cells. J Immunol. 2006;176:1465–1473. doi: 10.4049/jimmunol.176.3.1465. [DOI] [PubMed] [Google Scholar]
- 60.Pien GC, Nguyen KB, Malmgaard L, Satoskar AR, Biron CA. A unique mechanism for innate cytokine promotion of T cell responses to viral infections. J Immunol. 2002;169:5827–5837. doi: 10.4049/jimmunol.169.10.5827. [DOI] [PubMed] [Google Scholar]