

Abstract
Previously we demonstrated the expression of the long form of the leptin receptor in rodent pancreatic β-cells and an inhibition of insulin secretion by leptin via activation of ATP-sensitive potassium channels. Here we examine pancreatic islets isolated from pancreata of human donors for their responses to leptin. The presence of leptin receptors on islet β-cells was demonstrated by double fluorescence confocal microscopy after binding of a fluorescent derivative of human leptin (Cy3-leptin). Leptin (6.25 nM) suppressed insulin secretion of normal islets by 20% at 5.6 mM glucose. Intracellular calcium responses to 16.7 mM glucose were rapidly reduced by leptin. Proinsulin messenger ribonucleic acid expression in islets was inhibited by leptin at 11.1 mM, but not at 5.6 mM glucose. Leptin also reduced proinsulin messenger ribonucleic acid levels that were increased in islets by treatment with 10 nM glucagon-like peptide-1 in the presence of either 5.6 or 11.1 mM glucose. These findings demonstrate direct suppressive effects of leptin on insulin-producing β-cells in human islets at the levels of both stimulus-secretion coupling and gene expression. The findings also further indicate the existence of an adipoinsular axis in humans in which insulin stimulates leptin production in adipocytes and leptin inhibits the production of insulin in β-cells. We suggest that dysregulation of the adipoinsular axis in obese individuals due to defective leptin reception by β-cells may result in chronic hyperinsulinemia and may contribute to the pathogenesis of adipogenic diabetes.
Obesity and associated diabetes are epidemic throughout the world. The prevalence of morbid obesity is between 20–40% in populations of developing countries and predisposes to the development of diabetes, so-called adipogenic diabetes (1, 2). Moreover, obese individuals characteristically manifest insulin resistance and hyperinsulinemia, which predispose to glucose intolerance, diabetes, and cardiovascular disease (1). From 50–80% of subjects worldwide with diabetes are obese depending upon the ethnic background and the community (3–8). It has been estimated that for every kilogram of increase in weight, the risk for diabetes increases 4.5% (9). Remarkably, 97 million American adults (35% of the population) are overweight or obese (10). The marked increase in the prevalence of obesity is believed to be due in large part to a change in life-style favoring excessive caloric intake and a sedentary existence (11).
A major advance in understanding the hormonal causation of morbid obesity is the discovery of a hormone, leptin, produced by adipose tissue that modifies feeding behavior in rodents as a potent suppressor of food intake and stimulator of energy expenditure (12, 13). Leptin exerts its actions centrally on appetite and thermogenic control centers located in the hypothalamus. It is important to note that obesity in humans is believed to be due to a desensitization of leptin reception within the hypothalamus, resulting in hyperphagia (1, 12–14). Obese individuals have marked elevations in their plasma levels of leptin directly proportional to their body fat mass (1). Sustained elevated levels of plasma leptin are proposed to uncouple leptin actions on its receptors in the hypothalamus, thereby attenuating signal transduction pathways that exert the effects of the hormone on satiety and energy expenditure (12, 15). Inactivating mutations in the genes encoding leptin and the leptin receptor in the human population are rare and cannot account for the high prevalence of hyperphagia and obesity in the population (13).
Germane to the studies undertaken and described herein is that leptin also acts directly on receptors in pancreatic β-cells to suppress insulin secretion in rodents (16–30). Leptin suppresses insulin secretion, insulin messenger ribonucleic acid (mRNA) levels, and transcription from the rat insulin promoter (30a) in isolated islets, perfused pancreata of mice and rats, and rats studied in vivo. These findings led us to propose the existence of an adipoinsular axis in which insulin stimulates adipogenesis and leptin production, and leptin inhibits insulin secretion (21, 31). Leptin has been shown to suppress insulin secretion by activating the ATP-sensitive K+ channel (KATP) on β-cells and to thereby hyperpolarize the β-cells to resist depolarization required for insulin secretion (21, 27, 30). Most studies reported to date are limited to rodents (rats and mice). Two earlier reports described an inhibition by leptin of insulin secretion in cultured human islets (22, 32). The studies in rodents are informative, but beg the question of what the effects of leptin are on proinsulin gene expression in human islets. Effectors of glucose homeostasis in rodents may not always be equivalent to those operative in human islets (33, 34).
Here we report that the application of leptin to freshly obtained human islets results in a marked suppression of both insulin secretion and the expression of proinsulin mRNA within the islets. We also find that leptin inhibits the stimulation of proinsulin mRNA levels in human islets mediated by the insulinotropic hormone glucagon-like peptide-1 (GLP-1). These findings support further the existence of an antagonism between leptin and GLP-1 signaling on β-cells as indicated by the reported studies in mouse and rat islets (18, 21, 29). Based on these findings, we propose that in obese individuals leptin reception on β-cells is desensitized, similar to what is proposed to occur at the level of the hypothalamus. This desensitization of suppression of insulin secretion of β-cells contributes to the hyperinsulinemia of obesity. Hyperinsulinemia, in turn, leads to increased adipogenesis and insulin resistance, culminating in some individuals in the development of diabetes.
Materials and Methods
Human pancreatic islets
Human pancreases were procured by the National Disease Research Interchange and were obtained from multiorgan heart-beating donors. The organs were obtained after in situ perfusion of the abdominal aorta with cold University of Wisconsin solution and were kept on ice before processing. Islet isolation was performed essentially as previously described (35) at the Cell Transplant Center, Diabetes Research Institute, University of Miami School of Medicine (Miami, FL). Isolated islets were cultured for an average of 1 day at 24 C and after assessment of viability were shipped in CMRL 1066 medium at ambient temperature. The data presented are from islets obtained from six different donors on different occasions. Islets from different donors were not pooled. Several different types of experiments were performed on each shipment of islets from a single donor.
Detection of leptin binding to leptin receptor (Ob-R) on single dispersed primary human pancreatic β-cells
Human islets were dispersed into single cells using trypsinethyl-enediamine tetraacetate, seeded onto poly-L-lysine-coated glass microscope slides in RPMI 1640 medium supplemented with 10% FBS. Human leptin (PeproTech, Rocky Hill, NY) was coupled with iodocarbocyanine (Cy3) and purified as previously described (21). Receptor binding, immunocytochemistry for the detection of insulin, and microscopic image acquisition and processing were performed on the dispersed pancreatic islets as previously described (21), except that confocal microscopy was used.
Culturing of islets for measurements of insulin secretion
Islets (hand-picked) were placed 10/well (in a 12-well culture plate) with 1 mL/well 5.5 mM glucose RPMI medium containing 0.5% human serum albumin (Sigma Chemical Co., St. Louis, MO) and 1% aprotinin (Miles, Kankakee, IL). After incubation for 1 h, the medium was carefully removed and replaced with fresh medium at either 5.5 or 11.1 mM glucose, with or without recombinant human leptin (100 ng/mL or 6.25 nM; PeproTech). After a 1-h incubation, the medium was removed, centrifuged (15,000 rpm, 5 min), transferred to a fresh Eppendorf tube, and frozen at −20 C for subsequent insulin RIA.
Islet incubations for measurements of proinsulin mRNA levels by RT-PCR
Islets were incubated in 24-well plates at 200 islets/well in RPMI 1640 medium (Life Technologies, Gaithersburg, MD) supplemented with 0.1% human serum albumin (Sigma Chemical Co.) and 1% aprotinin (Miles) for the indicated intervals. Glucose concentrations in the incubation medium were adjusted to 5.6 and 11.1 mM, respectively. Recombinant human leptin (PeproTech) was added, where indicated, at a concentration of 6.25 nM (100 ng/mL). The vehicle for leptin was 10 mM Tris-buffer, pH 9.5. Recombinant GLP-1-(7–36)amide was obtained from Peninsula Laboratories, Inc. (Belmont, CA), and was used at a concentration of 10 nM. The vehicle for GLP-1 was PBS. To facilitate processing of the islets as a single series, incubations of leptin and GLP-1 were started first with the 48 h group, followed by the 24, 16, and 6 h points. In this way all incubation periods finished at the same time. All of the vehicle-treated islets serve a zero time point. All islet samples were harvested, and total RNA was extracted in a single series for RT-PCR.
Semiquantitative RT-PCR
Total RNA from islets was extracted by the single step guanidinium-isothiocyanate method with a commercial reagent (Trizol, Life Technologies). Extracted RNA was quantitated by OD260/280 measurement and gel electrophoresis. Equal amounts of isolated RNA were subjected to RT in triplicate samples by oligo(deoxythymidine) priming with Moloney murine leukemia virus reverse transcriptase (Superscript, Life Technologies), and 2 μL of each RT reaction were used as input for subsequent PCR. In each PCR reaction, endogenous β-actin was coamplified together with insulin complementary DNA as a control to adjust for differences in complementary DNA input. PCR amplification was performed using Taq-polymerase (TaKaRa, Takara Shuzo Co., Otsu, Japan) under the following conditions: 30 s at 94 C, 45 s at 54 C, and 45 s at 72 C (20 cycles). Primer oligonucleotides for insulin were: sense primer, 5′-CCAGCCGCAGCCTTTGTGA-3′; and antisense primer, 5′-GGTACAGCATTGTTCCACAATG-3′. Intron border-spanning oligonucleotides for β-actin were: sense primer, 5′-GATGACCCA GATCAT-GTTTG-3′; and antisense primer, 5′-GAGCAATGATCTTGATCTTC-3′. Cycle numbers were determined to cover the exponential range of the amplification kinetics. For each sample, a negative control reaction without template was included, and to control for residual DNA contamination in the RNA preparations, a RT reaction without reverse transcriptase (RT-minus) was amplified by PCR for each islet RNA sample. Only RT-PCR reactions with negative results in the corresponding RT-minus samples were used for quantification. PCR products were subjected to agarosegel electrophoresis. After staining with ethidium bromide, the band intensities were quantitated on a Fluorimager 575 using ImageQuant software (Molecular Dynamics, Inc., Sunnyvale, CA) by rectangle mode/local background/volume integration, and values were normalized to the intensity of the β-actin bands. Data are expressed as the means (±SEM) of three different independent RT-PCR reactions.
Effects of leptin on intracellular calcium ([Ca2+]i)
[Ca2+]i responses were measured in dispersed pancreatic β-cells seeded on concanavalin A-coated coverslips using fura-2/acetoxy-methyl ester (fura-2/AM, Molecular Probes, Inc., Eugene, OR) as previously described (21).
Results
The identification of leptin receptors on human β-cells was accomplished using fluorescently labeled leptin (Cy3-leptin) as a ligand and an antiserum to insulin on dispersed cells prepared from the islets. Representative β-cells immunostain with a green fluorescence, and Cy3-leptin bound to the β-cell provides a red fluorescence (Fig. 1). The leptin receptors, bound to the Cy3-leptin, give a punctate appearance characteristic of their aggregation in response to leptin (Fig. 1). As several isoforms of the leptin receptor exist, notably a short form (Ob-Ra) that is C-terminally truncated and deficient in conveying leptin signaling and a long form (Ob-Rb) that does signal, we used RT-PCR to determine the relative amounts of mRNAs encoding Ob-Ra and Ob-Rb. The ratios of the RT-PCR products formed were 4:1 Ob-Ra:Ob-Rb (data not shown).
Fig. 1.

Leptin binds to human pancreatic β-cells. Human leptin labeled with the fluorochrome Cy3 (red fluorescence) was incubated for 30 min with dispersed human islet cells that had been cultured overnight. The cells were costained with an antiserum to insulin and a secondary antibody coupled to fluorescein isothiocyanate (green fluorescence). Shown is a typical β-cell in phase contrast (upper left), stained with insulin antiserum (upper right), with Cy3-labeled leptin (lower left), and with insulin and Cy3-leptin images superimposed (lower right).
Leptin suppresses insulin secretion by cultured human islets
Incubations of human islets in the presence of leptin (6.25 nM) and normal concentrations of glucose (5.6 mM) for 1 h resulted in a 20% inhibition of insulin secretion compared to the control period of incubation in the absence of leptin (Table 1). A similar incubation of human islets with 6.25 nM leptin in the condition of high glucose (11.1 or 25 mM) failed to significantly suppress insulin secretion. These findings of a glucose dependence on leptin suppression of insulin secretion are in complete agreement with our findings reported previously on the glucose dependence of leptin suppression of insulin secretion on isolated mouse islets (21).
TABLE 1.
Suppression of insulin secretion by leptin in cultured human pancreatic islets
| n | Glucose (5.6 mmol/L) |
Glucose (11.1 mmol/L) |
|||
|---|---|---|---|---|---|
| Control | Leptina | Control | Leptin | ||
| Exp 1 | 3 | 7.06b | 5.87 | 15.06 | 13.2 |
| Exp 2 | 3 | 7.67 | 5.70 | 10.40 | 9.80 |
| Exp 3 | 6 | 9.24 | 7.36 | 14.44 | 16.5 |
| Compiled mean | 12 | 7.99 | 6.31 | 13.30 | 13.17 |
| SEMc | 0.65 | 0.53 | 1.46 | 1.93 | |
| P (by t test) | <0.01 | <0.46 | |||
Insulin released into culture media (nanograms per mL) after 1 hr of treatment with leptin or vehicle alone (control) was measured by RIA.
Leptin concentration of 6.25 nmol/L.
Means of the numbers (n) shown.
By Student’s paired t test.
Leptin inhibits glucose-dependent proinsulin mRNA expression in human islets
Studies of rodent islets provide evidence that leptin may reduce steady state levels of proinsulin mRNA (22, 24, 25). To determine whether leptin may alter levels of proinsulin gene expression in addition to inhibiting insulin secretion, aliquots of human islets were cultured for up to 48 h in medium containing normal (5.6 mM) or high (11.1 mM) concentrations of glucose with and without leptin (6.25 nM). Proinsulin and actin mRNA levels in extracts of islets obtained at 6, 16, 24, and 48 h of incubation were determined by semiquantitative RT-PCR. High glucose compared to low glucose typically stimulated proinsulin mRNA levels by 1.5-fold (Fig. 2A). Notably, incubation of the islets in the presence of 6.25 mM leptin resulted in a time-dependent decrease in proinsulin mRNA levels in the presence of high, but not low, glucose (Fig. 2B). No effects of leptin were observed on the levels of the control actin mRNA.
Fig. 2.

Leptin suppresses proinsulin mRNA levels in human islets in high (11.1 mM), but not normal (5.6 mM), concentrations of glucose. Aliquots of human islets were cultured for 48 h in the absence (A; vehicle) or the presence (B) of 6.25 nM human leptin. RNA concentrations were determined by semiquantitative RT-PCR as previously described (51). Actin mRNA served as a control. The bar graphs represent the relative amounts of RT-PCR products determined by fluorescence densitometry (see Materials and Methods). Values are the mean ± SEM (n = 3). The densitometric value for 5.6 mM glucose at 6 h was arbitrarily set at 1.0.
Because the established half-life of proinsulin mRNA in rat islets is 25 h (36), we assumed that it would be similar in human islets. Thus, the first time point analyzed was 6 h to allow the incubations to stabilize. By 16 h, the levels of proinsulin mRNA in islets treated with leptin and 11.1 mM glucose reached 50% of the 6 h value. The 6 h value in 5.6 mM glucose was arbitrarily set at 1.0 mRNA densitometric units. By 24 h, the level of proinsulin mRNA fell to 25% of the initial value. These findings indicate that in the presence of leptin and 11.1 mM glucose, and assuming a cessation of gene transcription, the half-life of the human proinsulin mRNA is approximately 16 h. It is also possible that leptin alters the stability of the proinsulin mRNA independent of gene transcription. Thus, the glucose-dependent inhibition of proinsulin gene expression by leptin, as determined by mRNA levels, and the glucose-dependent inhibition of insulin secretion display different sensitivities with respect to absolute glucose concentrations in human islets.
Leptin inhibits GLP-1-stimulated proinsulin mRNA levels in the presence of both low (5.6 mM) and high (11.1 mM) glucose concentrations
GLP-1 acts through cAMP-coupled receptors on β-cells to markedly stimulate glucose-dependent insulin secretion at both normal physiological and supraphysiological concentrations of glucose. GLP-1 also stimulates transcription of the rat insulin gene by 1.5- to 2-fold (37) and increases proinsulin mRNA levels by 2- to 3-fold in cultured insulinoma cells (38). Because leptin acts on a cytokine receptor and cytokine signaling is known to be antagonistic to cAMP signaling, and vice versa (39, 40), we sought to determine whether leptin might inhibit the increase in proinsulin mRNA stimulated by GLP-1 in human islets. The islets were incubated in the presence of GLP-1 (10 nM) in conditions of normal (5.6 mM) or high (11.1 mM) glucose with or without leptin (6.25 nM) for 16 h. The incubation period of 16 h was chosen because it is the time when proinsulin levels in response to leptin have fallen to the half-maximal value (Fig. 2).
GLP-1 stimulated proinsulin mRNA levels by 1.5-fold (5.6 mM glucose) to 2-fold (11.1 mM glucose) during the 16-h control incubation, findings similar to what was reported for rat islets treated with GLP-1 (Fig. 3). Remarkable, however, is the finding that leptin (6.25 mM) inhibits the GLP-1-stimulated expression of proinsulin mRNA in conditions of both 5.6 and 11.1 mM glucose (Fig. 3). At 16 h, a time at which leptin suppresses proinsulin mRNA accumulation by 50% (Fig. 2), the GLP-1-stimulated proinsulin mRNA levels are inhibited by 80% (Fig. 3).
Fig. 3.

Leptin suppresses GLP-1-stimulated proinsulin mRNA levels in human islets in the presence of both normal (5.6 mM) and high (11.1 mM) glucose. Human islets were incubated for 16 h in the presence of normal (5.6 mM) or high (11.1 mM) glucose with and without 6.25 nM leptin and 10 nM GLP-1. The amounts of proinsulin and actin mRNAs were evaluated as described in Fig. 2 and Materials and Methods.
Leptin application to single human β-cells inhibits Ca2+ influx consistent with activation of KATP
It has been shown previously that leptin activates KATP channels in β-cells of normal and ob/ob mouse islets (21) and in the insulinoma cell line CRI-G1 (30) as well as in hypothalamic neurons (41). The activation of KATP channels results in a hyperpolarization of β-cells and thus inhibits the secretion of insulin. When β-cells are depolarized by the inactivation (closing) of KATP channels, voltage-sensitive Ca2+ channels open, resulting in an influx of Ca2+ into the β-cell. Thus, an index of KATP channel activity is the change in the levels of intracellular Ca2+. Therefore, the effect of the application of leptin (6.25 mM) to single human β-cells was examined using the fura-2 dual wavelength calcium-imaging system. The β-cells were first preincubated in 16.7 mM glucose and then transfered to 5.6 mM glucose to demonstrate the glucose responsiveness of the cells. Then a depolarizing stimulus consisting of 16.7 mM glucose was reapplied to the cells in the presence of leptin (6.25 mM; Fig. 4). As can be seen, the addition of leptin markedly decreases [Ca2+]i compared to the addition of 16.7 mM glucose alone (Fig. 4). Thus, human pancreatic β-cells respond to leptin in a manner indistinguishable from β-cells of mouse or insulinoma cell origins (21, 30).
Fig. 4.
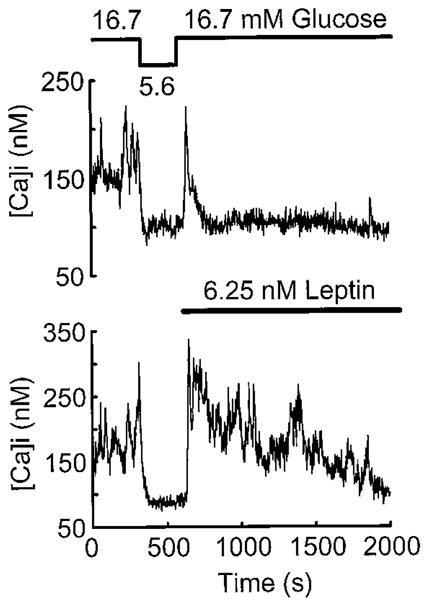
Leptin inhibits Ca2+ influx induced by 16.7 mM glucose in human β-cells. Two fura-2-loaded human β-cells were initially bathed in standard extracellular saline (SES containing 16.7 mM glucose) and exhibited irregular spikes of [Ca2+]i resulting from influx during Ca2+-dependent action potentials. A stable baseline of [Ca2+]i in 16.6 mM glucose was established visually and recorded for 5 min before lowering glucose to 5.6 mM and subsequent application of leptin. The bathing solution was then exchanged for 5.6 mM glucose SES, and this led to the inhibition of spiking activity, consistent with hyperpolarization of the β-cell membrane as the glucose concentration was reduced. Reintroduction of 16.7 mM glucose-SES plus 6.25 nM human leptin then led to an initial restoration of spiking activity that was immediately inhibited in the presence of leptin at this high glucose concentration. The upper and lower panels show two representative β-cell responses to leptin of eight cells so studied.
Discussion
These studies in human islets further add to a mounting body of evidence showing that leptin suppresses insulin secretion by pancreatic β-cells (16–30, 32) and does so by activating the ATP-sensitive K+ channel on β-cells (21, 27, 30). Further, these studies augment the available evidence that leptin inhibits not only insulin secretion, but also the expression of the proinsulin gene. It is worth noting that two published studies claim no effects of leptin on β-cells (42, 43), and two studies purport that leptin stimulates insulin secretion (44, 45). The explanation for these disparate experimental findings remains unknown. Notwithstanding these differences in experimental findings, the available evidence predominantly indicates that leptin exerts inhibitory influences on β-cells.
These results of our studies of leptin on the glucose dependence of proinsulin mRNA levels in the islets were unexpected because they are disparate with the effects of leptin on glucose-dependent insulin secretion in which suppression of insulin secretion by leptin is most pronounced in 5.6 mM glucose compared to 11.1 mM glucose. There are, however, at least two explanations for the differences in glucose dependence of leptin on suppression of insulin secretion and of proinsulin mRNA levels. The effects of leptin on insulin secretion are acute (1 h), whereas the effects of leptin on proinsulin mRNA levels take much longer. No decrease in mRNA levels was observed until 16 h after the addition of leptin (6 h was taken as the first time point to conserve islets, because in preliminary studies we found no difference in mRNA levels at 0 and 6 h). The effects of leptin on insulin secretion and proinsulin mRNA levels may involve different cellular pathways with differential sensitivity to glucose, e.g. KATP (21, 30) and the Janus kinase/STAT (signal transducer and activator of transcription) signaling pathway (46), respectively.
Our studies indicate that leptin appears to antagonize preferentially proinsulin mRNA levels that are induced by GLP-1. These findings of a leptin-mediated inhibition of proinsulin mRNA levels stimulated by GLP-1 in conditions of both normal (5.6 mM) and high (11.1 mM) glucose are at odds with earlier published studies in mouse islets showing that GLP-1 prevents inhibition of insulin secretion by leptin. Thus, we propose that the relative effects of leptin on GLP-1-dependent insulin secretion and levels of proinsulin mRNA are not concordant.
The findings of our studies strongly indicate that the mechanism of action of leptin on β-cells of rodents and humans are similar. Our studies further support a model of β-cell function in which the actions of leptin and GLP-1 are antagonistic at the level of modulation of regulation of the activity of KATP. It is well known that GLP-1 and glucose act synergistically to inactivate KATP (47). The data reported herein in human β-cells in conjunction with the previously reported findings in rodent β-cells strongly suggest that leptin activates KATP, antagonizes GLP-1 actions to inactivate KATP, and results in a situation in which leptin suppresses insulin secretion.
Leptin is produced predominantly by adipose tissue, and insulin is well known to stimulate adipogenesis in adipose tissue (48, 49). Our earlier findings of the suppression of insulin secretion by leptin in mouse islets led us to propose the existence of an adipoinsular axis in which insulin stimulates adipogenesis and leptin production, and leptin inhibits insulin secretion (21, 31). The adipoinsular axis is proposed to act in concert with the well established enteroinsular axis in which GLP-1 and other intestinal incretin hormones stimulate insulin secretion (50). The findings of the studies described herein further support the idea of the existence of an adipoinsular axis in humans as well as in rodents.
What are the implications of the findings that leptin suppresses insulin secretion and the expression of the insulin gene in human islets? Leptin, the obesity/satiety hormone produced by adipose tissue, is an important controller of food intake and energy expenditure by its actions on receptors located in regions of the hypothalamus that regulate feeding behavior. Normally, leptin suppresses feeding and increases energy expenditure. In obese individuals, however, who have elevated plasma levels of leptin, the hormone fails to produce satiety or curtail feeding. It is generally believed that this failure of leptin reception by the hypothalamus in obese individuals is due to a desensitization of the receptor system in response to the high plasma levels of leptin.
By analogy to leptin actions on the hypothalamus, we propose that in conditions of obesity and prolonged elevated plasma leptin levels, the receptor system in pancreatic β-cells becomes desensitized. Such a loss of leptin reception by β-cells results in a dysregulation of the adipoinsular axis and a corresponding failure to suppress insulin secretion, resulting in chronic hyperinsulinemia, a common accompaniment of obesity. Chronic hyperinsulinemia and failure of leptin reception set up a positive feedback loop leading to increased adipogenesis and further increases in plasma leptin. Thus, it seems reasonable to suggest that desensitization of leptin reception at the levels of the hypothalamus and the pancreatic β-cells, and resulting hyperphagia and hyperinsulinemia, respectively, may be important factors in the pathogenesis of adipogenic diabetes mellitus.
Acknowledgments
We thank Maurice Castonguay and Karen McManus for expert experimental assistance, and Townley Budde for preparation of the manuscript.
Footnotes
This work was supported in part by USPHS Grant DK30834 and the Juvenile Diabetes Foundation Human Islet Distribution Program at the Diabetes Research Institute, University of Miami (Miami, FL). J.S. is supported by Deutsche Forschungsgemeischaft (DFG). Grant number Se-787/1–2.
References
- 1.Smith SR. The endocrinology of obesity. Endocrinol Metab Clin North Am. 1996;25:921–942. doi: 10.1016/s0889-8529(05)70362-5. [DOI] [PubMed] [Google Scholar]
- 2.Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 3.Bray GA. Obesity. In: Fauci AS, Braunwald E, Isselbacher KJ, Wilson JD, Martin JB, Kasper DL, Hauser SL, Longo DL, editors. Harrison’s principles of internal medicine. 1998. pp. 454–462. [Google Scholar]
- 4.Cairney J, Wade TJ. Correlates of body weight in the 1994 National Population Health Survey. Int J Obes Relat Metab Disord. 1998;22:584–591. doi: 10.1038/sj.ijo.0800632. [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran A, Snehalatha C, Viswanathan V, Viswanathan M, Haffner SM. Risk of noninsulin dependent diabetes mellitus conferred by obesity and central adiposity in different ethnic groups: a comparative analysis between Asian Indians, Mexican Americans and Whites. Diabetes Res Clin Pract. 1997;36:121–125. doi: 10.1016/s0168-8227(97)00040-5. [DOI] [PubMed] [Google Scholar]
- 6.Seidell JC. Time trends in obesity: an epidemiological perspective. Horm Metab Res. 1997;36:155–158. doi: 10.1055/s-2007-979011. [DOI] [PubMed] [Google Scholar]
- 7.Hodge AM, Zimmet PZ. The epidemiology of obesity. Baillieres Clin Endocrinol Metab. 1994;8:577–599. doi: 10.1016/s0950-351x(05)80287-3. [DOI] [PubMed] [Google Scholar]
- 8.Turner R. The United Kingdom Prospective Diabetes Study. Barcelona: European Association for the Study of Diabetes; 1998. [Google Scholar]
- 9.Ford ES, Williamson DF, Liu S. Weight changes and diabetes incidence: findings from a national cohort of US adults. Am J Epidemiol. 1997;146:214–222. doi: 10.1093/oxfordjournals.aje.a009256. [DOI] [PubMed] [Google Scholar]
- 10.Bren L, editor. Clinical guidelines on treating overweight and obesity now available. Bethesda: Summer; WIN Notes; 1998. www.niddk.nih.gov/health/nutrit/related.htm. [Google Scholar]
- 11.O’Rahilly S. Non-insulin dependent diabetes mellitus: the fathering storm. Br Med J. 1997;314:955–959. doi: 10.1136/bmj.314.7085.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flier JS. Leptin expression and action: new experimental paradigms. Proc Natl Acad Sci USA. 1997;94:4242–4245. doi: 10.1073/pnas.94.9.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flier JS. Clinical review 94: what’s in a name? In search of leptin’s physiologic role. J Clin Endocrinol Metab. 1998;83:1407–1413. doi: 10.1210/jcem.83.5.4779. [DOI] [PubMed] [Google Scholar]
- 14.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 15.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 16.Chen NG, Swick AG, Romsos DR. Leptin constrains acetylcholine-induced insulin secretion from pancreatic islets of ob/ob mice. J Clin Invest. 1997;100:1174–1179. doi: 10.1172/JCI119629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emilsson V, Liu YL, Cawthorne MA, Morton NM, Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. doi: 10.2337/diab.46.2.313. [DOI] [PubMed] [Google Scholar]
- 18.Fehmann HC, Bode HP, Ebert T, Karl A, Göke B. Interaction of GLP-1 and leptin at rat pancreatic β-cells: effects on insulin secretion and signal transduction. Horm Metab Res. 1997;29:572–576. doi: 10.1055/s-2007-979103. [DOI] [PubMed] [Google Scholar]
- 19.Fehmann HC, Peiser C, Bode HP, et al. Leptin: a potent inhibitor of insulin secretion. Peptides. 1997;18:1267–1273. doi: 10.1016/s0196-9781(97)00135-6. [DOI] [PubMed] [Google Scholar]
- 20.Ishida K, Murakami T, Mizuno A, Lida M, Kuwajima M, Shima K. Leptin suppresses basal insulin secretion from rat pancreatic islets. Regul Pept. 1997;70:179–182. doi: 10.1016/s0167-0115(97)01002-1. [DOI] [PubMed] [Google Scholar]
- 21.Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes. 1997;46:1087–1093. doi: 10.2337/diab.46.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni RN, Wang ZL, Wang RM, et al. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest. 1997;100:2729–2736. doi: 10.1172/JCI119818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ookuma M, Ookuma K, York DA. Effects of leptin on insulin secretion from isolated rat pancreatic islets. Diabetes. 1998;47:219–223. doi: 10.2337/diab.47.2.219. [DOI] [PubMed] [Google Scholar]
- 24.Pallett AL, Morton NM, Cawthorne MA, Emilsson V. Leptin inhibits insulin secretion and reduces insulin mRNA levels in rat isolated pancreatic islets. Biochem Biophys Res Commun. 1997;238:267–270. doi: 10.1006/bbrc.1997.7274. [DOI] [PubMed] [Google Scholar]
- 25.Poitout V, Rouault C, Guerre-Milo M, Briaud I, Reach G. Inhibition of insulin secretion by leptin in normal rodent islets of Langerhans. Endocrinology. 1997;139:822–826. doi: 10.1210/endo.139.3.5812. [DOI] [PubMed] [Google Scholar]
- 26.Roduit R, Thorens B. Inhibition of glucose-induced insulin secretion by long-term preexposure of pancreatic islets to leptin. FEBS Lett. 1997;415:179–182. doi: 10.1016/s0014-5793(97)01115-0. [DOI] [PubMed] [Google Scholar]
- 27.Harvey J, Ashford MLJ. Insulin occludes leptin activation of ATP-sensitive K+ channels in rat CRI-G1 insulin secreting cells. J Physiol. 1998;511:695–706. doi: 10.1111/j.1469-7793.1998.695bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey J, Ashford ML. Role of tyrosine phosphorylation in leptin activation of ATP-sensitive K+ channels in the rat insulinoma cell line CRI-G1. J Physiol (Lond) 1998;510:47–61. doi: 10.1111/j.1469-7793.1998.047bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao AZ, Bornfeldt KE, Beavo JA. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J Clin Invest. 1998;102:869–873. doi: 10.1172/JCI3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harvey J, McKenna F, Herson PS, Spanswick D, Ashford MLJ. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J Physiol. 1997;504:527–535. doi: 10.1111/j.1469-7793.1997.527bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a.Seufert J, Kieffer TJ, Habener J. Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc Natl Acad Sci USA. 1999;96:674–679. doi: 10.1073/pnas.96.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kieffer TJ, Heller RS, Habener JF. Leptin receptors expressed on pancreatic beta-cells. Biochem Biophys Res Commun. 1996;224:522–527. doi: 10.1006/bbrc.1996.1059. [DOI] [PubMed] [Google Scholar]
- 32.Fehmann HC, Berghofer P, Brandhorst D, et al. Leptin inhibition of insulin secretion from isolated human islets. Acta Diabetol. 1997;34:249–252. doi: 10.1007/s005920050083. [DOI] [PubMed] [Google Scholar]
- 33.Eizirik DL, Pipeleers DG, Ling Z, Welsh N, Hellerstrom C, Andersson A. Major species differences between humans and rodents in the susceptibility to pancreatic β-cell injury. Proc Natl Acad Sci USA. 1994;91:9253–9256. doi: 10.1073/pnas.91.20.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zawalich WS, Zawalich KC. Species differences in the induction of time-dependent potentiation of insulin secretion. Endocrinology. 1996;137:1664–1669. doi: 10.1210/endo.137.5.8612499. [DOI] [PubMed] [Google Scholar]
- 35.Linetsky E, Bottino R, Lehmann R, Alejandro R, Inverardi L, Ricordi C. Improved human islet isolation using a new enzyme blend, liberase. Diabetes. 1997;46:1120–1123. doi: 10.2337/diab.46.7.1120. [DOI] [PubMed] [Google Scholar]
- 36.Welsh M, Nielsen DA, MacKrell AJ, Steiner DF. Control of insulin gene expression in pancreatic beta-cells and in an insulin-producing cell line, RIN-5F cells. II. Regulation of insulin mRNA stability. J Biol Chem. 1985;260:13590–13594. [PubMed] [Google Scholar]
- 37.Fehmann HC, Habener JF. Insulinotropic hormone glucagon-like peptide-I(7–37) stimulation of proinsulin gene expression and proinsulin biosynthesis in βTC-1 insulinoma cells. Endocrinology. 1992;130:159–166. doi: 10.1210/endo.130.1.1309325. [DOI] [PubMed] [Google Scholar]
- 38.Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA. 1987;84:3434–3438. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai CH, Hung LM, Cheng HP, Chen JK. Increased intracellular cyclic AMP levels suppress the mitogenic responses of human astrocytoma cells to growth factors. J Neurooncol. 1995;23:41–52. doi: 10.1007/BF01058458. [DOI] [PubMed] [Google Scholar]
- 40.Garrone P, Banchereau J. Agonistic and antagonistic effects of cholera toxin on human B lymphocyte proliferation. Mol Immunol. 1993;30:627–635. doi: 10.1016/0161-5890(93)90073-k. [DOI] [PubMed] [Google Scholar]
- 41.Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford MLJ. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- 42.Leclercq-Meyer V, Considine RV, Sener A, Malaisse WJ. Do leptin receptors play a functional role in the endocrine pancreas? Biochem Biophys Res Commun. 1996;229:794–798. doi: 10.1006/bbrc.1996.1882. [DOI] [PubMed] [Google Scholar]
- 43.Leclercq-Meyer V, Malaisse WJ. Failure of leptin to counteract the effects of glucose on insulin and glucagon release by the perfused rat pancreas. Med Sci Res. 1997;25:257–259. [Google Scholar]
- 44.Shimizu H, Ohtani K, Tsuchiya T, et al. Leptin stimulates insulin secretion and synthesis in HIT-T15 cells. Peptides. 1997;18:1263–1266. doi: 10.1016/s0196-9781(97)00137-x. [DOI] [PubMed] [Google Scholar]
- 45.Tanizawa Y, Okuya S, Ishihara H, Asano T, Yada T, Oka Y. Direct stimulation of basal insulin secretion by physiological concentrations of leptin in pancreatic beta cells. Endocrinology. 1997;138:4513–4516. doi: 10.1210/endo.138.10.5576. [DOI] [PubMed] [Google Scholar]
- 46.Darnell JE., Jr Reflections on STAT3, STAT5, and STAT6 as fat STATs. Proc Natl Acad Sci USA. 1996;93:6221–6224. doi: 10.1073/pnas.93.13.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holz GG, Kühtreiber WM, Habener JF. Pancreatic β-cells rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7–37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wabitsch M, Jensen PB, Blum WF, et al. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes. 1996;45:1435–1438. doi: 10.2337/diab.45.10.1435. [DOI] [PubMed] [Google Scholar]
- 49.Barr VA, Madide D, Zarnowski MJ, Taylor SI, Cushman SW. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology. 1997;138:4463–4472. doi: 10.1210/endo.138.10.5451. [DOI] [PubMed] [Google Scholar]
- 50.Habener JF. The incretin notion and its relevance to diabetes. Endocrinol Metab Clin North Am. 1993;22:775–794. [PubMed] [Google Scholar]
- 51.Seufert JS, Weir GC, Habener JF. Differential expression of the insulin gene transcriptional repressor C/EBPβ and transactivator IDX-1 in rat pancreatic β-cells during the development of diabetes mellitus. J Clin Invest. 1998;101:2528–2539. doi: 10.1172/JCI2401. [DOI] [PMC free article] [PubMed] [Google Scholar]