

Abstract
The L-Valine ester of antiviral agent cyclopropavir, valcyclopropavir (6), was synthesized and evaluated for antiviral properties. Prodrug (6) inhibited replication of HCMV virus (Towne and AD169 strain) in HFF cells to approximately the same extent as the parent drug cyclopropavir (5). Stability of 6 toward hydrolysis at pH 7.0 roughly corresponds to that of valganciclovir (2). Pharmacokinetic studies in mice established that the oral bioavailability of valcyclopropavir (6) was 95%.
Introduction
Efficacy of antiviral nucleoside analogues is often hampered by their low solubility and bioavailability. To address this deficiency, several types of prodrugs have been synthesized[1]. Amino acid esters were found especially advantageous. Thus, valine ester prodrugs of acyclovir (1, valacyclovir, Valtrex, Chart 1) [2–4] and ganciclovir (2, valganciclovir, Valcyte) [5.6] are already part of our armament for fighting infections caused by herpes virus 1 and 2 (HSV-1 and HSV-2), varicella zoster virus (VZV) and human cytomegalovirus (HCMV). Additional advantage of these prodrugs resides in their ability to facilitate the membrane transport by using peptide transporters [6,7] PEPT1 and PEPT2. L-Valine ester prodrugs are readily hydrolyzed by chemical or esterase-catalyzed hydrolysis and/or a biphenyl hydrolase-like protein (BPHL, valaciclovirase) [8]. More recently, this approach has been extended to nucleoside analogue 2′-deoxy-L-cytidine (L-dC) [9] effective against hepatitis B (HBV) and 2′-C-methylcytidine [10] which is an anti-hepatitis C (HCV) agent. Both L-valine esters, Val-L-dC (3, valtorcitabine) [11] and valopicitabine (4) [12] have improved pharmacokinetic profiles over the parent analogues.
Chart 1.

In our laboratory, we have developed a new class of antiviral agents -methylenecyclopropane analogues of nucleosides [13]. Among these analogues, cyclopropavir (5) exhibits antiviral parameters superior to ganciclovir [14–16] and it is a subject of current preclinical studies. In order to improve therapeutic efficacy of cyclopropavir (5) we have turned our attention to L-valine ester - valcyclopropavir (6). The synthesis, antiviral activity and pharmacokinetic parameters of analogue 6 is the subject of this communication.
Methods
Synthesis
The mass spectra (MS) were determined in an electrospray ionization (ESI) mode. Valcyclopropavir (6) was synthesized as described below. Cyclopropavir (5) was synthesized by GLSynthesis, Inc., Worcester, Massachusetts by previously published method [14]. All other chemicals were analytical reagent grade and all solvents were HPLC grade.
(Z)-9-{[2-(N-tert-Butoxycarbonyl-L-valyloxymethyl)-2-(tetrahydropyranyloxymethyl)-cyclopropylidene]methyl}guanine (8)
A mixture of N-(tert-butoxycarbonyl)-L-valine (BOCValOH, 955 mg, 4.4 mmol) and dicyclohexylcarbodiimide (DCC, 537 mg, 2.6 mmol) in dichloromethane (36 mL) was stirred at room temperature under nitrogen for 3 h. The insoluble portion was filtered off and solvent from the filtrate was evaporated in vacuo. The resultant N-(t-butoxycarbonyl)-L-valine anhydride/(BOCVal)2O/was dissolved in N,N-dimethylformamide (DMF, 50 mL) and compound 7 (0.7 g, 2.02 mmol) was added followed by 4-dimethylaminopyridine (DMAP, 25 mg, 0.3 mmol). The mixture was stirred at room temperature for 40 h, solvent was removed in vacuo and the crude product was chromatographed on a silica gel column using CH2Cl2 - MeOH (30: 1 to 8: 1) to give compound 8 (0.81 g, 73%) as a solid, mp 228–230 °C. UV λmax (ethanol) 274 nm (ε11,200), 231 nm (ε 27,100). ESI-MS (MeOH + NaCl) 547 (M + H, 10.9), 569 (M + Na, 100.0). The NMR spectra could not be obtained because of poor solubility.
(Z)-9-{[2-Hydroxymethyl-2-(L-valyloxymethyl)cyclopropylidene]methyl}guanine Hydrochloride (6)
A solution of compound 8 (0.5 g, 0.92 mmol) in 10% HCl/ethyl acetate (50 mL) was stirred at room temperature for 1.5 h. The volatile components were removed in vacuo, the crude product was chromatographed on a silica gel column using CH2Cl2 - MeOH (10: 1 to 5: 1). Evaporation of solvents afforded product 6 as a white solid after trituration of the residue with methanol and ethyl acetate (280 mg, 76.4%), mp >300 °C, [α]D20 21.0° (c 1.0, DMSO). UV (ethanol) λmax 273 nm (ε 12,600), 230 nm (ε 29,100). 1H NMR (DMSO-d6, 400 MHz) δ 10.92 (s, 1H, NH), 8.51 (bs, 3H, NH3(+) of Val), 8.22, 8.18 (2s, 1H, H8), 7.16 (s, 1H, H1′), 6.78 (s, 2H, NH2), 5.36 (poorly resolved t, 1H, OH), 4.38–4.25 (m, 2H, H5″), 3.82–3.73 (m, 2H, H5′), 3.46–3.42 (m, 1H, CHα of Val, partly overlapped with H2O), 2.15–2.12, 2.03–1.96/2m, 1H, CH(CH3)2/, 1.51–1.46 (m, 2H, H3′), 0.94, 0.89 (2d, J = 6.4, 6.8 Hz), 0.84–0.83 (poorly resolved apparent t, 6H, CH3); 13C NMR 169.5, 169.4 (C=O), 157.1 (C6), 155.0 (C2), 150.4, 150.3 (C4), 134.9, 134.8 (C8), 116.9, 116.5 (C2′), 116.4 (C2′, C5), 112.2 (C1′), 67.5, 66.9 (C5′), 63.2, 63.1 (C5″), 58.0 (Cα of Val), 29.93, 29.88 (C4′), 27.8, 27.7 (CH(CH3)2, 18.9, 18.7, 18.3, 18.2 (CH3), 12.4, 12.1 (C3′). ESI-MS (MeOH - KOAc) 363 (100.0, M + H), 401 (36.9, M + K). For analysis it was dried at 100 °C in vacuo. Anal. Calcd. for C16H22N6O4 x 1.2 HCl x 1.4 H2O: C, 44.55; H, 6.08; Cl, 9.86; N, 19.48. Found: C, 44.93; H, 5.87; Cl, 9.63; N, 19.13.
Kinetics of Hydrolysis of Valcyclopropavir (6) at pH 7.0
A solution of compound 6 (1 mg, 2.25 μmol) in 0.02 M Na2HPO4 (pH 7.0, 1 mL) was stirred at room temperature for 36 h. At selected time intervals, aliquots of the mixture was analyzed by HPLC on DuPont Zorbax NH column 250 × 4.6 mm, mobile phase 0.05 M KH2PO4 - acetonitrile (3: 1), flow rate 1 mL/min., UV detection at 273 nm, see Figure 1.
Figure 1.

Kinetics of hydrolysis of prodrug 6 at pH 7.0 and room temperature.
Antiviral Assays
Plaque reduction assays in human foreskin fibroblasts (HFF’s) were performed with HCMV strains Towne and AD169 as described [14, 15].
Stability of the Prodrug Valcyclopropavir (6) in Mouse Plasma and Whole Blood
Male Swiss-Webster mice (Taconic Farms, Germantown, Pennsylvania) were anesthetized and blood collected (ca. 1 ml per animal) by cardiac puncture into Monoject® No. 8881-311149 collection tubes (Tyco Healthcare, Mansfield, Massachusetts), containing a anticoagulant [0.04 ml of 7.5% EDTA(K3)]. The blood was cooled in ice and used immediately for plasma preparation and stability studies. For plasma preparation, half of the collected blood was centrifuged for 10 min. at 1100 × G in a refrigerated centrifuge (4°C). For stability at 0°C, ice cooled blood and plasma samples were spiked with a 10 mM solution of the prodrug (6) in DMSO to a final concentration of 50 μM. The samples were kept in ice water. For stability at 37°C, the plasma and blood samples were brought to 37°C by placing in warm water, and then in a thermostatic water bath. Aliquots of 200 μL plasma, and 400 μL blood were taken at the indicated times. The plasma samples were frozen immediately in a dry ice/acetone mixture. The blood samples were centrifuged in a refrigerated centrifuge (4°C) for 5 min. at 2200 × G. Plasma (150 μL) was removed, frozen in a dry ice/acetone mixture and stored at −20°C until analyzed. The samples were analyzed for valcyclopropavir (6), and cyclopropavir (5) by the HPLC method describe below.
Bioavailability of Cyclopropavir (5) after IV and PO Dosing of the Prodrug Valcyclopropavir (6) to Mice
1. Dosing and blood sampling of mice
Male Swiss-Webster mice were obtained from Taconic Farms, Germantown, Pennsylvania. Prodrug (6) was dissolved in physiological saline at 4.92 mg/mL. Mice (n=3/group/time point) were injected intravenously (IV) via the tail vein at a dose of 24.6 mg/kg (5 mL/kg, ca. 140 uL/animal), or given orally, by gavage (IO), at a dose of 49.2 mg/kg (10 mL/kg, ca. 300 μL/animal). Mice given the IV dose had a mean body weight of ca. 28 g. Mice given the IO dose were fasted overnight before dosing and had a mean body weight on the day of dosing of ca. 30 g. At the indicated sampling times, (10, 20, 40, 80, 160, and 240 min. for IV; and 20, 40, 60, 120, 240, and 320 min. for IO), animals were anesthetized, sacrificed, and blood collected by cardiac puncture. Blood samples were cooled on ice, and immediately subjected to centrifugation at 4°C for plasma preparation. The plasma samples were frozen on dry ice and stored at −20°C until analyzed.
2. Analytical methods
A. Preparation of standard solutions and spiked plasma calibration samples
Six standard solutions, containing a mixture of prodrug (6) and cyclopropavir (5), at concentrations of 0.6, 2.0, 6.0, 20.0 and 60.0 μg/mL, were prepared by diluting DMSO stock solutions (10 mg/mL) with acetonitrile. For spiked plasma calibration samples, aliquots of 250 μL of the standard solutions were mixed with equal volume of mouse plasma in polypropylene microfuge tubes. For extraction efficiency determination, another set of standard solutions, (250 μL), were mixed with an equal volume of water. All tubes were vortexed for 30 sec, maintained at room temperature for 30 min., vortexed again for 30 sec, and then centrifuged for 15 min. at 13,000 rpm. The supernatants (375 μL each) were pipetted from each tube into a new microfuge tube, and evaporated to dryness in a Speed-Vac (4–5 hours). DMSO (250 μL) was added to each residue, and the samples were vortexed for 60 min. The solutions were centrifuged for 15 min. at 13,000 rpm, and transferred to HPLC vials. Each sample was analyzed by HPLC by the method described below.
B. Preparation and extraction of plasma samples from dosed mice
The plasma samples were thawed on ice, and aliquots (250 μL) were immediately transferred to microfuge tubes, and deproteinated by addition of acetonitrile, (250 μL). The samples were further processed as described above for the spiked plasma standards.
C. HPLC method
The HPLC analysis was performed on a Varian ProStar® HPLC System consisting of two pumps, a variable wavelength detector, an autosampler, and a desktop computer for HPLC control, data acquisition and data analysis. The data analysis software was the Varian® Star Chromatography Workstation (version 6.0). Compound elution was performed on a 4.6 × 150 mm Ace 5 C18 HPLC column (Advanced Chromatography Technologies). The mobile phase consisted of reservoir A, containing 100% water with 0.1% TFA; and reservoir B, containing 100% methanol. Each solvent was filtered through a 0.45 μm nylon filter and degassed before use. Elution from the column was at a flow rate of 1 mL/min. using the following gradient program: a linear gradient of 0% B to 30% B from zero to 18 min., then a linear gradient from 30% to 100% B for 1 min., hold at 100% B for 2 min., followed by a linear gradient from 100% to 0% B for 1 min., and then equilibration by a hold at 0% B for 5 min.. Compound elution was monitored by UV absorbance at 274 nm. Total analysis time was 27 min. per sample. Retention times were 9.8 min. for cyclopropavir (5) and 14.6 min. for the prodrug (6).
D. Data analysis
Peak areas from HPLC analysis of plasma samples were measured, and concentrations of cyclopropavir (5) and prodrug (6) were calculated based on the standard curves generated with the plasma spiked samples. The extraction efficiency for each compound was determined by comparing the peak areas for spiked plasma samples with those for standard samples processed with water instead of plasma. The average extraction efficiency was 67% for cyclopropavir (5), and 88% for the prodrug (6). The limit of detection and the limit of quantification, determined as 3x and 5x the intensity of the matrix peaks in the regions of detection of the analytes were 0.24 μg/mL and 0.40 μg/ml for cyclopropavir (5), and 0.048 μg/mL and 0.080 μg/mL for the prodrug (6). The PK modeling and analysis was done using the PK modeling program WinNonLin® (Pharsight, Mountain View, CA).
Results
A. Antiviral activity
Prodrug 6 inhibited replication of HCMV in two strains of virus, Towne and AD169 (Table 1). These studies confirmed that its potency was comparable to ganciclovir, which was used as a control, and was also similar to cyclopropavir (5) reported previously [14]. These results confirmed that valcyclopropavir (6) was readily hydrolyzed to cyclopropavir (5) inside the virus-infected cells or in the culture medium that contains bovine plasma. However, its efficacy relative to cyclopropavir (5) cannot be discerned from these data.
Table 1.
Anti-HCMV activity of valcyclopropavir (6).
| Compound | HCMV/HFFa, EC50/CC50 (μM) |
|
|---|---|---|
| Towneb | AD169c | |
| Valcyclopropavir (6)d | 0.21/>100 | 2.9/>300 |
| Ganciclovir | 1.58±1.53/>100 | 3.6 ± 1.4/>100 |
| Cyclopropavir (5)e | 0.42±0.23/>100 | 1.2 ± 0.8/>380 |
Efficacy and cytotoxicity in human foreskin fibroblasts (HFF’s) as determined by plaque reduction assay.
Visual cytotoxicity in uninfected HFF’s used in plaque reduction assays.
Cytotoxicity by neutral red assay.
Results from single assays.
EC50 values for cyclopropavir (5) were reported previously for both strains of the virus [14].
B. Conversion of valcyclopropavir (6) to cyclopropavir (5) in vitro in mouse plasma and whole blood
The prodrug valcyclopropavir (6) was converted into the parent drug, cyclopropavir (5) when incubated at 37°C in mouse blood or plasma with a half-life of 1 h 20 min., and 40.4 min., respectively (Figure 2, Table 2). At 0°C the conversion was more than an order of magnitude slower, with estimated half-lives of 22 h (blood) and 12 h (plasma). In a control experiment, blood and plasma samples were processed immediately after spiking with the prodrug (no incubation). The level of the cyclopropavir peak detected in those samples corresponded to a 3.7% (plasma) and 9.0% (blood) conversion of valcyclopropavir (6) to cyclopropavir (5). This is assumed to take place during the plasma preparation and sample processing, and should not affect the overall kinetic parameters observed for the in vitro or in vivo metabolism of the prodrug (6), since it is the same for all incubation/sampling times. In our opinion, this is small conversion does not warrant correction of the dose-dependent PK parameters for the prodrug (6).
Figure 2.

Conversion of prodrug 6 into cyclopropavir (5) after incubation at 0 and 37 °C in mouse whole blood and plasma.
Table 2.
Conversion of valcyclopropavir (6) to cyclopropavir (5) in mouse whole blood and plasma at 0 and 37°C.
| Incubation conditions | First order rate constant, min.−1 | half live | R2 |
|---|---|---|---|
| min. | |||
| Plasma 0°C | 0.0010 | 727 | 0.9925 |
| Plasma 37°C | 0.0172 | 40.4 | 0.9998 |
| Blood 0°C | 0.0005 | 1310 | 0.9645 |
| Blood 37°C | 0.0087 | 80 | 0.9997 |
C. IV dosing
The average plasma concentration of the prodrug (6), and cyclopropavir (5) are plotted in Figure 3.
Figure 3.

Plasma concentrations of cyclopropavir (5), and prodrug 6 vs. time after single IV bolus injection of 24.6 mg/kg prodrug in mice.
Prodrug (6)
The plasma concentration data for prodrug (6) were fitted into a single compartment bolus IV first order elimination model:
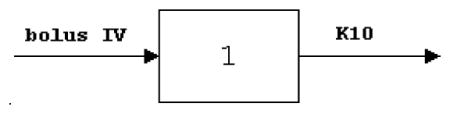
where
C is the plasma concentration, mg/L
D is the dose, mg/kg
V is the apparent volume of distribution, L
K10 is the elimination rate constant, L/min.
T is the time, min.
The best fit curve is presented in Figure 4A, and the best fit PK parameters are presented in Table 3. Analysis of the plasma concentration profile suggested that the prodrug (6) is converted by a fast process to cyclopropavir (5). At the same time, some amount of the prodrug (6) remains in a depot compartment, and is released by a slow process. This results in a large half-life and volume of distribution parameter in the single compartment model (Table 3). The data for the prodrug (6), therefore, was fitted to a two compartment bolus IV 1st order elimination model as described below:
Figure 4.

PK modeling of the experimental data for prodrug (6) after IV bolus injection in mice. A, Single compartment first order elimination model. B, Two compartments first order distribution and elimination model.
Table 3.
Single compartment first order elimination PK parameters for prodrug (6) after IV dosing in mice.
| Parameter | Best fit | CV% |
|---|---|---|
| Apparent volume of distribution, VD, L/kg | 93.5 | 11.9 |
| Area under the curve, AUC, mg.min/L | 61.2 | 7.33 |
| Rate constant elimination, K, min.−1 | 0.0043 | 14.8 |
| Half-life, T1/2, min. | 161 | 23.9 |
| Intercept (time zero concentration), Cmax, mg/L | 0.263 | 11.9 |
| Clearance (Cl), L/min. | 0.402 | 7.33 |
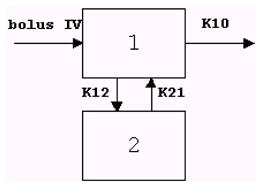
where
C is the plasma concentration, mg/L
A=D/V*(ALPHA-K21)/(ALPHA-BETA)
B=−D/V*(BETA-K21)/(ALPHA-BETA)
ALPHA and BETA are the roots of the quadratic equation r*r+(K12+K21+K10)*r+K21*K10=0
D is the dose, mg/kg
V is the apparent volume of distribution, L
K12 is the rate constant for transfer from compartment 1 to compartment 2, L/min.
K21 is the rate constant for transfer from compartment 2 to compartment 1, L/min..
K10 is the elimination rate constant, L/min.
T is the time, min.
The best fit graph and corresponding parameters are presented in Figure 4B and Table 4. The parameters are more consistent with the properties expected of the prodrug (6). The lack of very early time points resulted in high coefficients of variability, especially of the parameters highly influenced by K12.
Table 4.
Two compartments first order distribution and elimination PK parameters for prodrug (6) after bolus IV dosing in mice.
| Parameter | Best Fit | CV% |
|---|---|---|
| Volume of main compartment, V, L/kg | 0.0753 | 14.9 |
| Area under the curve, AUC, mg.min/L | 97.1 | 85.6 |
| Rate constant of distribution phase, α, min.−1 | 0.25 | 536 |
| Half-life of distribution phase, T1/2(α), min. | 2.73 | 535 |
| Rate constant of elimination phase, β, min.−1 | 0.00263 | 114 |
| Half-life of elimination phase, T1/2(β), min. | 301 | 114 |
| Intercept (time zero concentration) of distribution phase, C0 (A), mg/L | 36.5 | 1234 |
| Intercept (time zero concentration) of elimination phase, C0 (B), mg/L | 1.037 | 12.3 |
| Clearance (Cl), L/min. | 0.217 | 222 |
Cyclopropavir (5)
The plasma concentration of cyclopropavir (5) was first modeled using a single compartmental first order elimination model, in which the last three data points were used to estimate the terminal elimination rate constant, β (Figure 5A). The PK parameters based on this analysis are presented in Table 4.
Figure 5.

PK modeling of the experimental data for cyclopropavir (5) after IV bolus injection of prodrug 6 in mice ( observed,
observed,  predicted). A, Single compartment first order elimination modeling: Logarithmic plot of the average plasma concentration of cyclopropavir (5) vs. time. The straight line is the linear regression fit to the last three data points (1/Y weighting), which was used to estimate the terminal elimination rate constant β. B, Two compartmental analysis. See text for model description.
predicted). A, Single compartment first order elimination modeling: Logarithmic plot of the average plasma concentration of cyclopropavir (5) vs. time. The straight line is the linear regression fit to the last three data points (1/Y weighting), which was used to estimate the terminal elimination rate constant β. B, Two compartmental analysis. See text for model description.
The time dependence of the cyclopropavir (5) plasma concentration was also assessed by using a two compartmental model as described below:
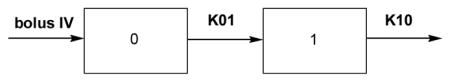
where:
0 is the valcyclopropavir (6) occupied compartment,
1 is the main, sampling compartment
C is the plasma concentration of 5 in the sampling compartment, mg/L;
D is the dose, mg/kg (for simplification of the calculation a dose of 16.2 mg/kg of cyclopropavir was used, which is equivalent to a valcyclopropavir dose of 24.6 mg/kg)
V is the apparent volume of distribution, L;
K01 is the first order input rate constant, i.e. the rate constant for conversion of prodrug (6) to cyclopropavir (5), L/min.;
K10 is the rate constant for elimination of cyclopropavir (5) from the plasma compartment, L/min.;
T is the time from the prodrug (6) dosing, min.
The best fit graph and corresponding parameters are presented in Figure 5B and Table 6. The PK parameters obtained by this analysis are in good overall agreement with the corresponding PK parameters, obtained by the single compartment method (Table 5). However, since the elimination of the prodrug (6) does not follow first order kinetics there is significant uncertainty in the estimation of the input rate constant K01.
Table 6.
Two compartmental analysis of cyclopropavir (5) plasma levels after IV bolus dosing of prodrug (6). See text for model description.
| Parameter | Best Fit | CV% |
|---|---|---|
| Volume of distribution, V, L/kg | 2.61 | 21.0 |
| Area under the curve, AUC, mg.min/L | 874 | 22.3 |
| Input rate constant, K01, min.−1 | 0.21 | 121 |
| Elimination rate constant, K10, min.−1 | 0.0071 | 32.7 |
| Elimination Half-life of T1/2K10, min. | 97 | 32.7 |
| Time of maximum plasma concentration, Tmax, min. | 16.7 | 85.8 |
| Maximum plasma concentration, Cmax, mg/L | 5.52 | 15.0 |
| Clearance (Cl), L/min. | 0.0185 | 22.3 |
A complete conversion of the prodrug (6) to cyclopropavir (5) is assumed, which would result in a equivalent dose of cyclopropavir (5) of 16.2 mg/kg.
Table 5.
Single compartment first order elimination PK parameters for cyclopropavir (5) after IV dosing of prodrug (6) in mice. The last three data points were used to model the elimination rate constant.a
| Parameter | Best Fit |
|---|---|
| Rate constant of elimination phase, β, min.−1 | 0.0092 |
| Half-life based on termin.al elimination phase, T1/2(β), min. | 75.5 |
| Time of maximum plasma concentration, Tmax, min. | 10 |
| Maximum plasma concentration, Cmax, mg/L | 5.59 |
| Volume of distribution based on the termin.al phase, Vz, L | 1.94 |
| Area under the curve at infinity, AUC∞, mg.min/L | 902 |
| Total plasma clearance (Cl), L/min. | 0.018 |
In this analysis a complete conversion of the prodrug (6) to cyclopropavir (5) was assumed, which would result in a equivalent dose of cyclopropavir (5) of 16.2 mg/kg.
D. Oral dosing
The results of the plasma sample analysis after a single oral dose of 49.2 mg of prodrug (6) in mice is presented in Figure 6. No prodrug (6) was detected in the oral samples (limit of detection = 0.048 μg/mL). The absorption phase took less than 10 min. and was not observed. The log10 of the observed experimental cyclopropavir (5) concentrations demonstrated an overall linear decline (Figure 6B, red line). The data was fitted into a first order elimination non-compartmental model. The model-predicted and experimental cyclopropavir concentrations are shown in Table 7, and shown plotted in semilog form in Figure 6B. The PK parameters derived from the above model are presented in Table 8. The prodrug (6) oral bioavailability, calculated according to the formula: OB%= (IVAUC/OralAUC)*(IV dose/Oral dose)*100, was found to be very high at 95.3%.
Figure 6.

Plot of plasma concentrations of cyclopropavir 5 vs. time in mice after a single oral dose of 49.2 mg/kg of prodrug 6. A, normal plot; B, log plots of the experimental (o—o—o) and modeled (—) plasma concentrations.
Table 7.
Experimental and model-predicted concentrations of cyclopropavir (5) and “area under the curve” for oral dosing of prodrug (6).
| Time, min. | Exp. conc. mg/L | Predicted conc. mg/L | Area under the curve, AUC mg.min/L |
|---|---|---|---|
| 20 | 15.71 | 157.1 | |
| 40* | 14.39 | 14.75 | 458.1 |
| 60* | 10.17 | 11.94 | 703.7 |
| 120* | 5.10 | 6.330 | 1161.8 |
| 240* | 2.82 | 1.780 | 1637.0 |
| 320* | 0.71 | 0.764 | 1778.2 |
Time point included in modeling of the elimination phase.
Table 8.
PK parameters of cyclopropavir (5) after oral dosing of prodrug (6).
| Parameter | Best Fit |
|---|---|
| Rate constant of oral elimination phase, λz, 1/min. | 0.0106 |
| Half-life of oral elimination phase, T1/2λz, min. | 65.6 |
| Time of maximum plasma concentration, Tmax, min. | ≤ 20 |
| Maximum plasma concentration, Cmax, mg/L | ≥ 15.7 |
| Volume of distribution based on the terminal phase, Vz, L | 2.52 |
| Area under the curve at infinity, AUC∞, mg.min/L | 1845 |
| Total body clearance (Cl), L/min. | 0.0267 |
| Oral bioavailability, % | 95.3 |
In this analysis, a complete conversion of the bioavailable prodrug (6) to cyclopropavir (5) was assumed, which would result in a dose equivalent to 32.4 mg/kg of cyclopropavir (5).
Discussion
Chemistry
The previously described [17] tetrahydropyranyl (THP) cyclopropavir (7) which is available from cyclopropavir in two steps (73% yield) served as a convenient starting material for synthesis of valcyclopropavir (6, Scheme 1). The method described [2] for synthesis of valacyclovir (1) was followed. Reaction of 7 with N-(t-butoxycarbonyl)-L-valine anhydride (BOCVal)2O in DMF under catalysis with 4-dimethylaminopyridine (DMAP)gave intermediate 8 in 73% yield. The BOC and THP group were removed in acid medium (10% HCl in ethyl acetate) to give valcyclopropavir (6) as a hydrochloride in 76% yield. Similar to valganciclovir (2) [3], prodrug (6) is a mixture of two stereoisomers. The stereoisomeric ratio of 2 determined from the 1H NMR spectrum is 2: 1.
Scheme 1.

Synthesis of valcyclopropavir (6)
a. (BOCVal)2O, DMAP, DMF. b. 10% HCl in AcOEt.
As expected, prodrug (6) has a limited stability at pH 7.0 (Figure 1) with the half-life of 22.5 h at room temperature. For comparison, the half-life of valganciclovir (2) [18] at pH 7.08 and 37 °C is 11 h.
Pharmacokinetic (PK) studies
Prodrug (6) is converted to cyclopropavir (5) in vitro in mouse whole blood and blood plasma, and in vivo after both intravenous (IV) and oral (PO) administration. Prodrug (6) is quickly and efficiently absorbed by the gastrointestinal tract, with PO bioavailability of 95%. The high apparent volume of distribution of 5 after both IV and PO dosing with prodrug (6), is suggestive of possible sequestering of either 6 or 5 in non-sampling compartments. The PK profile of prodrug (6) after IV dosing also supports the possibility of non-uniform distribution of 6 with sequestering outside of the plasma compartment.
Acknowledgments
The work described herein was supported by SBIR grant 5 R44 AI054135, contract NO1-AI30049 and research grant RO1-CA32779 from the National Institutes of Health, Bethesda, Maryland. We thank L. M. Hrihorczuk from the Central Instrumentation Facility, Department of Chemistry, Wayne State University for mass spectra, Kathy Borysko at University of Michigan for plaque assays, Sofya Dvoskin and George Wright from GLSynthesis for valuable assistance in bioavailability studies.
Footnotes
Disclosure statement
The authors declare no competing interest.
References
- 1.Mackman R, Cihlar T. Prodrug strategies in the design of nucleoside and nucleotide antiviral therapeutics. Ann Rep Med Chem. 2004;39:305–321. [Google Scholar]
- 2.Beauchamp LM, Orr GF, de Miranda P, Burnette T, Krenitsky TA. Amino acid ester prodrugs of acyclovir. Antiviral Chem Chemother. 1992;3:157–164. [Google Scholar]
- 3.Perry CM, Faulds D. Valaciclovir: A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs. 1996;52:754–772. doi: 10.2165/00003495-199652050-00009. [DOI] [PubMed] [Google Scholar]
- 4.Ormrod D, Scott LJ, Perry CM. Valaciclovir: a review of its long term utility in the management of genital herpes simplex virus and cytomegalovirus infections. Drugs. 2000;59:839–863. doi: 10.2165/00003495-200059040-00013. [DOI] [PubMed] [Google Scholar]
- 5.Nestor JJ, Womble SW, Maag H. 2-(2-Amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propandiol L-monovaline ester of (ganciclovir valine ester) as an antiviral with improved oral absorption. US Appl. 1994;281:893. [Google Scholar]; Chem Abstr. 1996;124:290266j. [Google Scholar]
- 6.Curran M, Noble S. Valganciclovir. Drugs. 2001;61:1145–1150. doi: 10.2165/00003495-200161080-00013. [DOI] [PubMed] [Google Scholar]
- 7.Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun. 1998;246:470–475. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- 8.Kim I, Song X, Vig BS, et al. A novel nucleoside prodrug-activating enzyme: Substrate specificity of biphenyl hydrolase-like protein. Mol Pharm. 2004;1:117–127. doi: 10.1021/mp0499757. [DOI] [PubMed] [Google Scholar]
- 9.Bryant ML, Bridges EG, Placidi L, et al. Antiviral L-nucleosides specific for hepatitis B virus infection. Antimicrob Agents Chemother. 2001;45:229–235. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bichko V, Tausek M, Qiu L, et al. Enhanced antiviral activity of NM 107, alone or in combination with interferon γ. J Hepatol. 2005;42 (Suppl 2):154. [Google Scholar]
- 11.Gosselin G, Pierra C, Benzaria S, et al. β-L-2′-Deoxythymidine (LdT) and β-L-2′-deoxycytidine (LdC): How simple structures can be potent, selective and specific anti-HBV drugs. In: Schinazi RF, Liotta DC, editors. Frontiers in Nucleosides and Nucleic Acids. Tucker, GA: IHL Press; 2005. pp. 309–317. [Google Scholar]
- 12.Pierra C, Amador A, Benzaria S, et al. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J Med Chem. 2006;49:6614–6620. doi: 10.1021/jm0603623. [DOI] [PubMed] [Google Scholar]
- 13.Zemlicka J. Methylenecyclopropane Analogues of Nucleosides as Anti-Herpes Agents. In: De Clercq E, editor. Advances in Antiviral Drug Design. Amsterdam, The Netherlands: Elsevier; 2007. pp. 113–165. [Google Scholar]
- 14.Zhou S, Breitenbach JM, Borysko KZ, et al. Synthesis and Antiviral Activity of (Z)- and (E)-2,2-[Bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: Second-Generation Methylenecyclopropane Analogues of Nucleosides. J Med Chem. 2004;47:566–575. doi: 10.1021/jm030316s. [DOI] [PubMed] [Google Scholar]
- 15.Kern ER, Kushner NL, Hartline CB, et al. In vitro Activity and Mechanism of Action of Methylenecyclopropane Analogs of Nucleosides against Herpesvirus Replication. Antimicrob Agents Chemother. 2005;49:1039–1045. doi: 10.1128/AAC.49.3.1039-1045.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kern ER, Bidanset DJ, Hartline CB, Yan Z, Zemlicka J, Quenelle DC. Oral Activity of a Methylenecyclopropane Analog, Cyclopropavir, in Animal Models for Cytomegalovirus Infections. Antimicrob Agents Chemother. 2004;48:4745–4753. doi: 10.1128/AAC.48.12.4745-4753.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Z, Kern ER, Gullen E, Cheng YC, Drach JC, Zemlicka J. Nucleotides and Pronucleotides of 2,2-Bis(hydroxymethyl)methylenecyclopropane Analogues of Purine Nucleosides: Synthesis and Antiviral Activity. J Med Chem. 2005;48:91–99. doi: 10.1021/jm040149b. [DOI] [PubMed] [Google Scholar]
- 18.Stefanidis D, Brandl M. Reactivity of Valganciclovir in Aqueous Solution. Drug Development Ind Pharm. 2005;31:879–884. doi: 10.1080/03639040500271951. [DOI] [PubMed] [Google Scholar]