

Abstract
Enzyme-mediated redox chain initiation involving glucose oxidase (GOX) was employed in an iterative solution dip-coating technique to polymerize multiple, three-dimensional hydrogel layers using mild aqueous conditions at ambient temperature and oxygen levels. To our knowledge, sequential enzyme-mediated dip-coating resulting in an interfacial radical chain polymerization and subsequent formation of three-dimensional hydrogel layers has not been previously explored. Conformal, micron-scale, uniform poly(ethylene glycol) (PEG)-based hydrogel layers were polymerized within seconds and remained securely associated after incubation in water for 16 weeks. Incorporation of either small molecules (i.e., rhodamine-B acrylate, fluorescein acrylate) or fluorescent nanoparticles into crosslinked hydrogel layers during the polymerization reaction was also achieved. The encapsulation of 0.2 μm- diameter nanoparticles into hydrogels during polymerization of a 2-hydroxyethyl acrylate (HEA)/PEG575 diacrylate monomer formulation, using the GOX-mediated initiation, resulted in minimal effects on polymerization kinetics, with final acrylate conversions of 95% (±1%) achieved within minutes. The temporal control and spatial localization afforded by this interfacial redox approach resulted in the polymerization of uniform secondary layers ranging between 150 (±10) μm and 650 (±10) μm for 15 and 120 s immersion times, respectively. Moreover, increasing the PEG575-fraction within the initial hydrogel substrate from 10% to 50% decreased the subsequent layer thicknesses from 690 (±30) μm to 490 (±10) μm owing to lowered glucose concentration at the hydrogel interface. The ability to sequentially combine differing initiation mechanisms with this coating approach was achieved by using GOX-mediated interfacial polymerization on hydrogel substrates initially photopolymerized in the presence of glucose. The strict control of layer thicknesses combined with the rapid, water soluble, and mild polymerization will readily benefit applications requiring formation of stratified, complex, and three-dimensional polymer structures.
Keywords: Hydrogels, Poly(ethylene glycol) (PEG), Redox chain initiation, Coatings, Nanoparticle hydrogel incorporation, Enzyme-mediated initiation, Glucose Oxidase
Introduction
The formation of polymer layers, that each exhibit distinct chemical and/or mechanical properties, is significant for a variety of applications including controlled drug delivery,1 tissue engineering,2 food stability,3 and surface modifications.4 Such applications benefit from multilayer fabrication since alterations in layer composition may ultimately yield a cohesive material stratified with assorted, desired characteristics (e.g., mechanical strength, size exclusion, hydrophobicity, cell type), generally infeasible to obtain using a single material composition.5
Due to the potential importance and heightened functionality of layered polymer structures, several distinct approaches have been taken to fabricate such structures. One prominent approach for generating nanoscale multi-layers involves the layer-by-layer (LBL) self-assembly of polyelectrolytes.6 LBL approaches have been used extensively and successfully for numerous biomedical applications such as the production of multilayers for pancreatic islet encapsulation7 and the production of antifungal surface modifications.4 Another beneficial multi-layering approach involves the crosslinking of natural polymers (e.g., polysaccharides), which is particularly advantageous for emulating native stratified tissues when the polymer layers are laden with cells. For example, millimeter-scale, layered alginate hydrogels have been employed to mimic stratified cartilage structure for tissue engineering applications. 8
In addition to the LBL approach and cross-linking of natural polymers, the formation of synthetic-based hydrogel layers offers many unique advantages owing to well-defined polymeric structures that are easily tailored for a particular application. For example, bi-layer hydrogels composed of differing high molecular weight poly(ethylene glycol) (PEG) polymers were employed for protein release to arterial media.9 Additionally, multi-laminated methacrylate-containing hydrogels have been used to investigate controlled drug delivery.10,11 To generate these synthetic crosslinked hydrogels, the photopolymerization of acrylated monomers is frequently used. For example, recent successful investigations sequentially photopolymerized hydrogel multilayers on an eosin-functionalized glass slide support to produce covalently coupled hydrogel layers.12 Additionally, sequential crosslinking reactions have been employed to control cell morphology in three-dimensional hydrogels.13 Photopolymerization offers many benefits including mild reaction conditions capable of maintaining the integrity of biological molecules and rapid polymerization rates. However, the formation of uniform, conformal coatings and multilayer structures in complex, three-dimensional structures would benefit significantly from a light-independent, interfacially triggered approach able to cure the entire conformal layer simultaneously without concerns of light attenuation (e.g., shadowing effects).
One such light-independent reaction involves a rapid enzyme-mediated initiation system capable of kinetically controlling the polymerization of water-soluble acrylate monomers at ambient temperatureand atmospheric conditions.14 This initiation system utilizes the catalytic turnover of β-D-glucose and oxygen by the glucose oxidase (GOX) enzyme to generate hydrogen peroxide (H2O2). By combining ferrous ions with this enzymatic H2O2 production, hydroxyl radicals, which function as polymerization initiators, are subsequently generated through Fenton chemistry. Due to the highly specific binding affinity between β-D-glucose and the GOX enzyme, spatially localizing the site where this binding event occurs also coincides with the spatial localization of initiator generation. Here, we employ a hydrogel as a physical barrier between the glucose and the GOX enzyme, thus spatially controlling the enzyme-glucose binding event, and subsequent H2O2 and hydroxyl radical generation, to the hydrogel-solution interface. This spatial control permits the polymerization to occur at the hydrogel surface, ultimately forming three-dimensional hydrogel layers (Figure 1).
Figure 1.

Generation of a three-dimensional layered hydrogel using GOX-mediated interfacial polymerization. A cylindrical crosslinked core hydrogel substrate (green), pre-swollen with glucose, is immersed into an aqueous precursor solution (pink) that contains the remaining components necessary for initiating polymerization (i.e., GOX enzyme, Fe+2 and vinyl monomer). The inset is a molecular depiction that illustrates the presence of glucose at the hydrogel substrate-precursor solution interface resulting from glucose diffusion out of the hydrogel into the precursor solution. Catalytic turnover of glucose by the GOX enzyme generates H2O2, which reacts further with Fe+2, to ultimately generate the hydroxyl radical initiator (·OH) capable of reacting with a vinyl monomer. The initiation components (e.g., H2O2, glucose) and monomer may also diffuse into the surface boundary of the hydrogel substrate (not shown here). Spatial localization of the initiation components to the hydrogel substrate boundary, as a consequence of glucose diffusion from the core hydrogel, promotes polymerization solely at this interface to ultimately form a polymer coating at this site. Here, the hydrogel substrate ultimately contains one layer but repeating this entire process could generate additional layers. Moreover, each layer composition may be altered independently by simply including differing moieties within the precursor solution (e.g., fluorescent monomers, nanoparticles, proteins, cells).
The current approach employs an in situ radical chain polymerization reaction to polymerize sequential crosslinked micron-scale hydrogel layers and differs fundamentally from the LBL polyelectrolyte approach that uses the adsorption of pre-formed polyelectrolytes to form nano-scale layers. The current GOX-mediated approach generates far thicker (i.e., micron-scale) layers, compared to LBL, and facilitates variations in polymer layer thicknesses simply through manipulation of reaction conditions, such as immersion time and glucose concentration. Additionally, the applications, the advantages, and the limitations differ between the LBL method and the current GOX-mediated approach. For example, multiple polymer layers, as generated by the GOX-mediated method, may hold potential for applications requiring micron-scale hydrogel films, such as investigating drug release profiles from synthetic, three-dimensional hydrogels.
Additionally, in comparison to the aforementioned photoinitiation approaches used to generate planar hydrogel slabs, the GOX initiation does not require a light source, thereby permitting hydrogel layer formation in a variety of three-dimensional geometries without concern of potential shadowing effects and light attenuation during the polymerization. In effect, the current approach combines the ease of a dip-coating type methodology with the benefits of an in situ interfacial chain polymerization to form complex, three-dimensional, crosslinked hydrogels that are comprised of multiple layers, each layer formed with independent control of their composition and structure. For example, by simply including a desired moiety (e.g., nanoparticles, acrylated small molecules, proteins) within each aqueous dip-coating precursor solution, these species can be readily incorporated into the complex hydrogel structures during the rapid dip-coating polymerization reaction.
Herein, we introduce the novel approach for the polymerization of three-dimensional, crosslinked hydrogel layers using GOX-mediated radical chain polymerization. This approach permits the formation of uniform, three-dimensional layers through a simple, rapid and light-independent iterative interfacial polymerization technique. Using this system, we investigate the generation of sequential three-dimensional multilayers, the incorporation of nanoparticle and small molecule within the hydrogel layers, and variations in layer thickness by manipulation of initiation conditions, including reactant concentration and time. Collectively, these investigations present a novel and facile approach to generate stable, layered hydrogels with relevance across a broad spectrum of applications requiring stratified polymer structures.
Experimental
Polymerization of hydrogel substrates
The three-dimensional (i.e., cylindrical) core hydrogel substrates (i.e., used as a platform for subsequent hydrogel layer formations) were formed using either photopolymerization or GOX-mediated polymerization. For UV curing of core hydrogel substrates, 0.1wt% of 4-(2-hydroxyethoxy) phenyl-(2-hydroxy-2-propyl) ketone (Irgacure 2959) (Ciba), 15wt% PEGDA575 and 0.1 M glucose were mixed, added to a cylindrical mold (dimensions 4mm X 1.5mm) and irradiated at 25mW/cm2 for 10 minutes using 320–390 nm light. After briefly rinsing in water and blotting, the UV-cured core hydrogel substrates were immediately utilized in the hydrogel layer formation reaction using GOX-mediated dip-coating. Unless otherwise specified in the text, all remaining core hydrogel substrates, which had dimensions of approximately 4 mm diameter, 4.8 cm height, were generated using the GOX-mediated polymerization. For GOX-mediated polymerization of core hydrogel substrates, 6.25 X 10−7 M of GOX from Aspergillus niger (Sigma-Aldrich) was added to a mixing solution of all other reaction components comprising 2.5 X 10−4 M iron (II) sulfate (Fe+2) (Sigma-Aldrich), 4.0 X 10−4 M glucose (Sigma-Aldrich), 1.0 X 10−2 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer pH=4.5 (Teknova) and appropriate monomer formulation. The D-(+)-glucose solution, as purchased from the vendor, was likely equilibrated as a stable mixture of α-glucose anomer and β-glucose anomer since mutarotation (i.e., optical rotation transformation) of glucose in aqueous solution equilibrates within hours. This stable and equilibrated glucose solution is important because the GOX enzyme selectively catalyzes β-D-glucose. After mixing, the solution was immediately transferred to a cylindrical mold and permitted to cure for approximately 5 minutes, rinsed with Milli-Q water (18megohm cm resistivity) and agitated in ~100mL of Milli-Q water overnight. Extensive washing of the core hydrogels was performed to remove residual initiation components. After washing, the core hydrogel substrates were incubated in an appropriate concentration of glucose overnight (at least 12 hours) before the coating reactions.
Polymerization reactions were monitored using Fourier transform infrared (FTIR) spectroscopy with a Nicolet 750 Magna FTIR instrument. The acrylate double bond conversion was followed in real time using the overtone absorption peak between 6212–6150 cm−1 corresponding to the C-H stretch of the acrylate functional group. After mixing the reaction components, the reaction solutions were immediately transferred to a 1mm thick, glass sample compartment and placed in a FTIR instrument equipped with a horizontal transmission apparatus.15
Hydrogel layer formation and characterization
For the hydrogel layer formation (i.e., coating) reactions, all hydrogel substrates, after overnight (at least 12 hours) incubation in an appropriate glucose concentration, were rinsed briefly in Milli-Q water, blotted to remove excess surface water, and immersed in 1mL of appropriate precursor coating solution at ambient temperature and atmosphere. During the immersion, the hydrogels did not make contact with the container walls to prevent abnormalities in the coating layer morphology. To prevent container contact, the cylindrical hydrogels (approximate dimensions 4mm diameter, 4.8 cm height) were partially submerged into the precursor solution, resulting in hydrogel coatings as shown in Figure 2. The smaller UV-cured core hydrogels (dimensions 4mm diameter X 1.5mm height) were punctured with a fine gauge wire and completely submerged into the precursor solution. After the appropriate immersion time, the coating polymerization reaction was stopped by transferring the gels into ~100mL of Milli-Q water and further rinsed for at least 24 hours prior to image analysis. For the multilayer coating experiments, the aforementioned process was repeated by re-swelling the hydrogel in glucose and performing the interfacial polymerization process. To permit image analysis using fluorescent microscopy, fluorescent acrylate monomers, including either acryloyloxy fluorescein (fluoroscein o-acrylate) (Sigma-Aldrich) or methacryloxyethyl thiocarbamoyl rhodamine B (rhodamine-B acrylate) (Polysciences) or fluorescent nanoparticles (Invitrogen) were included in the mixture. When using the fluorescent monomers, the final DMSO concentration in the polymerization mixture was between 0.5 and 1 volume%. Specifically, the fluorescent polystyrene nanoparticles used in these studies included amine-modified, 0.2 μm-diameter yellow-green nanoparticles (excitation/emission, 505/515) or carboxylate-modified, 0.02μm-diameter blue nanoparticles (excitation/emission, 365/415). The nanoparticles, as purchased from Invitrogen, are internally labeled with fluorescent dyes. The nanoparticle stock solutions were vortexed for two minutes immediately prior to use.
Figure 2.

The digital camera image displays the formation of a three-dimensional polymer layer on a cylindrical core hydrogel substrate (substrate dimensions: 4mm diameter, 4.8 cm height). A 15wt% PEGDA575, 0.04wt% fluorescein-acrylate core hydrogel substrate, swollen with 2.5 X 10−2 M glucose, (shown in yellow) was partially immersed into a precursor solution containing 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES pH=4.5, 0.005wt% rhodamine-B acrylate, 15wt% PEGDA700 for 60 seconds. The uniform three-dimensional intact coating (shown in red) remained attached to the hydrogel after agitation in water for 24 hours and incubation in water for 16 weeks.
The coating characterization was performed by sectioning the cylindrical hydrogel substrates and subsequently imaging the sections. The coated UV-cured core hydrogels (dimensions 4mm X 1.5 mm) were not sectioned before imaging. The fluorescence images were obtained using a Zeiss LSM 710 NLO confocal microscope or a Zeiss Pascal LSM 5 confocal microscope. The gels containing blue nanoparticles were imaged with a 405 nm laser diode with fluorescence monitored from 409–499 nm. Fluorescein-acrylate- or green-yellow nanoparticle-containing gels were excited using the 488 nm line from an argon ion laser and the fluorescence was monitored from 492–557 nm. The gels containing rhodamine-B acrylate were excited with a 543 nm helium neon laser and fluorescence was monitored from 547–680 nm. For certain images, dyes were imaged independently to reduce imaging bleed-through. Fluorescent images were analyzed using ImageJ software (i.e., freeware provided by the National Institutes of Health).
Results and Discussion
Formation of three-dimensional hydrogel layers
In general, the GOX-mediated redox initiation system permits rapid, light-independent, and aqueous solution chain polymerization at ambient temperature and oxygen levels in a manner that has been demonstrated to be sufficiently mild to enable simultaneous cell encapsulation.14 In this initiation system, the GOX enzyme catalyzes the oxidation of the β-D-glucose and subsequently utilizes oxygen to regenerate the flavin adenine dinucleotide (FAD) enzyme cofactor and produce H2O2. By combining ferrous ions with this enzymatic H2O2 production, primary hydroxyl radical species are produced that react further with acrylated monomers. Notably, an overall unique aspect of this GOX-mediated initiation system involves the enzymatic consumption of oxygen, a powerful inhibitor of radical chain polymerization.
Using the GOX-mediated initiation system, the current studies demonstrate the generation of stable and uniform three-dimensional hydrogel multilayers. This process occurs by immersing three-dimensional hydrogel substrates, pre-swollen with glucose, into an aqueous precursor solution containing the remaining components necessary for initiation (i.e., GOX, Fe+2, monomer). Here, the timescale for diffusion of glucose out of the glucose-swollen hydrogel substrate, together with diffusion of precursor solution components into the hydrogel substrate, limits radical generation to the hydrogel substrate-precursor solution interface (Figure 1). Thus, the polymerization reaction occurs at this interface site to generate a polymer layer, whereas the remainder of the precursor solution remains unpolymerized. For monomer formulations used in these studies, diffusion between hydrogel layers predominantly occurs with small molecule components of the reaction (e.g., H2O2, glucose). The GOX enzyme, with a molecular weight of 160 kDa, likely becomes physically entrapped within the hydrogel during the polymerization of the current monomer formulations. In general, micron thick layers were rapidly polymerized (i.e., between 15 and 120 seconds) using this approach.
To demonstrate the effectiveness of the GOX-mediated process in forming a uniform and conformal layer on a three-dimensional hydrogel substrate, a cylindrical hydrogel (4mm diameter, 4.8cm height) comprising 15wt% PEGDA575 was swollen in 2.5 X 10−2 M glucose, briefly rinsed in water, blotted to remove excess water, and immersed in a coating precursor solution containing 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES pH=4.5, 15wt% PEGDA700 for 60 seconds. A trace quantity of fluorescent monomer, either 0.005wt% rhodamine-B acrylate monomer for the hydrogel layer (present in the precursor solution) or 0.04wt% fluorescein acrylate monomer for the core hydrogel substrate was included during the polymerization reactions to demarcate the polymer layers during the image analysis. After removal from the rhodamine-B fluorescent precursor solution, the uniform attachment of a hydrogel layer coating the three-dimensional hydrogel substrate was immediately apparent, as illustrated in the digital camera image in Figure 2. These polymer coatings remained attached to the intact hydrogel substrates after incubation in water for 16 weeks. Moreover, any mechanical stresses present as the coated cylindrical hydrogels were agitated in water during the washing steps led to no discernible separation of the polymer layers. However, the qualitative observation involving durability of polymer layers was not further supplemented with mechanical testing. Additional investigations would be necessary to understand further the adhesive strength between these polymer layers. The observed durability of the polymer coatings, however, implies that strong molecular interactions exist between the layers attributed to physical forces (e.g., chain entanglement, hydrogen bonding) and/or to covalent attachment during the polymerization process. In the case of covalent interactions, the presence of unreacted and available acrylate groups on the hydrogel substrate surface may react during chain propagation, consequently anchoring the newly formed layer to the substrate. Additionally, chain-transfer to polymer events would also likely contribute to the covalent attachment of the hydrogel layers. For example, chain transfer to polymer would involve radical displacement from a propagating chain, in the precursor solution, to the hydrogel substrate. A radical generated on the hydrogel substrate from chain transfer would react further with monomers from the precursor solution, thereby covalently attaching the newly formed polymer layer to the original substrate.
Further investigation of the hydrogel layer morphology was conducted by examining cross-sections of fluorescent cylindrical hydrogels using confocal microscopy. Fluorescent images of cylindrical hydrogel cross-sections (Figure 3) display core hydrogel substrates composed of 15wt% PEGDA575 and 0.04wt% fluorescein-acrylate that were swollen in either 2.5 X 10−2 M glucose or with 0 M glucose, as a control. These core hydrogels were subsequently immersed in a precursor solution comprising 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 15wt% PEGDA575, 20wt% HEA, and 0.005wt% rhodamine-B acrylate for 120 seconds. The reaction resulted in the generation of a 400μm polymer coating layer (shown in red) attached to the core hydrogel substrate (shown in green) for the gel swollen in glucose. In its role as a control, the core hydrogel substrate without glucose failed to form any polymer coating layer, demonstrating the specificity of the initiation reaction to the interface and the lack of non-specific staining from the rhodamine-B fluorescent precursor mixture.
Figure 3.

These fluorescent images display cross-sections of cylindrical hydrogels generated by GOX-mediated polymerization. The core hydrogel substrates (green) comprise 15wt% PEGDA575 and 0.04wt% of fluorescein-acrylate and were swollen with either (A) 0mM glucose or (C) 2.5 X 10−2 M glucose. The hydrogels were briefly rinsed in water, blotted, and subsequently immersed in a precursor solution containing 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 15wt% PEGDA575, 20wt% HEA, and 0.005wt% rhodamine-B acrylate for 120 seconds. A 400μm coating layer (red) was generated on the hydrogel swollen with glucose whereas no coating layer was present on the hydrogel without glucose. The fluorescent intensities were measured by placing an arbitrary line across the fluorescent images, (A) and (C), to extend radially from the direction of the approximate center of the hydrogel cross-section on the image. The resulting fluorescent intensity profiles demonstrate an approximate 50μm overlap in fluorescent intensities for the single coating layer (D), whereas the negative control (B) shows no fluorescence emission for rhodamine B.
To examine further the interfacial regions of the bi-layer hydrogel, the pixel intensities (i.e., here corresponding to the fluorescence intensities) were measured along an arbitrary line located within the fluorescent images using ImageJ software. Figure 3B and Figure 3D show the fluorescent intensity profile for each position along the arbitrary line placed in Figure 3A and Figure 3C, respectively. Notably, examining these fluorescent images indicates that the fluorescein emission intensities (492–557nm) and rhodamine-B emission intensities (547–680nm) overlap in approximately a 50 μm region at the interface between the hydrogel layers (Figure 3D). Since the hydrogels were extensively washed after the coating reactions, the overlap in fluorescent profiles suggests that molecular interactions exist between the layers. This layer overlap potentially could be readily controlled by manipulating the parameters that affect the interdiffusion of the precursor solution components into the hydrogel substrate boundary (e.g., hydrogel substrate crosslinking density, glucose concentration, monomer molecular weight, etc.). However, in these studies, here particularly above pH=7, the fluorescein acrylate derivative was subject to diffusion post-polymerization, in accordance with previous reports.12 Therefore, to examine further the interface between hydrogel layers, fluorescent nanoparticle incorporation into the hydrogels was investigated, as discussed in the following section.
Nanoparticle incorporation into multilayered three-dimensional hydrogels
A valuable characteristic of the GOX-mediated interfacial coating method involves the ability to sequentially generate three-dimensional hydrogel multilayers by simple iterative immersions of glucose-swollen hydrogels into precursor solutions. Furthermore, incorporation of desired molecular or particulate moieties into the hydrogel layer is achieved simply by including these components in the precursor solution. Here, for instance, we encapsulate nanoparticles into the sequential hydrogel layers, but this approach holds potential for encapsulating other, biological moieties into the layers, such as proteins or cells. For example, the cellular encapsulation approach may be particularly advantageous since the GOX system was sufficiently mild to enable simultaneous cell encapsulation into PEG hydrogels.14
Here, to demonstrate the formation of sequential multilayer coatings, a cylindrical core hydrogel substrate (approximately 4mm diameter, 4.8cm height) comprising 15wt% PEGDA575, 20wt% HEA and 0.1wt% yellow/green fluorescent nanoparticles (0.2μm diameter) was swollen in 2.5 X 10−2 M glucose. To form the second layer, this glucose-swollen hydrogel was subsequently submerged into a precursor solution (15wt% PEGDA700, 0.005 wt% rhodamine-B acrylate, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES) for 60 seconds. To form a third layer, the coated gel was again swollen with 2.5 X 10−2 M glucose and subsequently submerged into precursor solution (15wt% PEGDA575, 20wt% HEA, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES) containing either 0.1 wt% yellow/green nanoparticles (0.2μm diameter) or 0.2 wt% of blue nanoparticles (0.02μm diameter). The fluorescent images (Figure 4A and 4C) display the resultant cross-sections of these cylindrical core hydrogels with three layers, as just described. Here, the demonstration of sequential multilayer coatings involved formation of three hydrogel layers (i.e., core hydrogel substrate with two hydrogel coating layers). Although not shown here, we anticipate this process could be repeated multiple times to generate a desired number of polymer layers (e.g., 4–5 layers) by re-swelling the layered substrate with glucose and exposing the hydrogel to a precursor solution (i.e., GOX, Fe+2 and monomer).
Figure 4.
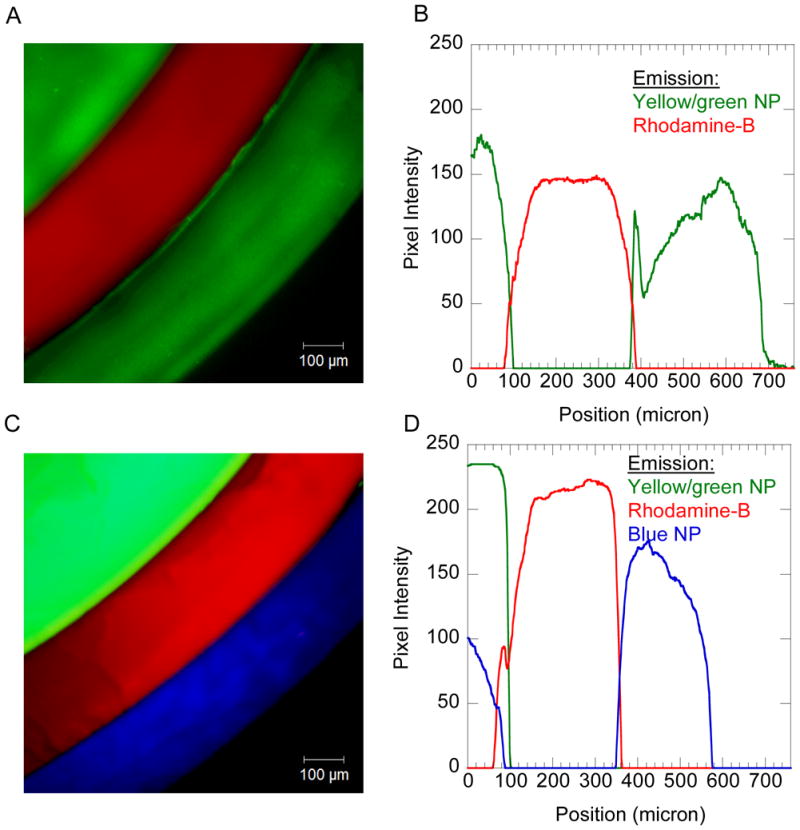
The fluorescent images of a cylindrical hydrogel cross-section (A, C) demonstrate the feasibility of forming multiple three-dimensional hydrogel layers. The green color displays the incorporation of 0.1wt% of yellow/green 0.2μm-diameter nanoparticles (NP); the blue color displays the incorporation of 0.2wt% of blue 0.02μm-diameter nanoparticles (NP); the red color displays the incorporation of 0.005wt% Rhodamine-B acrylate. Each layer was generated by immersing the hydrogel substrate into an appropriate precursor solution for 60 seconds at ambient atmosphere and temperature. The fluorescent intensity (pixel intensity) profiles were measured (B, D) by placing an arbitrary line across the fluorescent images A and C, respectively, to extend radially from the direction of the approximate center of the hydrogel cross-section on the image.
For the gels formed using the yellow/green nanoparticles (n=4 gels examined), as shown for one gel in Figure 4B, the fluorescent intensities of the layer interfaces overlap for 30μm ±5 μm between the core hydrogel and the rhodamine-B first layer. The fluorescent intensities overlap for 20μm ±5 μm for the interface between the rhodamine-B first layer (red) and the second layer containing yellow/green nanoparticles. This overlap in fluorescent intensities implies that molecular associations exist between the hydrogel layers, possibly from a combination of physical and covalent interactions. However, the nanoparticles (0.2μm diameter, 0.02μm diameter) are likely unable to extensively diffusion through a 15wt% PEGDA700 hydrogel owing to limitations in the PEGDA700 hydrogel mesh size.16,17 These experiments do not eliminate the potential for contributions to fluorescent overlap from optical artifacts from hydrogel slice orientation during fluorescent measurements. However, the durability of intact multi-layered hydrogels, in combination with the likely covalent attachment mechanism (i.e., chain transfer to polymer) does suggest strong, stable molecular associations between these hydrogel layers.
In addition, a general important feature in Figure 4 involves the entrapment of nanoparticles within the crosslinked hydrogel layers during the GOX-mediated polymerization reaction. For example, the green colored regions in Figure 4A,B represent the incorporation of the 0.2μm-diameter green/yellow amine-modified nanoparticles, whereas the blue regions (Figure 4C,D) indicate the incorporation of the 0.02μm-diameter blue carboxylate-modified nanoparticles. Since the GOX reactions were performed in slightly acidic conditions favorable for enzymatic activity (i.e., pH=4.5), the amine-modified particles remain positively charged and dispersed, thus readily incorporated into the gels. Although the fluorescent images indicate that the blue nanoparticles were also entrapped within the hydrogel (Figure 4C and 4D), nanoparticle agglomeration was visually observed within the precursor solution when using these blue carboxylate-modified nanoparticles. The agglomeration of carboxylate-modified nanoparticles was likely due to the slightly acidic pH of the solution and the presence of divalent iron. Moreover, the nanoparticle agglomeration often resulted in layers with a tendency to delaminate from the hydrogel substrate.
To investigate further the effects of including nanoparticles during the GOX-mediated polymerization reaction, the acrylate double bond conversions profiles were monitored in real-time using near-infrared spectroscopy (Figure 5). The addition of 0.1wt% of 0.2μm-diameter amine-modified yellow/green nanoparticles to a comonomer solution (15wt% PEGDA575, 20wt% HEA, 4.0 X 10−4 M glucose, 2.5 X 10−4 M Fe+2, 6.25 X 10−7 M GOX, 10mM MES pH=4.5) resulted in a slightly lower final acrylate conversion (95% ±1%) as compared to identical reactions without nanoparticles (99% ± 1%). Although the addition of 0.2wt% of 0.02μm-diameter blue carboxylate-modified nanoparticles to the same comonomer formulation produced high final acrylate conversion, these polymer samples appeared opaque from nanoparticle agglomeration and their associated inhomogeneous distribution. Reductions in polymerization rates and final acrylate conversions were previously reported when incorporating 20nm-diameter Nile Red (532/575nm) fluorescent nanoparticles into 25wt/vol% PEGDA575 using the eosin photoinitiator.18 In the current studies, however, the polymerization occurs without light indicating that fluorescent nanoparticles may affect the polymerization process in a light-independent manner, such as potentially terminating propagating radicals or potentially denaturing GOX during enzyme adsorption to the nanoparticles.
Figure 5.

Double bond conversion profiles for GOX-mediated initiation in the presence of (a) 0wt% nanoparticles, (b) 0.2 wt% of 0.02μm-diameter blue carboxylate-modified nanoparticles and (c) 0.1 wt% 0.2μm-diameter yellow/green amine-modified nanoparticles. Reactions contained 15wt% PEGDA575, 20wt% HEA, 2.5 X 10−4 M Fe+2, 6.25 X 10−7 M GOX, 4.0 X 10−4 M glucose and 10mM MES pH=4.5.
Overall, the slight reduction in the polymerization reaction rates in the presence of these nanoparticles would suggest the necessity to increase the reaction times when incorporating nanoparticles into the hydrogel layers using the GOX-mediated dip-coating technique. Interestingly, however, the presence of nanoparticles within the glucose containing core hydrogel substrate did not affect the subsequent layer thicknesses. For example, using a precursor solution comprising (15wt% PEGDA700, 0.005 wt% rhodamine-B acrylate, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES) the PEGDA700 layer thickness formed on a 15wt%PEGDA575, 20wt% HEA hydrogel substrate swollen with 2.5 X 10−2 M glucose and containing 0.1wt% of 0.2μm-diameter yellow-green nanoparticles was 270 μm (± 20), whereas without nanoparticles the PEGDA700 layer thickness was 240 μm (±10).
Controlling the thickness of three-dimensional hydrogel layers
The polymerization rates of the GOX-mediated system are highly predictable and regulated through simple adjustments in reaction conditions. For example, an increase in the GOX enzyme concentration or the glucose concentration increases the polymerization rates of water-soluble acrylate monomers at ambient temperature and atmosphere.14 Here, this kinetic control with the GOX-mediated initiation translates to the facile control of three-dimensional hydrogel layer thicknesses through the simple manipulation of reaction conditions, such as polymerization time and initiator concentration.
Investigations were performed that evaluated the dependence of layer thicknesses on polymerization conditions by using cylindrical corehydrogel substrates (approximate dimensions 4mm diameter X 4.8cm height) comprising 15wt% PEGDA 575. The core hydrogels were swollen in differing concentrations of glucose, rinsed briefly in water, blotted, and immersed into a PEGDA700 precursor solution containing 15wt% PEGDA700, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES (pH=4.5), and 0.005 wt% of rhodamine-B acrylate. A digital camera image (Figure 6A) displays the cross-sections of these cylindrical hydrogel substrates swollen with 0.1 M glucose and subsequently coated for different polymerization times. Controlling the precursor solution immersion times of the core hydrogel substrates resulted in thickness variation while simultaneously maintaining the uniformity of the three-dimensional polymer layer. For example, a 15 second immersion resulted in a coating thickness of 150 μm (±10 μm) whereas a 120 second immersion produced a coating thickness of 650 μm (±10 μm).
Figure 6.

(A) The digital camera image displays the cross-sections of cylindrical hydrogel substrates (15wt% PEGDA575) that were swollen with 0.1M glucose and subsequently immersed in a precursor solution (15wt% PEGDA700, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES (pH=4.5), and 0.005 wt% of rhodamine-B acrylate) for differing times. The picture illustrates that increasing time results in thicker coatings (coatings shown in red) while maintaining hydrogel layer uniformity. (B) The coating thickness may be controlled by immersion time or by swelling the hydrogel substrates in differing concentration of glucose as illustrated for 15wt% PEGDA575 gels swollen with 0.4 M (●), 0.1 M (◇) or 2.5 X 10−2 M (■) glucose and subsequently immersed into the aforesaid PEGDA700 coating precursor solution formulation.
The graph in Figure 6B further illustrates that the coating thicknesses increase linearly between 15 and 120 second immersion times, indicating a nearly constant polymerization rate throughout this period. It is expected, however, that these coating thickness values will eventually plateau owing to the depletion of reaction components (e.g., glucose, monomer). Under the current reaction conditions, notably, this plateau was not reached, and the hydrogel layer ultimately reached a limit caused by the size restrictions of the container into which the hydrogel was immersed. For instance, it was observed that 15wt% PEGDA575 hydrogels swollen in 0.1 M glucose and immersed in the aforementioned 15wt% PEGDA700 precursor solution formulation generated a millimeter-scale hydrogel layer conforming to the shape of the cylindrical container containing the precursor solution (container with dimensions of 12mm diameter X 38mm height) after seven minutes. Although polymers conforming to the container shape may be avoided by simply employing differing sized immersion containers, shorter immersion times, or lower glucose/GOX concentrations, this characteristic may also benefit applications requiring the formation of unique three-dimensional multilayered hydrogels. For example, utilizing complex shaped containers may enable hydrogel formation of desired, unique polymer shapes with a controllable polymer core of distinct composition.
Furthermore, the thickness of the layers is controlled by manipulating the glucose concentration within the hydrogel substrate (Figure 6). For instance, increasing the glucose concentration within the hydrogel from 0.025 M to 0.4 M resulted in a ~ 40% increase in film thickness from 180μm (±10) to 310 μm (±10) for a 30 second immersion. Higher glucose levels result in more glucose diffusion from the hydrogel, thus increasing the polymerization rates at the hydrogel substrate-precursor solution interface and ultimately producing thicker polymer layers. Similar behavior was previously observed when monitoring the acrylate conversions of monomer solutions wherein an increase in glucose concentrations increased the polymerization rates.14 Insufficient glucose quantities swollen within the hydrogel substrate resulted in non-uniform polymer layers. For example, 15wt% PEGDA575 hydrogels swollen in 6.25 × 10−3 M of glucose and immersed in the PEGDA700 precursor formulation (as used in Figure 6) generated non-uniform hydrogel layers after a 120 second immersion. This non-uniformity is likely attributed to the low concentration of hydroxyl radical initiating species at the hydrogel-solution interface, being unable to uniformly overcome termination reactions and initiate polymerization.
Additionally, increasing the polymer fraction in the core hydrogel substrate resulted in a decrease in the layer thicknesses. As shown in Figure 7, the layer thicknesses decrease logarithmically with increasing polymer content of the core hydrogel substrates. For example, an increase in the weight percent of the core hydrogel substrates from 10wt% PEGDA575 to 50wt% PEGDA575 results in a corresponding decrease in coating thicknesses from 690μm (±30 μm) to 490 μm (± 10 μm). Since the higher polymer content in the core hydrogel results in increased crosslinking, 19 this trend is expected. The glucose diffusivity may decrease in gels with higher polymer content, thereby reducing the initiator concentration at the hydrogel-solution interface, ultimately decreasing the initiation rates and decreasing the hydrogel layer thicknesses.
Figure 7.
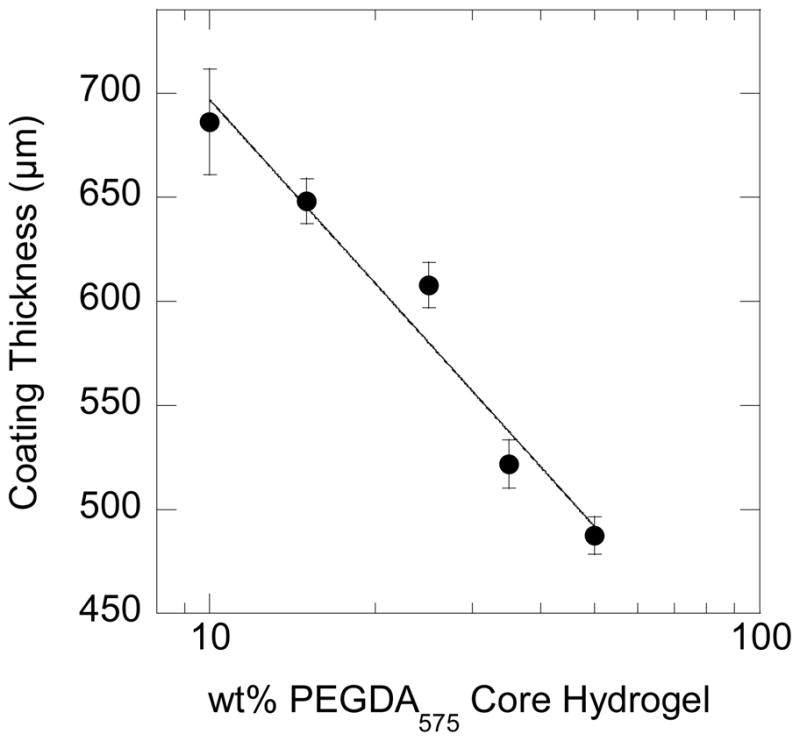
The coating thicknesses generated on core hydrogels composed of differing weight percents of PEGDA575. The various PEGDA575 hydrogels were each swollen in 0.1 M glucose and submerged into a solution containing 15wt% PEGDA700, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 0.005 wt% rhodamine-B acrylate, and 10mM MES pH=4.5 for 120 seconds. Increasing the polymer fraction in the core hydrogel substrate resulted in a decrease in the polymer layer thicknesses due to mass transfer limitations.
Combining photoinitiation and GOX-mediated layer formation
An additional, useful aspect of this coating method involves the capacity to employ differing core hydrogel formation mechanisms. Ultimately, this interfacial polymerization approach may be applicable to any core hydrogel substrate, whether a natural or synthetic polymer, into which a GOX-mediated initiation component can be swollen (e.g., glucose, GOX). Here, we demonstrate this concept by forming the core hydrogel by a distinct photoinitiation method. Specifically, 15wt% PEGDA575 hydrogels disks (4mm diameter X 1.5mm height) were generated by irradiating the monomer at 25mW/cm2 for 10 minutes with 320–390 nm light using 0.1wt% of I2959 photoinitiator in the presence of 0.1 M glucose. Here, the glucose is not used for the photopolymerization reaction but instead is incorporated into the core hydrogel substrate during polymerization. After incorporation, the glucose is immediately ready for the subsequent layer-formation reaction via GOX-mediated dip coating. After photopolymerization, the core hydrogel substrate was rinsed, blotted, and immediately transferred to a PEGDA700 monomer precursor solution (15wt% PEGDA700, 3.1 X 10−6 M GOX, 2.5 X 10−4 M Fe+2, 10mM MES (pH=4.5), and 0.005 wt% of rhodamine-B acrylate) for differing times. The resultant coating sizes were 100μM (±20), 230μM (±30), 450μM (±50), 540μM (±10) for immersion times of 15, 30, 75, and 120 seconds, respectively. These coating thicknesses were comparable of coatings formed on core hydrogel substrates formed by the GOX-mediated initiation system. In the case of using the photopolymerization method to form the core hydrogel substrates, the hydrogels do not require additional incubation in glucose, thus decreasing the overall time required for coating formation.
This current approach, namely combining photoinitiation with GOX-mediated interfacial polymerization, may prove beneficial for applications where it is critical to have both the benefits of the spatial and temporal control afforded by photoinitiation and the light independent benefits of forming conformal and uniform hydrogel coating with the enzyme-mediated (redox) system. For example, photopolymerization reactions are frequently employed for encapsulating cells in hydrogel scaffolds.20,21 Here, by simply including the biocompatible glucose molecule during the photopolymerization of a core cell-laden hydrogel scaffold, the cell-laden gel could be readily coated to form additional polymer layers (potentially with differing polymer and biological properties) to meet the goal of mimicking native, stratified tissue.8,9 In another example, the GOX-mediated dip-coating approach could be used to generate functionalized hydrogel layers that serve as protective barriers for cell-laden core hydrogels (e.g., pancreatic islet laden-core gels).22 In general, the ability to couple photoinitiated cellular encapsulation with a GOX-mediated layer formation system may provide a new approach for generating multiple, three dimensional hydrogel cellular scaffolds. Although this manuscript details utilization of low molecular weight monomers (e.g., PEGDA700) for the polymerization of hydrogel layers, we anticipate the current method could be utilized with higher molecular-weight monomers, termed macromers, employed for tissue engineering. For example, utilization of macromers for polymerization of hydrogel layers may be important for adequate diffusion of cellular nutrients through the polymer layer. Further investigations, however, would be required to assess the reaction conditions required for layer formation with macromers, owing to concentration dependence on the GOX-mediated reaction components on polymerization rates.14 Currently, work is underway to investigate these promising approaches for tissue engineering application.
Overall, this GOX-mediated coating scheme for polymerizing three-dimensional hydrogel layers holds potential for differing applications. For example, the capacity to incorporate desired moieties into the polymer layers by simple inclusion in a precursor solution may offer a facile method for investigating controlled drug release profiles. In particular, given the development of nanoparticles for drug delivery vehicles23 and the recent use of nanoparticles embedded within hydrogels for insulin release,24 the incorporation of the nanoparticles during the hydrogel layer formation may represent new avenues for investigating release profiles from synthetic, three-dimensional hydrogels.
Although this work describes the formation of polymer layers using glucose-swollen hydrogels with a GOX-mediated reaction, this process could readily be generalized and extended to other arrangements. For example, hydrogels swollen with other components, such as GOX or Fe+2 coupled with the appropriate precursor solution, could potentially be used for this interfacial process. Moreover, this generalized interfacial hydrogel-forming approach, namely the spatial localization of redox reactions to a hydrogel-solution interface, holds potential for extension to other beneficial redox reaction types and processes.25–28 In this manner, the generalized approach of utilizing redox reactions for light-independent formation of multiple hydrogel layers could be customized for any particular reaction requirements.
Conclusions
We introduce a novel, light-independent approach for generating multiple, stable three-dimensional hydrogel layers using GOX-mediated chain initiation. The polymerization of micron-scale, uniform hydrogel layers occurred within seconds under ambient temperature and atmospheric conditions via a simple interfacial polymerization technique. The incorporation both of small molecules (i.e., rhodamine-B, fluorescein) and of differing sized nanoparticles into the crosslinked hydrogel layers was achieved; the nanoparticle incorporation showed minimal effects on the polymerization kinetics. Further, the thickness of PEG hydrogel layers was easily manipulated, while simultaneously maintaining uniformity, by simply changing the immersion times and showed variations between 150 (±10) μm (15sec. immersion) and 650 (±10) μm (120 sec immersion) for 15wt% PEGDA575 hydrogels swollen with 0.1 M glucose. The hydrogel layer thicknesses were further controlled by manipulating the glucose quantity present at the hydrogel substrate-precursor solution interface. For example, decreasing the polymer fraction in the core hydrogel substrate or increasing the glucose concentration swollen within the hydrogel substrate resulted in thicker hydrogel layers. Finally, the ability to sequentially employ differing initiation mechanisms was achieved by using GOX-mediated dip coating on core hydrogels photopolymerized in the presence of glucose. Combining photoinitiation with GOX-mediated dip coating may prove advantageous for applications (e.g. tissue engineering) requiring both the spatial and temporal control of photoinitiation and the light independent benefits of the enzyme-mediated (redox) system.
Acknowledgments
This work has been supported by the National Institutes of Health Grant No. R21 CA 127884 and by NSF CBET 0626023. L.M.J. and C.A.D. acknowledge Graduate Assistantship in Areas of National Need Fellowship from the U.S. Department of Education. The authors thank S. Bryant and A. Lynn for their discussion of this work.
References
- 1.Abdul S, Poddar SS. J Controlled Release. 2004;97:393–405. doi: 10.1016/j.jconrel.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 2.Ng KW, Wang CCB, Mauck RL, Kelly TN, Chahine NO, Costa KD, Ateshian GA, Hung CT. Journal of Orthopedic Research. 2005;23:134–141. doi: 10.1016/j.orthres.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 3.Weiss J, Takhistov P, McClements DJ. J Food Sci. 2006;71:R107–R116. [Google Scholar]
- 4.Etienne O, Gasnier C, Taddei C, Voegel J, Aunis D, Schaaf P, Metz-Boutigue M, Bolcato-Bellemin A, Egles C. Biomaterials. 2005;26:6704–6712. doi: 10.1016/j.biomaterials.2005.04.068. [DOI] [PubMed] [Google Scholar]
- 5.Schneider S, Feilen PJ, Slotty V, Kampfner D, Preuss S, Berger S, Beyer J, Pommersheim R. Biomaterials. 2001;22:1961–1970. doi: 10.1016/s0142-9612(00)00380-x. [DOI] [PubMed] [Google Scholar]
- 6.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 7.Teramura Y, Kaneda Y, Iwata H. Biomaterials. 2007;28:4818–4825. doi: 10.1016/j.biomaterials.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 8.Lee CSD, Gleghorn JP, Choi NW, Cabodi M, Stroock AD, Bonassar LJ. Biomaterials. 2007;28:2987–2993. doi: 10.1016/j.biomaterials.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 9.An Y, Hubbell JA. J Controlled Release. 2000;64:205–215. doi: 10.1016/s0168-3659(99)00143-1. [DOI] [PubMed] [Google Scholar]
- 10.Lu S, Anseth KS. J Controlled Release. 1999;57:291–300. doi: 10.1016/s0168-3659(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 11.Watkins AW, Southard SL, Anseth KS. Acta Biomaterialia. 2007;3:439–448. doi: 10.1016/j.actbio.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kizilel S, Sawardecker E, Teymour F, Perez-Luna VH. Biomaterials. 2006;27:1209–1215. doi: 10.1016/j.biomaterials.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 13.Khetan S, Katz JS, Burdick JA. Soft Matter. 2009;5:1601–1606. [Google Scholar]
- 14.Johnson LM, Fairbanks BD, Anseth KS, Bowman CN. Biomacromolecules. 2009;10:3114–3121. doi: 10.1021/bm900846m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lovell LG, Berchtold KA, Elliott JE, Lu H, Bowman CN. Polymers for Advanced Technologies. 2001;12:335–345. [Google Scholar]
- 16.Lin C, Metters AT. Advanced Drug Delivery Reviews. 2006;58:1379–1408. doi: 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Watkins AW, Anseth KS. Macromolecules. 2005;38:1326–1334. [Google Scholar]
- 18.Hansen RR, Avens HJ, Shenoy R, Bowman CN. Anal Bioanal Chem. 2008;392:167–175. doi: 10.1007/s00216-008-2259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott JE, Macdonald M, Nie J, Bowman CN. Polymer. 2004;45:1503–1510. [Google Scholar]
- 20.Rice MA, Anseth KS. Tissue Eng. 2007;13:683–691. doi: 10.1089/ten.2006.0142. [DOI] [PubMed] [Google Scholar]
- 21.Bryant SJ, Nuttelman CR, Anseth KS. J Biomater Sci, Polym Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 22.Cheung CY, Anseth KS. Bioconjugate Chem. 2006;17:1036–1042. doi: 10.1021/bc060023o. [DOI] [PubMed] [Google Scholar]
- 23.Williams J, Lansdown R, Sweitzer R, Romanowski M, LaBell R, Ramaswami R, Unger E. J Controlled Release. 2003;91:167–172. doi: 10.1016/s0168-3659(03)00241-4. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Zhang SM, Chen PP, Cheng L, Zhou W, Tang WX, Chen ZW, Ke CM. J Mater Sci: Mater Med. 2007;18:2205–2210. doi: 10.1007/s10856-007-3010-0. [DOI] [PubMed] [Google Scholar]
- 25.Kitagawa M, Tokiwa Y. Carbohydr Polym. 2006;64:218–223. [Google Scholar]
- 26.Temenoff JS, Shin H, Conway DE, Engel PS, Mikos AG. Biomacromolecules. 2003;4:1605–1613. doi: 10.1021/bm030056w. [DOI] [PubMed] [Google Scholar]
- 27.Barros JAG, Fechine GJM, Alcantara MR, Catalani LH. Polymer. 2006;47:8414–8419. [Google Scholar]
- 28.Durand A, Lalot T, Brigodiot M, Marechal E. Polymer. 2000;41:8183–8192. [Google Scholar]