

Abstract
BACKGROUND AND PURPOSE
Dexmedetomidine, an α2-adrenoceptor agonist, exhibits anti-nociceptive actions at the spinal cord and enhances the effect of local anaesthetics in the peripheral nervous system. Although the latter action may be attributed in part to inhibition of nerve conduction produced by dexmedetomidine, this has not been fully examined yet.
EXPERIMENTAL APPROACH
We examined the effects of various adrenoceptor agonists including dexmedetomidine, and tetracaine, a local anaesthetic, on compound action potentials (CAPs) recorded from the frog sciatic nerve, using the air-gap method.
KEY RESULTS
Dexmedetomidine reversibly and concentration-dependently reduced the peak amplitude of CAPs (IC50 = 0.40 mmol·L−1). This action was not antagonized by two α2-adrenoceptor antagonists, yohimbine and atipamezole; the latter antagonist itself reduced CAP peak amplitude. Clonidine and oxymetazoline, two other α2-adrenoceptor agonists, also inhibited CAPs; the maximum effect of clonidine was only 20%, while oxymetazoline was less potent (IC50 = 1.5 mmol·L−1) than dexmedetomidine. On the other hand, (±)-adrenaline, (±)-noradrenaline, α1-adrenoceptor agonist (-)-phenylephrine and β-adrenoceptor agonist (-)-isoprenaline (each 1 mmol·L−1) had no effect on CAPs. Tetracaine reversibly reduced CAP peak amplitude (IC50 of 0.014 mmol·L−1).
CONCLUSIONS AND IMPLICATIONS
Dexmedetomidine reduced CAP peak amplitude without α2-adrenoceptor activation (at concentrations >1000-fold higher than those used as α2 adrenoceptor agonist), with a lower potency than tetracaine. CAPs were inhibited by other α2 adrenoceptor agonists, oxymetazoline and clonidine, and also an α2 adrenoceptor antagonist atipamezole. Thus, some drugs acting on α2 adrenoceptors are able to block nerve conduction.
Keywords: dexmedetomidine, tetracaine, local anaesthetic, compound action potential, sciatic nerve, frog, adrenoceptors
Introduction
α2-Adrenoceptor agonists have a variety of clinical actions including vasoconstriction, sedation, analgesia, anaesthesia and anaesthetic-sparing effects (for review see Yaksh, 1985; Kamibayashi and Maze, 2000). For example, intrathecal or epidural administration of α2 adrenoceptor agonists such as clonidine and dexmedetomidine ((+)-(S)-4-[1-(2,3-dimethylphenyl)-ethyl]-1H-imidazole; see the inset of Figure 1A for its chemical structure; also see Bhana et al., 2000) produces analgesia in humans (Filos et al., 1994) and animals (Fisher et al., 1991; Takano and Yaksh, 1991), possibly as a result of inhibiting glutamatergic excitatory synaptic transmission in spinal dorsal horn neurones (Sullivan et al., 1992). Peripheral or central administration of α2 adrenoceptor agonists, combined with local anaesthetics, extends the duration of peripheral nerve block in humans (Eisenach et al., 1996; Singelyn et al., 1996; Tschernko et al., 1998; Madan et al., 2001; Memişet al., 2004; Kanazi et al., 2006) and animals (Calasans-Maia et al., 2005; Brummett et al., 2008), possibly due to a constriction of local vessels by the agonists, resulting in a decrease in the clearance of the anaesthetics from the subarachnoid space (Concepcion et al., 1984; Vaida et al., 1986). Moreover, α2 adrenoceptor agonists inhibit nerve action potential (AP) conduction and thus have a local anaesthetic effect, contributing to the enhancement of local anaesthetic effect (Gaumann et al., 1992). Clonidine inhibits excitatory transmission in substantia gelatinosa (SG; lamina II of Rexed) neurones of the rat spinal cord (Pan et al., 2002; Kawasaki et al., 2003) and blocks AP conduction in peripheral nerves (Starke et al., 1972; Gaumann et al., 1992; Butterworth et al., 1993); the latter action requiring a much higher concentration of clonidine than the former. It has been recently reported that dexmedetomidine as well as clonidine depresses excitatory transmission in rat SG neurones (Ishii et al., 2008). Intracutaneously injected dexmedetomidine or clonidine together with lidocaine into the back of guinea-pigs enhances a degree of the local anaesthesia of lidocaine (Yoshitomi et al., 2008). It is possible that, like clonidine, dexmedetomidine has an inhibitory action on AP conduction, because dexmedetomidine depresses voltage-gated Na+-channel currents (Oda et al., 2007). To our knowledge, however, whether nerve conduction is affected by dexmedetomidine has not been fully examined yet.
Figure 1.
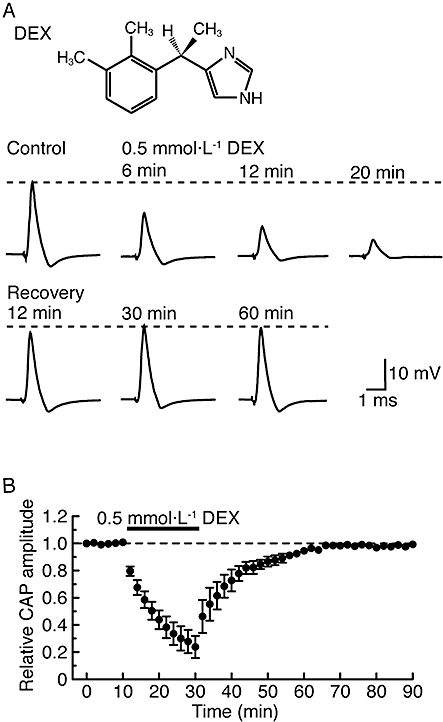
α2-Adrenoceptor agonist dexmedetomidine (DEX; 0.5 mmol·L−1) reversibly reduces the peak amplitude of compound action potentials (CAPs) recorded from frog sciatic nerve fibres. (A) Recordings of CAPs in the control, after 6, 12 and 20 min of exposure to dexmedetomidine and thereafter 12, 30 and 60 min in the absence of dexmedetomidine. Upper inset shows the chemical structure of dexmedetomidine (see Bhana et al., 2000). (B) Average time course of changes in CAP peak amplitudes following exposure to dexmedetomidine for 20 min, relative to that before the soaking, obtained from five sciatic nerves. The dexmedetomidine effect was close to a steady effect after 20 min of exposure.
We have recently revealed that a variety of opioids having different chemical structures inhibit AP conduction in the frog sciatic nerve without opioid-receptor activation and that a difference in the extent of this inhibition among opioids may be attributed to a distinction in their chemical structures (Katsuki et al., 2006; Mizuta et al., 2008). In order to know whether dexmedetomidine affected AP conduction and if so what were the mechanisms for this action, we examined the effects of dexmedetomidine and various drugs related to adrenoceptors on a fast-conducting and Na+-channel blocker tetrodotoxin (TTX)-sensitive compound action potential (CAP) recorded from the frog sciatic nerve by using the air-gap method. These effects were quantitatively compared with those of a well-known local anaesthetic tetracaine (Starke et al., 1972; Concepcion et al., 1984; see Catterall and Mackie, 2006), and also of other local anaesthetics (lidocaine, ropivacaine and cocaine) on frog sciatic nerve CAP, as reported previously (Katsuki et al., 2006; Mizuta et al., 2008).
Methods
Animals
This study was approved by the Animal Care and Use Committee of Saga University, and was conducted in accordance with the Guiding Principles for the Care and Use of Animals in the field of Physiological Science of the Physiological Society of Japan. All efforts were made to minimize animal suffering and the number of animals used.
Preparation of frog sciatic nerves
The method used for obtaining frog sciatic nerve preparation has been described previously (Katsuki et al., 2006; Mizuta et al., 2008). In brief, frogs (Rana nigromaculata; either sex) were decapitated and then pithed; thereafter the sciatic nerve was dissected from the lumbar plexus to the knee in Ringer solution. The isolated sciatic nerve was carefully desheathed under a binocular microscope and then loosely placed in five platinum wires that were glued to a Lucite plate (transparent synthetic resin), where the two ends of the nerve were tied to the wires by using threads. The plate was put on a beaker having Ringer solution in which the sciatic nerve was soaked. Throughout experiments, the Ringer solution was continuously stirred at a rate of about 350 rpm with a Teflon-covered magnetic stirrer bar in order to maintain a uniform composition of Ringer solution around the sciatic nerve. The composition of Ringer solution used was (mmol·L−1): NaCl, 115.5; KCl, 2.0; CaCl2, 1.8; Na2HPO4, 1.3; and NaH2PO4, 0.7 (pH = 7.0). Before the start of the experiment, the sciatic nerve was preincubated for at least 15 min with Ringer solution.
Recordings of CAPs from frog sciatic nerve fibres
As performed previously (Katsuki et al., 2006; Mizuta et al., 2008), the Lucite plate having platinum wires attached with the sciatic nerve was moved from the beaker containing Ringer solution to an empty one and then CAPs were recorded in air using a preamplifier. Here, two of the platinum wires were used to record CAPs, and other two were for stimulating the sciatic nerve. The stimulation was performed at a frequency of 1 Hz with a stimulator, where rectangular pulses having 0.1 ms duration and various strengths were used. In order not to dry the sciatic nerve in air, this procedure was quickly (20 sec at the most) performed at a time interval of 2 min. When the effects of drugs on CAPs were examined, the nerve was put back into the soaking solution with drugs in between two measurements. The data were monitored on a storage oscilloscope while being recorded on a thermal array recorder having a wave form storage module and stored on magnetic tape with a PCM tape recorder for later analyses. In several cases, the data were analysed with a personal computer by using pCLAMP 8.0 software (Molecular Devices, Foster City, CA, USA). Stimulating the sciatic nerve produced a CAP following a stimulus artefact. The peak amplitude of the CAP was measured as a difference between baseline and CAP peak level and the half-peak duration (HPD) as a time of CAP duration at a half of the peak amplitude, as previously described (Katsuki et al., 2006; Mizuta et al., 2008). The peak amplitude of the CAP depended on the strength of stimulus given to the sciatic nerve such that the CAP peak amplitude enhanced with an increase in stimulus strength and attained a maximal value (Katsuki et al., 2006; Mizuta et al., 2008). In the present study, we analysed the peak amplitude of the maximal CAP, although a further increase in stimulus strength resulted in producing a CAP which had smaller values of peak amplitude and conduction velocity (CV) than those of CAPs elicited at less stimulus strength. Such a fast-conducting CAP was completely blocked by TTX (1 µmol·L−1) in a reversible manner. Each of sciatic nerves was used only once to examine the effect of a drug on CAPs unless otherwise mentioned. A CV value was determined by using the fifth electrode as an additional stimulation site and then by measuring a change in time between stimulus artefact and the peak of CAP. All experiments were carried out at room temperature (22–27°C).
Data analysis
Concentration-dependence curve for the reduction of the peak amplitude of CAP in the sciatic nerve soaked with a drug was analysed using the following Hill equation:
where [Drug] is drug concentration, IC50 is the concentration of drug for half-maximal inhibition and nH is the Hill coefficient.
Data are shown as mean ± SEM and statistical significance was set at P < 0.05 using a paired Student's t-test. In all cases n refers to the number of sciatic nerves studied.
Materials
Drugs used were dexmedetomidine hydrochloride, atipamezole hydrochloride (given kindly by ORION PHARMA, Turku, Finland), yohimbine hydrochloride, (-)-phenylephrine, tetracaine hydrochloride, (±)-adrenaline (Wako, Osaka, Japan), (±)-noradrenaline (Aldrich Chem. Co., Milwaukee, WI, USA), (-)-isoprenaline hydrochloride, clonidine hydrochloride, oxymetazoline hydrochloride and tetraethylammonium (TEA) chloride (Sigma, St. Louis, MO, USA). All of drugs were directly dissolved in Ringer solution which was made up of inorganic salts of extra pure grade. The pH of Ringer solution containing dexmedetomidine, (-)-phenylephrine, (±)-adrenaline, (±)-noradrenaline, (-)-isoprenaline and oxymetazoline was adjusted to 7.0 with 1 mol·L−1-NaOH, because it is well-known that the action of local anaesthetics on nerve conduction depends on the pH of Ringer solution used (Skou, 1954). Nomenclature of all receptors, drugs, enzymes and ion channels follows Alexander et al. (2009).
Results
Effects of drugs on fast-conducting TTX-sensitive CAPs were examined in a total of 185 sciatic nerves, and when measured in some of the nerves, the CAPs had a CV of 32 ± 1 m·s−1 (range: 20–47 m·s−1; n = 32), values comparable to those reported previously (Katsuki et al., 2006; Mizuta et al., 2008).
Effects of dexmedetomidine on frog sciatic nerve CAPs
As seen in the lower traces of Figure 1A, soaking the sciatic nerve in dexmedetomidine (0.5 mmol·L−1)-containing Ringer solution reversibly reduced the peak amplitude of the CAP; this reduction was accompanied by an increase in its HPD. Figure 1B demonstrates an average of the time courses of a change in CAP peak amplitude following this treatment with dexmedetomidine (0.5 mmol·L−1), relative to control, which are obtained from five sciatic nerves. The dexmedetomidine-induced reduction in CAP peak amplitude was close to a steady effect at 20 min of exposure, where the peak amplitude of CAP was 24 ± 8% (P < 0.05) of control (18 ± 4 mV; n = 5) and its HPD was 142 ± 15% (P < 0.05) of control (0.49 ± 0.09 ms; n = 5). After 30 min of exposure of the sciatic nerve to dexmedetomidine-free solution, the CAP amplitude recovered to control level, as shown in Figure 1. Figure 2 demonstrates the effect of dexmedetomidine (0.5 mmol·L−1) on CAPs elicited at various stimulus strengths given to the sciatic nerve. The inhibitory effect of dexmedetomidine was seen for CAPs evoked at a maximal stimulus strength while a threshold to elicit CAPs was not changed by dexmedetomidine. Such a result was obtained in six other nerves. Figure 3A demonstrates the time courses of changes in CAP peak amplitude after treatment with dexmedetomidine at various concentrations ranging from 0.01 to 1 mmol·L−1. The rate of the reduction of CAP peak amplitude produced by dexmedetomidine was concentration-dependent as was the magnitude of the reduction at 20 min (Figure 3Ba). The corresponding concentration-response curve (Figure 3Bb) yielded an IC50 value of 0.40 mmol·L−1 with a nH of 3.9.
Figure 2.
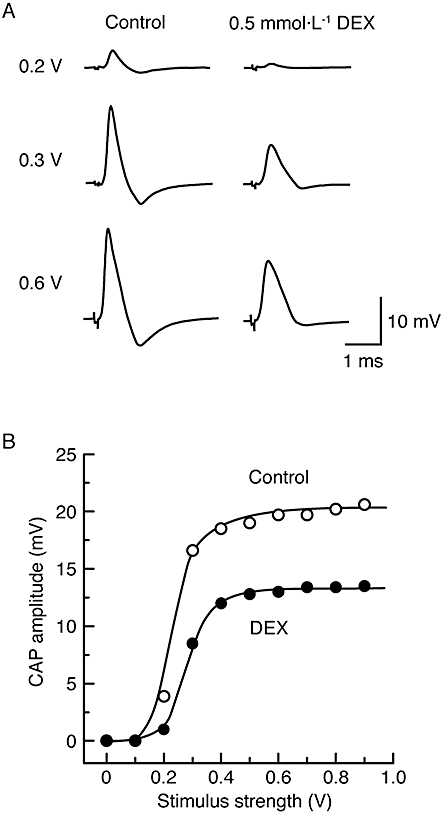
The reduction in compound action potential (CAP) peak amplitude by dexmedetomidine (DEX; 0.5 mmol·L−1) is seen without a change in a threshold to elicit CAPs and with a decrease in maximal responses. (A) Recordings of CAPs elicited at 0.2, 0.3 and 0.6 V in the control (left) and after the action of dexmedetomidine for a period of 20 min (right). (B) The peak amplitudes of CAP before and after dexmedetomidine are plotted against stimulus strength used to elicit the CAP. The solid lines were drawn by eye.
Figure 3.
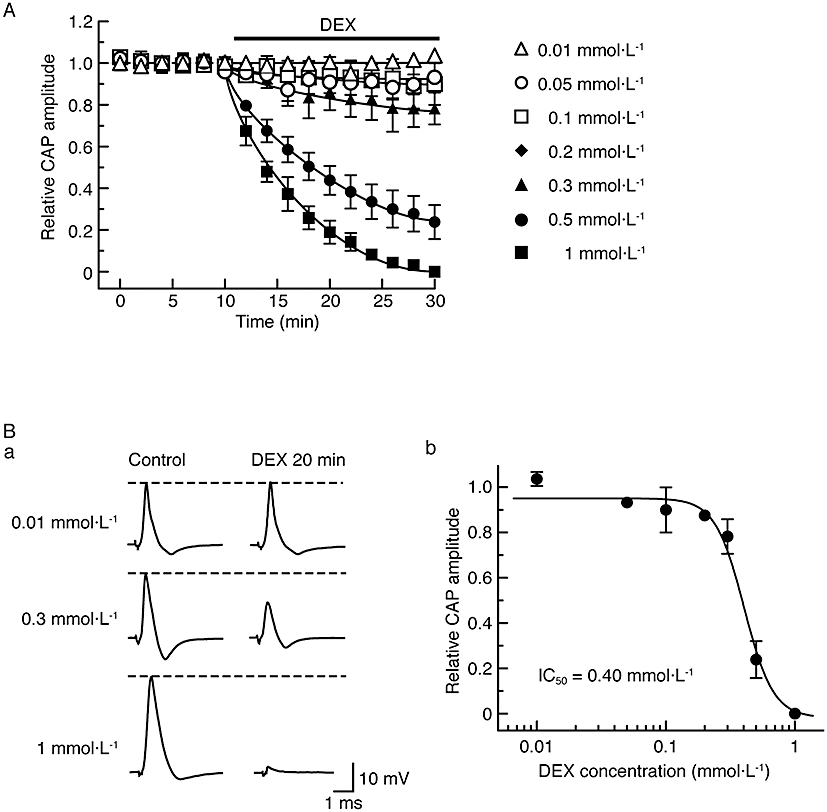
Compound action potential (CAP) peak amplitude is reduced by dexmedetomidine (DEX) in a concentration-dependent manner. (A) Comparison in average time course among CAP peak amplitude reductions produced by dexmedetomidine at 0.01–1 mmol·L−1, obtained from 23 sciatic nerves. The solid lines were drawn by eye. (B) Concentration dependence for CAP peak amplitude reduction by dexmedetomidine. (Ba) Recordings of CAPs in the control (left) and after 20 min of exposure to dexmedetomidine at 0.01, 0.3 and 1 mmol·L−1 (right); these were obtained from different sciatic nerves. (Bb) The peak amplitude of CAP recorded from sciatic nerve fibres treated with dexmedetomidine at various concentrations for 20 min, relative to control, was plotted against dexmedetomidine concentration. Each of the data points was obtained from 3–5 sciatic nerves. The concentration-response curve was drawn according to the Hill equation (IC50 = 0.40 mmol·L−1, nH = 3.9).
As dexmedetomidine exhibits a high affinity for α2 adrenoceptors (Bhana et al., 2000), we next investigated whether this effect of dexmedetomidine was mediated by α2 adrenoceptors. Figure 4 demonstrates how the effect of dexmedetomidine (0.5 mmol·L−1) on sciatic nerve CAPs was affected by two α2-adrenoceptor antagonists, yohimbine and atipamezole (Coughlan et al., 1992; Sjöholm et al., 1992; Sullivan et al., 1992; see Virtanen, 1989; MacDonald et al., 1997; their chemical structures are shown in the insets of Figure 4Aa and Ba respectively). Pretreatment of sciatic nerves with yohimbine (20 min; 0.01 mmol·L−1, a concentration enough to antagonize dexmedetomidine-induced membrane hyperpolarization mediated by α2 adrenoceptors in rat hypothalamic neurones; see Shirasaka et al., 2007), did not affect CAPs [peak amplitude, 100 ± 2% (n = 4; P > 0.05) of control; Figure 4A]. Adding dexmedetomidine (0.5 mmol·L−1) to yohimbine reduced peak amplitude to 28 ± 8% (P < 0.05) of that (22 ± 1 mV; n = 4) with yohimbine alone (Figure 4Aa and Ab). This value was not significantly different from that (24 ± 8%; n = 5; see above; P > 0.05) obtained by treatment with dexmedetomidine alone.
Figure 4.
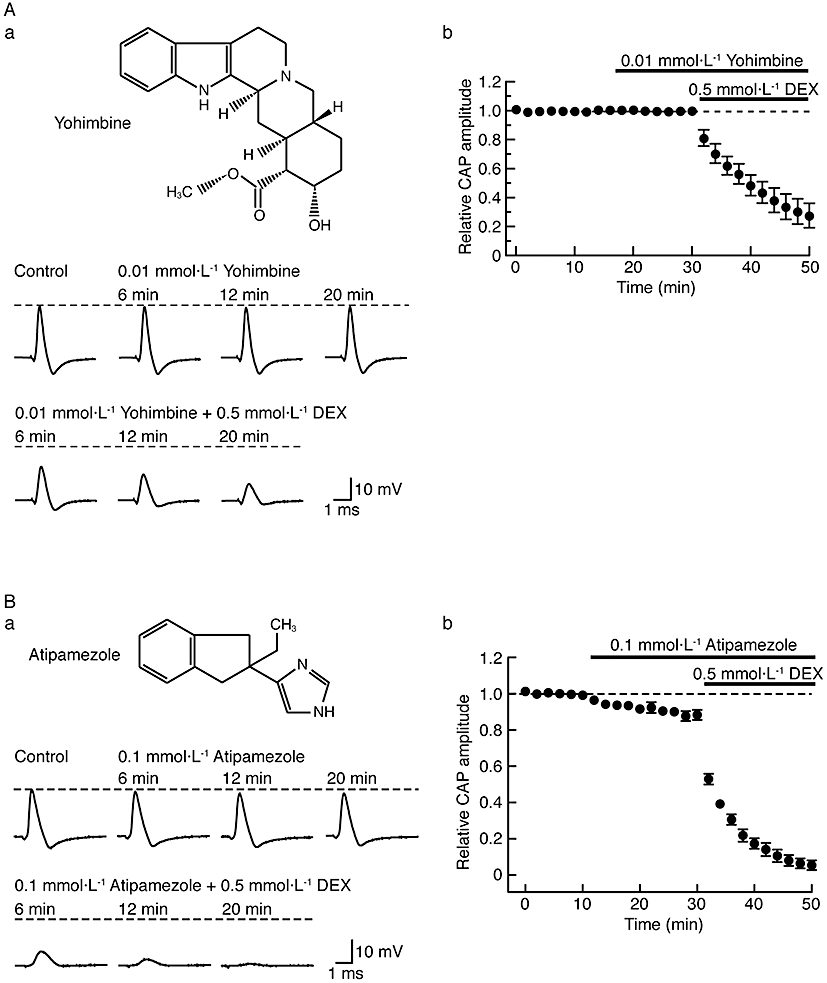
Dexmedetomidine (DEX; 0.5 mmol·L−1)-induced reduction in compound action potential (CAP) peak amplitude is resistant to α2-adrenoceptor antagonists, yohimbine (0.01 mmol·L−1) and atipamezole (0.1 mmol·L−1). (A, B) Effects of DEX on CAP peak amplitude in the presence of α2 antagonists (A: yohimbine; B: atipamezole). (Aa, Ba) Recordings of CAPs in the control, after 6, 12 and 20 min of exposure to α2 antagonists (Aa: yohimbine; Ba: atipamezole) and thereafter 6, 12 and 20 min in the presence of DEX together with α2 antagonists (Aa: yohimbine; Ba: atipamezole). Upper insets in (Aa) and (Ba) show the chemical structures of yohimbine and atipamezole (see Virtanen, 1989; Westfall and Westfall, 2006), respectively. (Ab, Bb) Average time courses of changes in CAP peak amplitudes following treatment with α2 antagonists (Ab: yohimbine; Bb: atipamezole) and with both DEX and α2 antagonists (Ab: yohimbine; Bb: atipamezole), relative to that before drug treatment, obtained from four (Ab) or five (Bb) sciatic nerves. Note that atipamezole (0.1 mmol·L−1) itself reduces CAP peak amplitudes.
Atipamezole was used at 0.1 mmol·L−1, a concentration enough to antagonize dexmedetomidine-induced hypnotic responses, mediated by α2 adrenoceptors in the rat locus coeruleus (Correa-Sales et al., 1992). As noted from the traces in Figure 4Ba, pretreatment with this drug itself for 20 min reduced the peak amplitude of CAP, but only slightly to 88 ± 3% (P < 0.05) of control (21 ± 2 mV; n = 5). Adding dexmedetomidine (0.5 mmol·L−1) to atipamezole markedly inhibited the CAP, as summarized in Figure 4Bb. The peak amplitude after 20 min with atipamezole and dexmedetomidine was 6 ± 3% (P < 0.05) of that (19 ± 2 mV; n = 5) just before the co-treatment and was significantly less than that (24 ± 8%; n = 5; see above; P < 0.05) obtained with dexmedetomidine only. This difference may be due to an interaction between the effects of dexmedetomidine and atipamezole. At a higher concentration (0.2 mmol·L−1), atipamezole by itself reduced CAP peak amplitude to 40 ± 5% (P < 0.05) of control (21 ± 2 mV; n = 5).
Effects of various adrenoceptor agonists on frog sciatic nerve CAPs
Next, we investigated whether CAPs are affected by other α2-adrenoceptor agonists, oxymetazoline (more selective for α2A than α2B and α2C adrenoceptors; see Bylund et al., 1994; MacDonald et al., 1997) and clonidine (their chemical structures are shown in the insets of Figure 5Aa and Ba, respectively). As seen with dexmedetomidine, oxymetazoline (1 mmol·L−1) reduced CAP peak amplitude in a reversible manner, as shown by the traces in Figure 5Aa. Figure 5Ab summarizes the time courses of a change in CAP peak amplitude following treatment with oxymetazoline (1 mmol·L−1). More rapidly than dexmedetomidine, oxymetazoline (1 mmol·L−1) reduced CAP peak amplitude to a steady state of 76 ± 2% (P < 0.05) of control (20 ± 2 mV; n = 4) with a slight tendency to increase the HPD of CAP [106 ± 9% (P > 0.05) of control (0.45 ± 0.06 ms; n = 4)].
Figure 5.
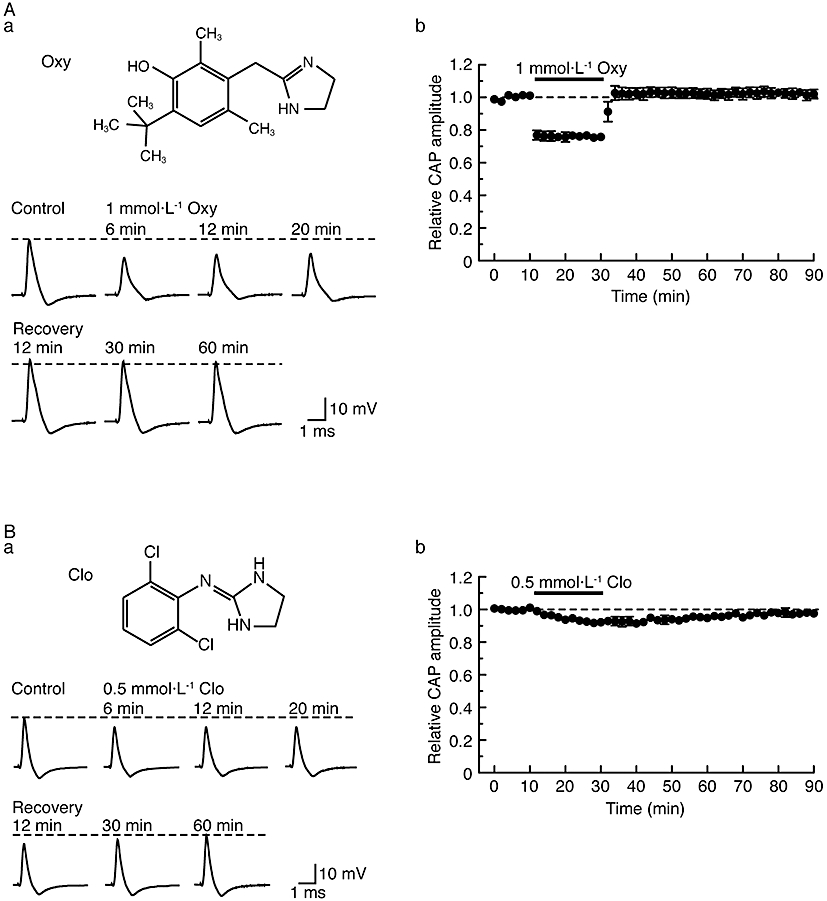
Other α2-adrenoceptor agonists, oxymetazoline (Oxy; 1 mmol·L−1) and clonidine (Clo; 0.5 mmol·L−1), also reversibly reduce compound action potential (CAP) peak amplitudes. (A, B) Effects of α2 agonists (A: Oxy; B: Clo) on CAP peak amplitudes. (Aa, Ba) Recordings of CAPs in the control, after 6, 12 and 20 min of exposure to α2 adrenoceptor agonists (A: Oxy; B: Clo) and thereafter 12, 30 and 60 min in the absence of α2 adrenoceptor agonists (A: Oxy; B: Clo). Upper insets in (Aa) and (Ba) show the chemical structures of oxymetazoline and clonidine, respectively (see Westfall and Westfall, 2006). (Ab, Bb) Average time courses of changes in CAP peak amplitudes following exposure to α2 adrenoceptor agonists (Ab: Oxy; Bb: Clo) for 20 min, relative to that before exposure, obtained from four (Ab) or five (Bb) sciatic nerves.
As there was a remarkable difference between the time taken for dexmedetomidine- and oxymetazoline-induced CAP inhibition to attain a steady state (see Figures 1B and 5Ab), we further examined the effects of both dexmedetomidine (0.5 mmol·L−1) and oxymetazoline (1 mmol·L−1) on CAPs in the same sciatic nerve. The CAP inhibition produced by oxymetazoline attained a steady level at 2 min following drug treatment; the peak amplitude of CAP at this time was the same (101 ± 2%; n = 3) as that at 20 min (100%). On the other hand, CAP inhibition produced by dexmedetomidine was clearly dependent on the time of exposure. Thus, CAP peak amplitudes at 2, 6 and 16 min following drug treatment were, respectively, 259 ± 36, 224 ± 35 and 118 ± 3% (n = 3) of that at 20 min (100%). Figure 6A and B show that the effects of oxymetazoline on CAPs were concentration-dependent over a wide range (0.02–5 mmol·L−1). At 2 mmol·L−1, oxymetazoline also increased the HPD of CAP [158 ± 18% (n = 4; P < 0.05) of control]. The full concentration-response curve is shown in Figure 6Bb and provided an IC50 value of 1.5 mmol·L−1 with a nH of 1.5.
Figure 6.
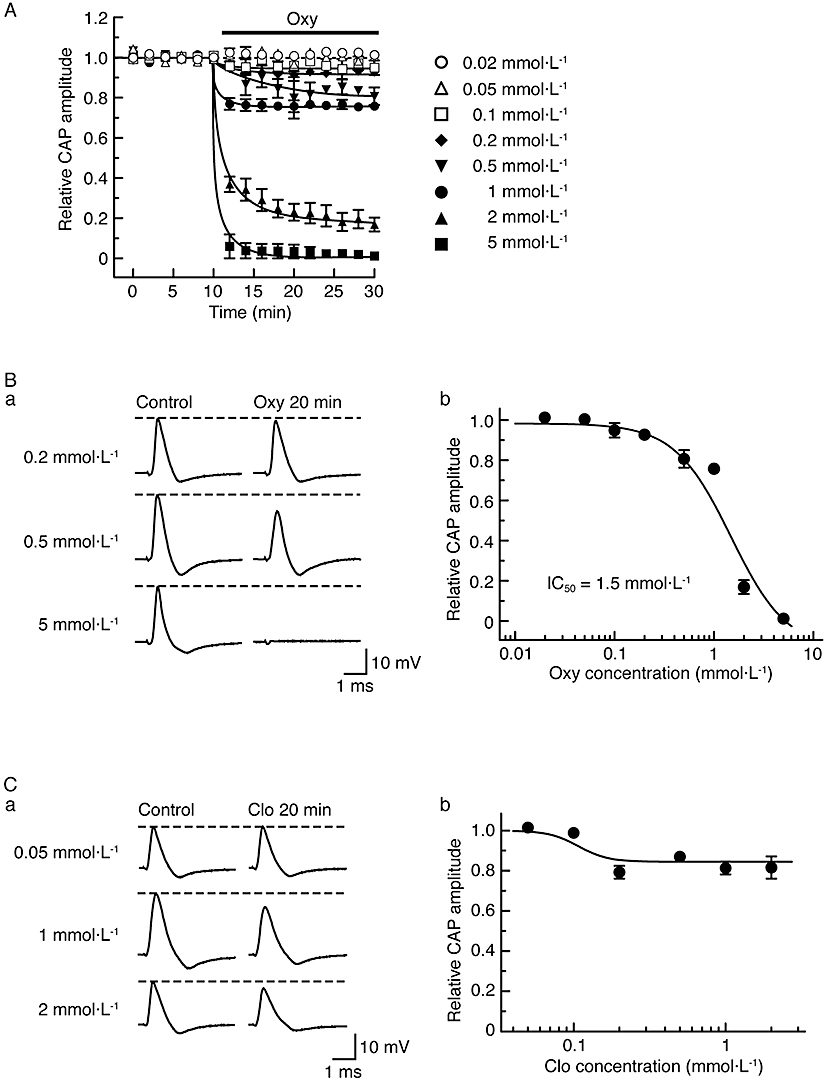
α2-Adrenoceptor agonists, oxymetazoline (Oxy) and clonidine (Clo), reduce compound action potential (CAP) peak amplitudes in a concentration-dependent manner. (A) Comparison in average time course among CAP peak amplitude reductions produced by oxymetazoline at 0.02–5 mmol·L−1, obtained from 25 sciatic nerves. The solid lines were drawn by eye. (B) Concentration dependence for the effect of oxymetazoline on CAPs. (Ba) Recordings of CAPs in the control (left) and after 20 min of exposure to oxymetazoline at 0.2, 0.5 and 5 mmol·L−1 (right); these were obtained from different sciatic nerves. (Bb) The peak amplitude of CAP recorded from sciatic nerve fibres treated with oxymetazoline at various concentrations for 20 min, relative to control, which was plotted against oxymetazoline concentration. Each of the data points was obtained from 3–4 sciatic nerves. The concentration-response curve in (Bb) was drawn according to the Hill equation (IC50 = 1.5 mmol·L−1, nH = 1.5). (C) Concentration dependence for CAP peak amplitude reduction by clonidine. (Ca) Recordings of CAPs in the control (left) and after 20 min of exposure to clonidine at 0.05, 1 and 2 mmol·L−1 (right); these were obtained from different sciatic nerves. (Cb) The peak amplitude of CAP recorded from sciatic nerve fibres treated with clonidine at various concentrations for 20 min, relative to control, was plotted against clonidine concentration. Each of the data points was obtained from 3–5 sciatic nerves.
Although clonidine (0.5 mmol·L−1) also reduced CAP peak amplitude in a reversible manner (Figure 5B), this effect of clonidine was less marked than that of dexmedetomidine or oxymetazoline. At a higher concentration (2 mmol·L−1), clonidine was no more effective than at 0.5 mmol·L−1 (Figure 6Ca) and the corresponding concentration-response curve for clonidine on CAP amplitude (Figure 6Cb) showed a maximum reduction by clonidine of about 20%.
Yohimbine (0.01 mmol·L−1) did not affect the inhibitory actions of oxymetazoline (1 mmol·L−1) and clonidine (0.5 mmol·L−1) on CAPs (peak amplitudes at 20 min: oxymetazoline only, 76 ± 2%; n = 4; yohimbine and oxymetazoline, 79 ± 7%, n = 3; P > 0.05: clonidine only, 93 ± 2%; n = 9; yohimbine and clonidine, 89 ± 5%, n = 3; P > 0.05).
We next examined the effects of various adrenoceptor agonists, (±)-adrenaline, (±)-noradrenaline, α1-adrenoceptor agonist (-)-phenylephrine and β-adrenoceptor agonist (-)-isoprenaline (their chemical structures are shown in the insets of Figure 7Aa, Ba, Ca and Da respectively) on CAPs. As seen from the traces in Figure 7Aa, Ba, Ca and Da, (±)-adrenaline, (±)-noradrenaline, (-)-phenylephrine and (-)-isoprenaline (each 1 mmol·L−1) did not affect CAPs. Figure 7Ab, Bb, Cb and Db summarize the time courses of changes in CAP peak amplitudes following treatment with these agonists and in Figure 7E the mean values of CAP peak amplitudes after 20 min treatment all demonstrate no effects of the adrenoceptor agonists (1 mmol·L−1).
Figure 7.
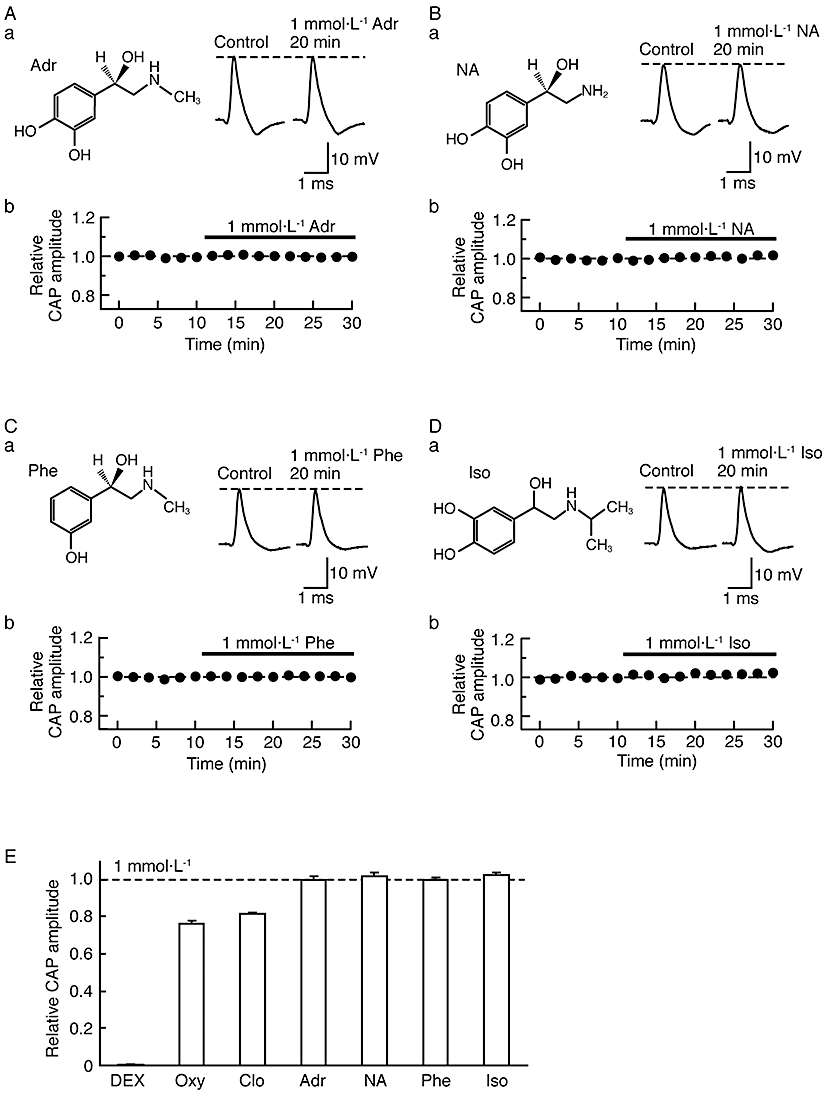
Effects of various adrenoceptor agonists on compound action potentials (CAPs). (Aa, Ba, Ca, Da) Recordings of CAPs in the control (left) and after 20 min of exposure to (±)-adrenaline (Adr; Aa), (±)-noradrenaline (NA; Ba), α1-adrenoceptor agonist (-)-phenylephrine (Phe; Ca) or β-adrenoceptor agonist (-)-isoprenaline (Iso; Da) at 1 mmol·L−1 (right); these were obtained from different sciatic nerves. Insets in (Aa), (Ba), (Ca) and (Da) show the chemical structures of adrenaline, noradrenaline, phenylephrine and isoprenaline, respectively (see Westfall and Westfall, 2006). (Ab, Bb, Cb, Db) Average time courses of changes in CAP peak amplitudes following treatment with adrenaline (Ab; n = 3), noradrenaline (Bb; n = 3), phenylephrine (Cb; n = 3) or isoprenaline (Db; n = 3), relative to that before drug treatment. (E) CAP amplitude (Relative CAP amplitude) after 20 min of exposure to adrenoceptor agonists [dexmedetomidine (DEX), Oxy, Clo, Adr, NA, Phe and Iso; each 1 mmol·L−1], relative to control. The relative CAP amplitudes under the actions of DEX, oxymetazoline and clonidine were taken from Figures 3Bb, 6Bb and Cb, respectively.
Effect of tetracaine on frog sciatic nerve CAPs
In order to compare the inhibitory effect of dexmedetomidine on CAPs with that of local anaesthetics, we next examined the effect of a well-known local anaesthetic tetracaine on CAPs. Tetracaine at a concentration of 0.02 mmol·L−1 reversibly reduced CAP peak amplitude, as seen in Figure 8Aa and summarized in Figure 8Ab. The maximal effect of tetracaine (0.02 mmol·L−1) on CAP amplitude was reached within 20 min of exposure, reducing peak amplitude to 27 ± 7% (P < 0.05) of control (20 ± 2 mV; n = 7) with an increase in the HPD of CAP [136 ± 11% (P < 0.05) of control (0.63 ± 0.06 ms; n = 7)]. The concentration-dependence of the effects of tetracaine (0.0005–0.05 mmol·L−1) on CAPs is shown in Figure 8B and C. The rate of the reduction of CAP peak amplitude produced by tetracaine was concentration-dependent as was the magnitude of the reduction at 20 min. Analysis of the concentration-response curve provided an IC50 value for tetracaine of 0.014 mmol·L−1 with a nH of 1.4.
Figure 8.
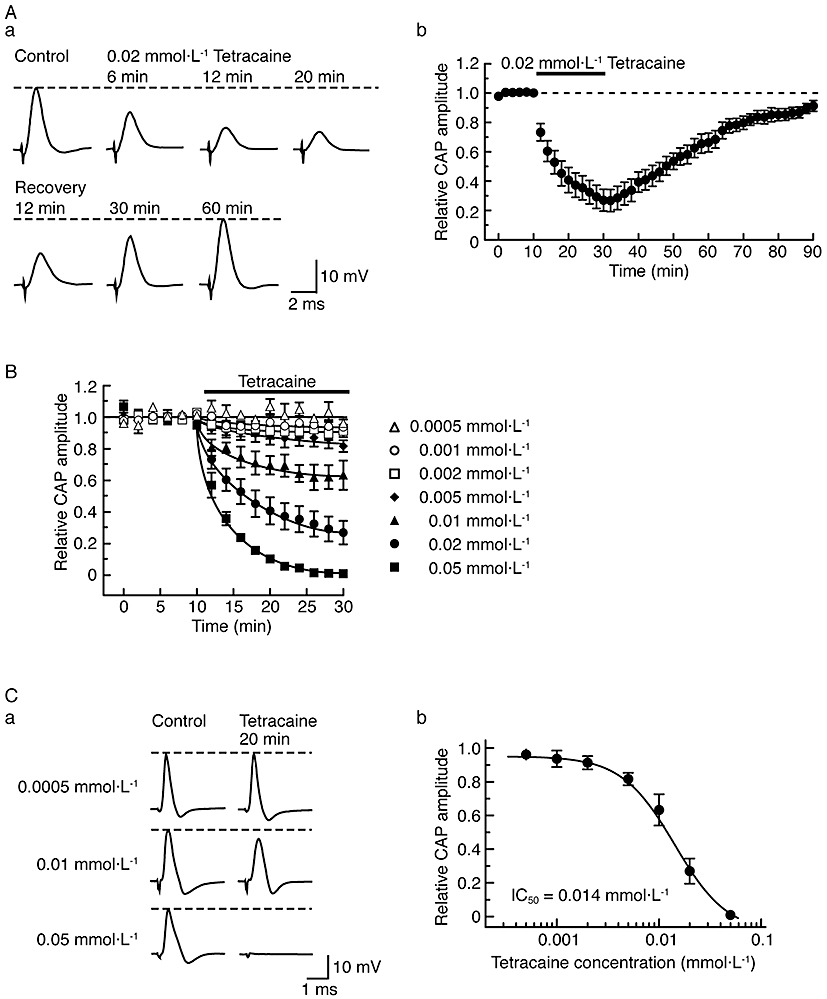
Effect of tetracaine on compound action potentials (CAPs) recorded from frog sciatic nerve fibres. (A) Tetracaine at a concentration of 0.02 mmol·L−1 reversibly reduces CAP peak amplitudes. (Aa) Recordings of CAPs in the control, after 6, 12 and 20 min of exposure to tetracaine and thereafter 12, 30 and 60 min in the absence of tetracaine. (Ab) Average time course of changes in CAP peak amplitudes following exposure to tetracaine for 20 min, relative to that before treatment, obtained from seven sciatic nerves. (B) Comparison in average time course among CAP peak amplitude reductions produced by tetracaine at 0.0005–0.05 mmol·L−1, obtained from 28 sciatic nerves. The solid lines were drawn by eye. (C) Concentration dependence for CAP peak amplitude reduction by tetracaine. (Ca) Recordings of CAPs in the control (left) and after 20 min of exposure to tetracaine at 0.0005, 0.01 and 0.05 mmol·L−1 (right); these were obtained from different sciatic nerves. (Cb) The peak amplitude of CAP recorded from sciatic nerve fibres treated with tetracaine at various concentrations for 20 min, relative to control, was plotted against tetracaine concentration. Each of the data points was obtained from 3–7 sciatic nerves. The concentration-response curve was drawn according to the Hill equation (IC50 = 0.014 mmol·L−1, nH = 1.4).
In order to define the channels affected by dexmedetomidine, we examined how the inhibitory action of dexmedetomidine (0.5 mmol·L−1) on CAPs was altered by treating the nerve trunk with tetracaine (0.01 mmol·L−1; which is well-known to inhibit voltage-gated Na+ channels; Bräu et al., 1998) or an inhibitor of delayed-rectifier K+-channels TEA (1 mmol·L−1) for 20 min. Tetracaine reduced CAP peak amplitude [62 ± 5% (n = 4; P < 0.05) of control] and increased the HPD of CAP [156 ± 12% (n = 4; P < 0.05) of control]; adding dexmedetomidine completely blocked the CAPs. TEA increased the HPD of CAP [120 ± 4% (n = 4; P < 0.05) of control] without a change in CAP peak amplitude [100 ± 1% (n = 4; P > 0.05) of control; see Mizuta et al., 2008]; after adding dexmedetomidine, the HPD of CAP was increased further (186 ± 12%; n = 3; P < 0.05) and the CAP peak amplitude markedly reduced (6 ± 2%; n = 4; P < 0.05).
Discussion
The present study demonstrated that the α2-adrenoceptor agonist dexmedetomidine reversibly and concentration-dependently reduced CAP peak amplitude with an IC50 value of 0.40 mmol·L−1 in the frog sciatic nerve. A reduction in amplitude was produced by other α2-adrenoceptor agonists, oxymetazoline (IC50 = 1.5 mmol·L−1) and clonidine (by 20% at 2 mmol·L−1). Inhibition of CAPs by clonidine has been reported in the frog and rat sciatic nerve (Starke et al., 1972; Butterworth et al., 1993), although their results are quantitatively different from ours (see below). The higher potency of dexmedetomidine than clonidine may indicate that the CAP inhibition is mediated by α2 adrenoceptors, because dexmedetomidine is about eightfold more potent than clonidine in binding to the α2 adrenoceptors of rat brain membrane preparations (Virtanen et al., 1988; Savola and Virtanen, 1991). Dexmedetomidine acts as a full agonist at α2 adrenoceptors while clonidine is a partial agonist in diminishing halothane anaesthetic requirement in rats (Maze et al., 1987; Segal et al., 1988), observations similar to ours on frog CAP (see Figures 3Bb and 6Cb). The activity of dexmedetomidine (0.5 mmol·L−1) in the present study, however, was resistant to an α2-adrenoceptor antagonist yohimbine (0.01 mmol·L−1). The activities of oxymetazoline (1 mmol·L−1) and clonidine (0.5 mmol·L−1) were also unaffected by yohimbine. Although another α2-adrenoceptor antagonist atipamezole (0.1 mmol·L−1) itself slightly reduced CAP amplitude, the dexmedetomidine-induced CAP inhibition was almost unaffected by atipamezole. Atipamezole-induced CAP inhibition was concentration-dependent in a range of 0.1–0.2 mmol·L−1. These results indicate no involvement of α2 adrenoceptors. Unlike α2 adrenoceptor-related drugs, (±)-adrenaline, (±)-noradrenaline, α1-adrenoceptor agonist (-)-phenylephrine and β-adrenoceptor agonist (-)-isoprenaline at 1 mmol·L−1 had no effect on CAPs. Thus, the dexmedetomidine effect was not mediated by adrenoceptors, in general.
In our study, the clonidine-induced reduction of CAP amplitude was concentration-dependent in a range of 0.05–0.2 mmol·L−1 with a maximal value of 20%; a further reduction was not seen at > 0.2 mmol·L−1 (Figure 6C). In contrast to this result, Starke et al. (1972) have reported a concentration-dependent inhibition of frog sciatic nerve CAP in a very narrow concentration range around 0.2 mmol·L−1 with 80% inhibition at 0.3 mmol·L−1. The reason for the discrepancy between these results is unknown. The CAPs of Aα and C fibres in the rat sciatic nerve were inhibited by clonidine with different IC50 values (2.0 and 0.45 mmol·L−1 respectively; Butterworth et al., 1993). The concentration dependency for clonidine-mediated CAP inhibition in the present study may be due to the presence of clonidine-sensitive and -insensitive fast-conducting (possibly A-type) fibres in the frog sciatic nerve. This issue remains to be further examined.
It may be of interest to note a difference in onset and offset time between CAP peak amplitude reductions produced by dexmedetomidine and oxymetazoline (see Figures 1B and 5Ab). Although dexmedetomidine was more effective than oxymetazoline in reducing CAP amplitude, oxymetazoline had shorter onset and offset times than dexmedetomidine. This may be due to a difference between their chemical structures, in that oxymetazoline has more methyl groups and is thus more lipophilic than dexmedetomidine (see Figures 1A and 5Aa). The α2 antagonist atipamezole appeared to be more effective in inhibiting CAP than dexmedetomidine and oxymetazoline. A chemical structure related to α2-adrenoceptor drugs, particularly dexmedetomidine and atipamezole, may play an important role in producing the CAP inhibition.
Although dexmedetomidine inhibits hyperpolarization-activated K+ channels and thus blocks nerve conduction by activating α2 adrenoceptors (Dalle et al., 2001; Shirasaka et al., 2007), this is unlikely in the present study. The dexmedetomidine-induced CAP inhibition in our study was resistant to α2-adrenoceptor antagonists and was not accompanied by a change in the threshold to elicit CAPs (see Figure 2B) which may be expected when hyperpolarization-activated K+ channels are inhibited (Dalle et al., 2001). Our preparation is the dissected sciatic nerve which lacked the neuronal cell body and the neuronal terminals. The inhibitory effect on nerve conduction is thus less likely to be related to G-protein coupled membrane receptors such as α2 adrenoceptors.
The dexmedetomidine action in the present study would be due to an inhibition of TTX-sensitive voltage-gated Na+ channels and/or TEA-sensitive (delayed-rectifier) K+ channels which are involved in producing frog CAPs (Mizuta et al., 2008). In support of this idea, Oda et al. (2007) have reported an inhibition by dexmedetomidine of voltage-gated Na+ channels in rat dorsal root ganglia (DRG) neurones in a manner resistant to yohimbine, although this type of Na+ channels is resistant to TTX. Dexmedetomidine acting on rat DRG neurones had an IC50 value of 0.058 mmol·L−1, about 10-fold smaller than that (0.40 mmol·L−1) we found for inhibition of frog CAPs. These values were >100-fold larger than that (0.62 µmol·L−1) in producing membrane hyperpolarization by activating α2 adrenoceptors in rat SG neurones (Ishii et al., 2008). The rat TTX-resistant Na+ channel was also inhibited by clonidine, yohimbine and lidocaine (IC50: 0.26, 0.0026 and 0.073 mmol·L−1, respectively; Oda et al., 2007). Dessaint et al. (2004) have reported that yohimbine inhibited not only TTX-resistant Na+ channels (IC50 = 0.078 mmol·L−1) but also TTX-sensitive Na+ channels (Nav1.2; IC50 = 0.047 mmol·L−1; by about 10% at 0.01 mmol·L−1) in rat DRG neurones. On the contrary, frog CAPs were not inhibited by yohimbine (0.01 mmol·L−1) and were more sensitive to dexmedetomidine than lidocaine by about twofold (see below). Very recently, it has been found that, in NG108-15 neuronal cells the delayed-rectifier K+-channel current amplitudes are reduced by dexmedetomidine with an IC50 value of 0.0046 mmol·L−1 and TTX-sensitive Na+-channel current amplitudes are attenuated by about 20% by dexmedetomidine (0.01 mmol·L−1) in a manner insensitive to yohimbine (Chen et al., 2009). These differences in potency among drugs may be due to a distinction in either animal species or Na+-channel types. When dexmedetomidine acts on TTX-sensitive Na+ channels and delayed-rectifier K+ channels in the frog sciatic nerve, the binding sites of dexmedetomidine in the channels appear to be different from those of tetracaine and TEA, because these drugs do not prevent reduction of CAP peak amplitude and increase of HPD, both of which are produced by dexmedetomidine.
Dexmedetomidine exhibits an affinity for not only α2 adrenoceptors but also imidazoline receptors (see Savola and Savola, 1996; Khan et al., 1999). Dahmani et al. (2008) have recently reported that dexmedetomidine increased the extent of phosphorylated extracellular signal-regulated protein kinase without α2-adrenoceptor activation in the rat hippocampus, possibly by activating imidazoline I1 receptors. On the other hand, the reduction of halothane anaesthetic requirement produced by dexmedetomidine in rats was not mediated by imidazoline receptors and was due to α2-adrenoceptor activation (Kagawa et al., 1997). Clonidine, which exhibits a high affinity for imidazoline I1 receptors (see Farsang and Kapocsi, 1999), was less potency in inhibiting frog CAP. Atipamezole does not activate imidazoline I2 receptors (Diaz et al., 1997) but was more effective in inhibiting CAPs than α2 adrenoceptor agonists. Thus, the dexmedetomidine-mediated inhibition of frog CAPs appeared not to be mediated by imidazoline receptors.
Comparison between dexmedetomidine and local anaesthetics in reducing CAP peak amplitudes
Frog CAP peak amplitude was reversibly and concentration-dependently reduced by tetracaine with an IC50 of 0.014 mmol·L−1 (Figure 8C). This value is not so different from that (0.0063 mmol·L−1) of frog sciatic nerve fibres, reported previously (Starke et al., 1972), and also from that (0.009 mmol·L−1) of rabbit A nerve fibres (Gissen et al., 1980). This value of tetracaine was about 30-fold smaller than that of dexmedetomidine (IC50 = 0.40 mmol·L−1). When compared with the actions of other local anaesthetics, as reported previously (Katsuki et al., 2006; Mizuta et al., 2008), this dexmedetomidine action was similar to that of ropivacaine (IC50 = 0.34 mmol·L−1) while being greater than those of lidocaine and cocaine (IC50 = 0.74 and 0.80 mmol·L−1 respectively).
Clinical significance of the effect of dexmedetomidine on nerve CAPs
In humans, dexmedetomidine produces sedation and analgesia, and decreases heart rate, cardiac output and memory; each of these dexmedetomidine actions depends on its plasma concentrations in a different manner (Ebert et al., 2000). In patients, sedation is rapidly produced by 0.2 to 0.7 µg·kg−1·h−1 i.v. (Bhana et al., 2000). In veterinary use, when injected intramuscularly in cats, 40 µg·kg−1 is a usual dose for sedation/analgesia (Slingsby and Taylor, 2008). Our study demonstrated a detail of the local anaesthetic effect of dexmedetomidine. The concentrations of dexmedetomidine, enough to inhibit nerve conduction, are mostly over 1000-fold higher than those used for dexmedetomidine as α2 adrenoceptor agonist, as clinical doses of dexmedetomidine give plasma levels below 0.05 µmol·L−1 (see Ebert et al., 2000). Thus, any effects of dexmedetomidine on nerve conduction is quite separate from the most current use of dexmedetomidine for sedation or analgesia.
α2-Adrenoceptor agonists including dexmedetomidine, combined with a local anaesthetic, have been used to extend the duration of peripheral nerve conduction block (Singelyn et al., 1996; Madan et al., 2001; Memişet al., 2004; Calasans-Maia et al., 2005; Kanazi et al., 2006). This action is suggested to be due to a local vasoconstriction leading to a delay of the absorption of the local anaesthetic and/or a direct inhibition of nerve conduction by α2 agonists (Tschernko et al., 1998). The latter mechanism may be supported by a nerve conduction inhibition by dexmedetomidine, as revealed in the present study.
In conclusion, dexmedetomidine produces a conduction block with a higher potency than that of the local anaesthetic lidocaine. This action is relevant when considering the topical application of dexmedetomidine on nerves, but is not related to dexmedetomidine use for sedation, anaesthesia or analgesia by systemic administration. Conduction block was observed after other α2-adrenoceptor agonists, oxymetazoline and clonidine, and also the α2-adrenoceptor antagonist atipamezole. A chemical structure related to the α2 adrenoceptor drugs may play an important role in producing nerve conduction block. The molecular targets of the α2 adrenoceptor drugs, in this context, remain to be determined.
Glossary
Abbreviations
- AP
action potential
- CAP
compound action potential
- CV
conduction velocity
- DRG
dorsal root ganglia
- HPD
half-peak duration
- SG
substantia gelatinosa
- TTX
tetrodotoxin
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhana N, Goa KL, McClellan KJ. Dexmedetomidine. Drugs. 2000;59:263–268. doi: 10.2165/00003495-200059020-00012. [DOI] [PubMed] [Google Scholar]
- Bräu ME, Vogel W, Hempelmann G. Fundamental properties of local anesthetics: half-maximal blocking concentrations for tonic block of Na+ and K+ channels in peripheral nerve. Anesth Analg. 1998;87:885–889. doi: 10.1097/00000539-199810000-00026. [DOI] [PubMed] [Google Scholar]
- Brummett CM, Norat MA, Palmisano JM, Lydic R. Perineural administration of dexmedetomidine in combination with bupivacaine enhances sensory and motor blockade in sciatic nerve block without inducing neurotoxicity in rat. Anesthesiology. 2008;109:502–511. doi: 10.1097/ALN.0b013e318182c26b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth JF, IV, Strichartz GR. The α2-adrenergic agonists clonidine and guanfacine produce tonic and phasic block of conduction in rat sciatic nerve fibers. Anesth Analg. 1993;76:295–301. [PubMed] [Google Scholar]
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, et al. International union of pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- Calasans-Maia JA, Zapata-Sudo G, Sudo RT. Dexmedetomidine prolongs spinal anaesthesia induced by levobupivacaine 0.5% in guinea-pigs. J Pharm Pharmacol. 2005;57:1415–1420. doi: 10.1211/jpp.57.11.0006. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Mackie K. Local anesthetics. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11th edn. New York: McGraw-Hill, Medical Publishing Division; 2006. pp. 369–386. [Google Scholar]
- Chen B-S, Peng H, Wu S-N. Dexmedetomidine, an α2-adrenergic agonist, inhibits neuronal delayed-rectifier potassium current and sodium current. Br J Anaesth. 2009;103:244–254. doi: 10.1093/bja/aep107. [DOI] [PubMed] [Google Scholar]
- Concepcion M, Maddi R, Francis D, Rocco AG, Murray E, Covino BG. Vasoconstrictors in spinal anesthesia with tetracaine – a comparison of epinephrine and phenylephrine. Anesth Analg. 1984;63:134–138. [PubMed] [Google Scholar]
- Correa-Sales C, Rabin BC, Maze M. A hypnotic response to dexmedetomidine, an α2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology. 1992;76:948–952. doi: 10.1097/00000542-199206000-00013. [DOI] [PubMed] [Google Scholar]
- Coughlan MG, Lee JG, Bosnjak ZJ, Schmeling WT, Kampine JP, Warltier DC. Direct coronary and cerebral vascular responses to dexmedetomidine. Significance of endogenous nitric oxide synthesis. Anesthesiology. 1992;77:998–1006. doi: 10.1097/00000542-199211000-00024. [DOI] [PubMed] [Google Scholar]
- Dahmani S, Paris A, Jannier V, Hein L, Rouelle D, Scholz J, et al. Dexmedetomidine increases hippocampal phosphorylated extracellular signal-regulated protein kinase 1 and 2 content by an α2-adrenoceptor-independent mechanism: evidence for the involvement of imidazoline I1 receptors. Anesthesiology. 2008;108:457–466. doi: 10.1097/ALN.0b013e318164ca81. [DOI] [PubMed] [Google Scholar]
- Dalle C, Schneider M, Clergue F, Bretton C, Jirounek P. Inhibition of the Ih current in isolated peripheral nerve: a novel mode of peripheral antinociception? Muscle Nerve. 2001;24:254–261. doi: 10.1002/1097-4598(200102)24:2<254::aid-mus110>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Dessaint J, Yu W, Krause JE, Yue L. Yohimbine inhibits firing activities of rat dorsal root ganglion neurons by blocking Na+ channels and vanilloid VR1 receptors. Eur J Pharmacol. 2004;485:11–20. doi: 10.1016/j.ejphar.2003.11.039. [DOI] [PubMed] [Google Scholar]
- Diaz A, Mayet S, Dickenson AH. BU-224 produces spinal antinociception as an agonist at imidazoline I2 receptors. Eur J Pharmacol. 1997;333:9–15. doi: 10.1016/s0014-2999(97)01118-7. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93:382–394. doi: 10.1097/00000542-200008000-00016. [DOI] [PubMed] [Google Scholar]
- Eisenach JC, De Kock M, Klimscha W. α2-Adrenergic agonists for regional anesthesia. A clinical review of clonidine (1984–1995) Anesthesiology. 1996;85:655–674. doi: 10.1097/00000542-199609000-00026. [DOI] [PubMed] [Google Scholar]
- Farsang C, Kapocsi J. Imidazoline receptors: from discovery to antihypertensive therapy (facts and doubts) Brain Res Bull. 1999;49:317–331. doi: 10.1016/s0361-9230(99)00057-x. [DOI] [PubMed] [Google Scholar]
- Filos KS, Goudas LC, Patroni O, Polyzou V. Hemodynamic and analgesic profile after intrathecal clonidine in humans. A dose-response study. Anesthesiology. 1994;81:591–601. doi: 10.1097/00000542-199409000-00011. [DOI] [PubMed] [Google Scholar]
- Fisher B, Zornow MH, Yaksh TL, Peterson BM. Antinociceptive properties of intrathecal dexmedetomidine in rats. Eur J Pharmacol. 1991;192:221–225. doi: 10.1016/0014-2999(91)90046-s. [DOI] [PubMed] [Google Scholar]
- Gaumann DM, Brunet PC, Jirounek P. Clonidine enhances the effects of lidocaine on C-fiber action potential. Anesth Analg. 1992;74:719–725. doi: 10.1213/00000539-199205000-00017. [DOI] [PubMed] [Google Scholar]
- Gissen AJ, Covino BG, Gregus J. Differential sensitivities of mammalian nerve fibers to local anesthetic agents. Anesthesiology. 1980;53:467–474. doi: 10.1097/00000542-198012000-00006. [DOI] [PubMed] [Google Scholar]
- Ishii H, Kohno T, Yamakura T, Ikoma M, Baba H. Action of dexmedetomidine on the substantia gelatinosa neurons of the rat spinal cord. Eur J Neurosci. 2008;27:3182–3190. doi: 10.1111/j.1460-9568.2008.06260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa K, Mammoto T, Hayashi Y, Kamibayashi T, Mashimo T, Yoshiya I. The effect of imidazoline receptors and α2-adrenoceptors on the anesthetic requirement (MAC) for halothane in rats. Anesthesiology. 1997;87:963–967. doi: 10.1097/00000542-199710000-00032. [DOI] [PubMed] [Google Scholar]
- Kamibayashi T, Maze M. Clinical uses of α2-adrenergic agonists. Anesthesiology. 2000;93:1345–1349. doi: 10.1097/00000542-200011000-00030. [DOI] [PubMed] [Google Scholar]
- Kanazi GE, Aouad MT, Jabbour-Khoury SI, Al Jazzar MD, Alameddine MM, Al-Yaman R, et al. Effect of low-dose dexmedetomidine or clonidine on the characteristics of bupivacaine spinal block. Acta Anaesthesiol Scand. 2006;50:222–227. doi: 10.1111/j.1399-6576.2006.00919.x. [DOI] [PubMed] [Google Scholar]
- Katsuki R, Fujita T, Koga A, Liu T, Nakatsuka T, Nakashima M, et al. Tramadol, but not its major metabolite (mono-O-demethyl tramadol) depresses compound action potentials in frog sciatic nerves. Br J Pharmacol. 2006;149:319–327. doi: 10.1038/sj.bjp.0706868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kumamoto E, Furue H, Yoshimura M. α2 Adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98:682–689. doi: 10.1097/00000542-200303000-00016. [DOI] [PubMed] [Google Scholar]
- Khan ZP, Ferguson CN, Jones RM. Alpha-2 and imidazoline receptor agonists. Their pharmacology and therapeutic role. Anaesthesia. 1999;54:146–165. doi: 10.1046/j.1365-2044.1999.00659.x. [DOI] [PubMed] [Google Scholar]
- MacDonald E, Kobilka BK, Scheinin M. Gene targeting-homing in on α2-adrenoceptor-subtype function. Trends Pharmacol Sci. 1997;18:211–219. doi: 10.1016/s0165-6147(97)01063-8. [DOI] [PubMed] [Google Scholar]
- Madan R, Bharti N, Shende D, Khokhar SK, Kaul HL. A dose-response study of clonidine with local anesthetic mixture for peribulbar block: a comparison of three doses. Anesth Analg. 2001;93:1593–1597. doi: 10.1097/00000539-200112000-00056. [DOI] [PubMed] [Google Scholar]
- Maze M, Birch B, Vickery RG. Clonidine reduces halothane MAC in rats. Anesthesiology. 1987;67:868–869. doi: 10.1097/00000542-198711000-00061. [DOI] [PubMed] [Google Scholar]
- Memiş D, Turan A, Karamanlioglu B, Pamukçu Z, Kurt I. Adding dexmedetomidine to lidocaine for intravenous regional anesthesia. Anesth Analg. 2004;98:835–840. doi: 10.1213/01.ane.0000100680.77978.66. [DOI] [PubMed] [Google Scholar]
- Mizuta K, Fujita T, Nakatsuka T, Kumamoto E. Inhibitory effects of opioids on compound action potentials in frog sciatic nerves and their chemical structures. Life Sci. 2008;83:198–207. doi: 10.1016/j.lfs.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Oda A, Iida H, Tanahashi S, Osawa Y, Yamaguchi S, Dohi S. Effects of α2-adrenoceptor agonists on tetrodotoxin-resistant Na+ channels in rat dorsal root ganglion neurons. Eur J Anaesthesiol. 2007;24:934–941. doi: 10.1017/S0265021507000543. [DOI] [PubMed] [Google Scholar]
- Pan Y-Z, Li D-P, Pan H-L. Inhibition of glutamatergic synaptic input to spinal lamina IIo neurons by presynaptic α2-adrenergic receptors. J Neurophysiol. 2002;87:1938–1947. doi: 10.1152/jn.00575.2001. [DOI] [PubMed] [Google Scholar]
- Savola MKT, Savola J-M. [3H]Dexmedetomidine, an α2-adrenoceptor agonist, detects a novel imidazole binding site in adult rat spinal cord. Eur J Pharmacol. 1996;306:315–323. doi: 10.1016/0014-2999(96)00224-5. [DOI] [PubMed] [Google Scholar]
- Savola J-M, Virtanen R. Central α2-adrenoceptors are highly stereoselective for dexmedetomidine, the dextro enantiomer of medetomidine. Eur J Pharmacol. 1991;195:193–199. doi: 10.1016/0014-2999(91)90535-x. [DOI] [PubMed] [Google Scholar]
- Segal IS, Vickery RG, Walton JK, Doze VA, Maze M. Dexmedetomidine diminishes halothane anesthetic requirements in rats through a postsynaptic alpha2 adrenergic receptor. Anesthesiology. 1988;69:818–823. doi: 10.1097/00000542-198812000-00004. [DOI] [PubMed] [Google Scholar]
- Shirasaka T, Kannan H, Takasaki M. Activation of a G protein-coupled inwardly rectifying K+ current and suppression of Ih contribute to dexmedetomidine-induced inhibition of rat hypothalamic paraventricular nucleus neurons. Anesthesiology. 2007;107:605–615. doi: 10.1097/01.anes.0000281916.65365.4e. [DOI] [PubMed] [Google Scholar]
- Singelyn FJ, Gouverneur J-M, Robert A. A minimum dose of clonidine added to mepivacaine prolongs the duration of anesthesia and analgesia after axillary brachial plexus block. Anesth Analg. 1996;83:1046–1050. doi: 10.1097/00000539-199611000-00025. [DOI] [PubMed] [Google Scholar]
- Sjöholm B, Voutilainen R, Luomala K, Savola J-M, Scheinin M. Characterization of [3H]atipamezole as a radioligand for α2-adrenoceptors. Eur J Pharmacol. 1992;215:109–117. doi: 10.1016/0014-2999(92)90615-b. [DOI] [PubMed] [Google Scholar]
- Skou JC. Local anesthetics. I. The blocking potencies of some local anesthetics and of butyl alcohol determined on peripheral nerves. Acta Pharmacol Toxicol. 1954;10:281–291. doi: 10.1111/j.1600-0773.1954.tb01344.x. [DOI] [PubMed] [Google Scholar]
- Slingsby LS, Taylor PM. Thermal antinociception after dexmedetomidine administration in cats: a dose-finding study. J Vet Pharmacol Ther. 2008;31:135–142. doi: 10.1111/j.1365-2885.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- Starke K, Wagner J, Schümann HJ. Adrenergic neuron blockade by clonidine: comparison with guanethidine and local anesthetics. Arch Int Pharmacodyn. 1972;195:291–308. [PubMed] [Google Scholar]
- Sullivan AF, Kalso EA, McQuay HJ, Dickenson AH. The antinociceptive actions of dexmedetomidine on dorsal horn neuronal responses in the anaesthetized rat. Eur J Pharmacol. 1992;215:127–133. doi: 10.1016/0014-2999(92)90617-d. [DOI] [PubMed] [Google Scholar]
- Takano Y, Yaksh TL. Relative efficacy of spinal alpha-2 agonists, dexmedetomidine, clonidine and ST-91, determined in vivo by using N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline, an irreversible antagonist. J Pharmacol Exp Ther. 1991;258:438–446. [PubMed] [Google Scholar]
- Tschernko EM, Klepetko H, Gruber E, Kritzinger M, Klimscha W, Jandrasits O, et al. Clonidine added to the anesthetic solution enhances analgesia and improves oxygenation after intercostal nerve block for thoracotomy. Anesth Analg. 1998;87:107–111. doi: 10.1097/00000539-199807000-00023. [DOI] [PubMed] [Google Scholar]
- Vaida GT, Moss P, Capan LM, Turndorf H. Prolongation of lidocaine spinal anesthesia with phenylephrine. Anesth Analg. 1986;65:781–785. [PubMed] [Google Scholar]
- Virtanen R. Pharmacological profiles of medetomidine and its antagonist, atipamezole. Acta Vet Scand. 1989;85:29–37. [PubMed] [Google Scholar]
- Virtanen R, Savola J-M, Saano V, Nyman L. Characterization of the selectivity, specificity and potency of medetomidine as an α2-adrenoceptor agonist. Eur J Pharmacol. 1988;150:9–14. doi: 10.1016/0014-2999(88)90744-3. [DOI] [PubMed] [Google Scholar]
- Westfall TC, Westfall DP. Adrenergic agonists and antagonists. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11th edn. New York: McGraw-Hill, Medical Publishing Division; 2006. pp. 237–295. [Google Scholar]
- Yaksh TL. Pharmacology of spinal adrenergic systems which modulate spinal nociceptive processing. Pharmacol Biochem Behav. 1985;22:845–858. doi: 10.1016/0091-3057(85)90537-4. [DOI] [PubMed] [Google Scholar]
- Yoshitomi T, Kohjitani A, Maeda S, Higuchi H, Shimada M, Miyawaki T. Dexmedetomidine enhances the local anesthetic action of lidocaine via an α-2A adrenoceptor. Anesth Analg. 2008;107:96–101. doi: 10.1213/ane.0b013e318176be73. [DOI] [PubMed] [Google Scholar]