

Abstract
MSMEG_0220 from Mycobacterium smegmatis, the ortholog of the Rv0183 gene from M. tuberculosis, recently identified and characterized as encoding a monoacylglycerol lipase, was cloned and expressed in Escherichia coli. The recombinant protein (rMSMEG_0220), which exhibits 68% amino acid sequence identity with Rv0183, showed the same substrate specificity and similar patterns of pH-dependent activity and stability as the M. tuberculosis enzyme. rMSMEG_0220 was found to hydrolyze long-chain monoacylglycerol with a specific activity of 143 ± 6 U mg−1. Like Rv0183 in M. tuberculosis, MSMEG_0220 was found to be located in the cell wall. To assess the in vivo role of the homologous proteins, an MSMEG_0220 disrupted mutant of M. smegmatis (MsΔ0220) was produced. An intriguing change in the colony morphology and in the cell interaction, which were partly restored in the complemented mutant containing either an active (ComMsΔ0220) or an inactive (ComMsΔ0220S111A) enzyme, was observed. Growth studies performed in media supplemented with monoolein showed that the ability of both MsΔ0220 and ComMsΔ0220S111A to grow in the presence of this lipid was impaired. Moreover, studies of the antimicrobial susceptibility of the MsΔ0220 strain showed that this mutant is more sensitive to rifampin and more resistant to isoniazid than the wild-type strain, pointing to a critical structural role of this enzyme in mycobacterial physiology, in addition to its function in the hydrolysis of exogenous lipids.
Tuberculosis, which is caused by Mycobacterium tuberculosis, is a major public health issue worldwide. Because of the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains and the high incidence of HIV and tuberculosis coinfection (16), it is becoming increasingly difficult to combat the spread of this disease, and the global health burden of tuberculosis is extremely heavy. The reasons for the persistence of the tubercle bacillus include not only its ability to enter into a state of dormancy in its host for decades, evading the immune system by forming structures called granulomas (17), but also its unique and complex cell wall composed of specific lipids (8). These characteristics are thought to be good focus points for drug development. In granulomas, during the nonreplicative stage, the bacteria have been found to accumulate lipids in the form of intracellular lipid inclusion bodies (LIBs) (13). These lipids are composed mainly of triacylglycerols (TAG) (9, 13) and may originate from the lipolysis of host lipids and/or fatty acid uptake. In fact, M. tuberculosis in the granuloma center can even accumulate lipids originating from the degradation of immune cells (20). In addition, it has been reported that M. tuberculosis internalized by foamy macrophages accumulated LIBs when it joined cell lipid droplets composed of neutral lipids (32). Lipid storage may provide the bacillus with energy via the β-oxidation pathway followed by the glyoxylate cycle, during the chronic phase and the reactivation step (3, 17). These lipids may also supply precursors for the synthesis of bacterial cell membrane lipids, which play a key role in the pathogenicity of M. tuberculosis (4, 23). To investigate the molecular basis of the virulence and pathogenicity of M. tuberculosis, it was therefore proposed to study the lipid metabolism and cell wall remodeling processes in this bacterium.
The enzymes involved in the lipid degradation processes induced by this bacterium have attracted considerable attention during the last few years. Based on the complete M. tuberculosis H37Rv genome sequence (6), several open reading frames (ORFs) encoding proteins potentially involved in the lipid metabolism of this strain have been identified, among which are the two lipases from M. tuberculosis that have been purified and characterized so far. Deb et al. identified an enzyme, Rv3097c (LipY), belonging to the hormone-sensitive lipase family, which is able to hydrolyze long-chain TAG (10). A study of LIB mobilization in a lipY-deficient mutant has shown that LipY was involved in TAG hydrolysis under nutriment-deprived conditions (10). LipY may therefore be involved in the degradation of TAG stored during the dormant stage and the subsequent reactivation of the pathogen. In addition, electron microscopy immunolabeling studies of LipY clearly showed that the enzyme had a cell surface localization, thus in direct contact with the host immune system (28). The last identified lipase to date is a monoacylglycerol lipase annotated Rv0183 (7). Like LipY, Rv0183 is located in the cell wall, but its exact physiological function has not yet been elucidated. One hypothesis could be that, like some mammalian cells (e.g., adipocytes), M. tuberculosis expresses several lipolytic enzymes sequentially involved in the lipolysis of TAG (37). The Rv0183 enzyme is conserved in M. bovis (Mb0189) and M. leprae (ML2603), as well as in M. smegmatis (MSMEG_0220), a nonpathogenic mycobacterium which provides a useful model organism and a surrogate host for molecular analysis of M. tuberculosis (19). In order to decipher the cellular role of Rv0183 in M. tuberculosis H37Rv and its contribution to the lipid metabolism of this bacterium, biochemical studies were performed on the homologue MSMEG_0220. For this purpose, the MSMEG_0220 gene from M. smegmatis, encoding a protein showing 68% amino acid sequence identity with Rv0183, was cloned, and the recombinant MSMEG_0220 enzyme (rMSMEG_0220) was produced in Escherichia coli, purified, and biochemically characterized. An M. smegmatis mutant with an MSMEG_0220 disrupted gene was produced to investigate the physiological role of MSMEG_0220.
MATERIALS AND METHODS
Materials.
Pfx DNA polymerase and pDest14 and pUC18 plasmids were purchased from Invitrogen. E. coli Rosetta(DE3)/pLysS cells were purchased from Novagen. A Ni2+-nitrilotriacetic acid (NTA) agarose gel was obtained from Amersham Biosciences. Horseradish anti-rabbit immunoglobulins conjugated to peroxidase, dioctanoin, dimyristin, monooctanoin, monomyristin, and sodium taurodeoxycholate (NaTDC) were purchased from Sigma-Aldrich. Kanamycin (Km), hygromycin (Hyg), and Tween 80 were obtained from Euromedex. Middlebrook 7H9 and 7H11 culture media and Middlebrook albumin-dextrose-catalase (ADC) enrichment were purchased from BD. Pure mono- and diolein were purified from low-grade commercial dl-α-monoolein from Fluka. pPR27 and pVV16 plasmids and the M. smegmatis mc2155 strain were a kind gift from the Pasteur Institute (Paris, France).
Bacterial strains and growth conditions.
E. coli Rosetta(DE3)/pLysS was grown in Terrific broth (TB; Invitrogen). M. smegmatis mc2155, MsΔ0220 (an MSMEG_0220 disrupted mutant), ComMsΔ0220 (a complemented mutant containing an active MSMEG_0220 enzyme), and ComMsΔ0220S111A (a complemented mutant containing an inactive MSMEG_0220 enzyme) strains were routinely cultured, for 3 to 5 days, on Middlebrook 7H9 broth containing ADC enrichment and 0.05% (vol/vol) Tween 80 and on solid Middlebrook 7H11. Alternatively, M. smegmatis mc2155, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains were grown on Middlebook 7H9 broth and 7H9 agar (15 g liter−1) supplemented with 0.02% or 1% (vol/vol) monoolein in the presence or absence of 0.05% (vol/vol) Tween 80. Wild-type (WT) M. smegmatis mc2155 was also cultured on Luria-Bertani (LB) agar and LB broth supplemented with 0.05% (vol/vol) Tween 80 for disrupted mutant selection. When required, sucrose was added to the culture medium at a final concentration of 5% (wt/vol). Both Km and Hyg were included at 50 μg ml−1. All cultures were incubated at 37°C, except during M. smegmatis mutant selection, and liquid cultures were incubated with shaking at 220 rpm.
Mycobacterial genomic DNA extraction.
Mycobacterial genomic DNA was isolated as follows. M. smegmatis mc2155 cells were harvested from 5 ml of saturated cell culture (final optical density at 600 nm [OD600] of ≈3) by centrifugation at 3,000 × g for 10 min and resuspended in 400 μl of solution I (50 mM Tris-HCl [pH 8.0], 50 mg ml−1 lysozyme, 0.25 mg ml−1 RNase). After a 2-h incubation step at 37°C, 750 μl of solution II (150 mM Tris-HCl [pH 8.0], 100 mM EDTA, 1% [wt/vol] SDS, 2 mg ml−1 proteinase K) was added to the reaction mixture. The mixture was then incubated at 45°C for 16 h. The mycobacterial DNA was extracted with 5 ml phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol/vol). After centrifugation at 3,000 × g for 15 min, the upper phase was washed twice with 5 ml chloroform-isoamyl alcohol (24:1, vol/vol). The mycobacterial DNA was precipitated by 0.7 vol isopropanol in the presence of 0.1 vol sodium acetate (3 M), resuspended in distilled water, and stored at −20°C.
Cloning of the MSMEG_0220 gene.
PCR was performed to amplify the MSMEG_0220 gene (954 bp) using Pfx DNA polymerase from M. smegmatis mc2155 genomic DNA and P1 and P2 primers (see Table S1 in the supplemental material). P1 contained the attB1 recombination site, the Shine-Dalgarno sequence, and the sequence coding for the His6 tail and the tobacco etch virus (TEV) NIa protease site, while P2 contained the attB2 recombination site. The PCR product was cloned into the pDest14 expression vector following the manufacturer's instructions (Gateway; Invitrogen). The DNA sequence of the MSMEG_0220 ORF was confirmed by performing DNA sequencing (GATC, Germany). The rMSMEG_0220 protein without the His6 tail and the TEV site was composed of 280 residues and had a calculated molecular mass of 29,882 Da, a theoretical isoelectric point of 6.62, and a theoretical molar absorption coefficient of 0.867 M−1 cm−1 at 280 nm.
Construction of the M. smegmatis mc2155 MSMEG_0220 disrupted mutant.
An M. smegmatis mc2155 mutant containing a disrupted MSMEG_0220::Km gene on the chromosome was constructed by performing allelic exchange using the temperature-sensitive sacB procedure (33, 35). A 1,761-bp DNA fragment containing the MSMEG_0220 gene of M. smegmatis mc2155 flanked by 521 and 400 bp at the 5′ and 3′ ends, respectively, was amplified by PCR from genomic DNA using oligonucleotides P3 and P4 (see Table S1 in the supplemental material) and cloned into the XbaI site of the pUC18 plasmid to obtain pRD220. A kanamycin resistance cassette obtained from the pUC4K plasmid was then inserted between an original and a created SalI site in the MSMEG_0220 gene fragment, generating a 186-bp deletion in the coding sequence of this gene and yielding pRD221. This plasmid was subsequently digested by XbaI, and the 2,775-bp fragment that contains the disrupted MSMEG_0220::Km gene and its flanking regions was purified and inserted into the XbaI site of pPR27 (31), a mycobacterial thermosensitive suicide plasmid harboring the counterselectable marker sacB, resulting in plasmid pRD222. M. smegmatis mc2155 cells were transformed by electroporation (Eppendorf electroporator 2510 pulser) with a single electric pulse of 2.5 kV using the pRD222 vector, and the transformants were selected on LB agar plates containing Km (50 μg ml−1) after being incubated for 3 to 5 days at 30°C. Three clones were selected and grown in 5 ml of LB broth with 0.05% Tween 80 and Km for 3 or 4 days. Several dilutions of these cultures were then plated onto LB agar containing Km and sucrose (5%, wt/vol) and incubated at 42°C for 3 days.
PCR screening to detect the disruption of the MSMEG_0220 gene was performed with primers P3, P4, P5, and P6 (see Table S1 in the supplemental material) after extraction of the genomic DNA from 42 Km- and sucrose-resistant colonies. As the M. smegmatis genomic DNA is a template richly endowed with GCs, the PCR conditions were optimized to provide greater primer specificity. Nonspecific amplifications were prevented by using dimethyl sulfoxide (DMSO) at 3% (vol/vol) in the reaction mixture. A clone named MsΔ0220, with the pattern corresponding to disruption of the MSMEG_0220 gene, was selected for further analysis.
Complementation of the M. smegmatis mc2155 MSMEG_0220 disrupted mutant.
PCR amplification was carried out on a region covering the MSMEG_0220 gene from M. smegmatis mc2155 genomic DNA using oligonucleotides P5 and P6 (see Table S1 in the supplemental material). The PCR product devoid of the original stop codon of M. smegmatis was digested with NdeI and HindIII and inserted into the pVV16 expression vector harboring Km and Hyg resistance markers, the hsp60 promoter, and a His6 tag for the expression of a C-terminal His6-tagged fusion protein to generate the pRD223 plasmid. The sequence encoding the catalytic serine residue (S111) in the MSMEG_0220 gene was mutated, using a QuikChange II site-directed mutagenesis kit (Stratagene), into a sequence coding for an alanine residue. The resulting plasmid, named pRD224, allows the production of an inactive MSMEG_0220 enzyme. The pRD223 and pRD224 plasmids were transferred to MsΔ0220 cells by electroporation, and transformants were selected on LB agar plates containing Km and Hyg. Two clones, named, respectively, ComMsΔ0220 and ComMsΔ0220S111A, were selected for further analysis.
Production and purification of rMSMEG_0220.
E. coli Rosetta(DE3)/pLysS cells were transformed with the pDest14-His6-MSMEG_0220 plasmid. MSMEG_0220 was produced and purified as previously described in the case of Rv0183, the homologue of MSMEG_0220 from M. tuberculosis (7). Briefly, the cell pellet recovered from TB cell cultures (1 liter) was resuspended in ice-cold lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% [vol/vol] Triton X-100, 0.25 mg ml−1 lysozyme; 30 ml per initial liter of culture). To purify rMSMEG_0220, the supernatant obtained after cell lysis and centrifugation at 17,000 × g for 30 min was loaded (3 ml min−1) onto a Ni2+-NTA agarose column (1 ml/5 mg of recombinant protein) previously equilibrated with buffer A (10 mM Tris-HCl [pH 8.0], 150 mM NaCl) containing 10 mM imidazole, using a fast-performance liquid chromatography (FPLC) system (Amersham Biosciences). The column was then washed with 15 column volumes of buffer A. Proteins were eluted with a fraction volume of 1.350 ml by application of a linear imidazole concentration gradient ranging from 10 to 500 mM in tubes containing 150 μl of 2-methyl-2,4-pentanediol (MPD) to obtain a final concentration of 10% (vol/vol). After SDS-PAGE analysis on a 12% polyacrylamide gel (24), the eluted fractions containing rMSMEG_0220 were pooled to achieve a protein concentration of around 1 mg ml−1, and 6 ml was loaded onto a HiLoad 26/60 Superdex 200 chromatography column (Amersham Biosciences) and eluted at a rate of 1.5 ml min−1 with buffer A containing 10% (vol/vol) MPD. The His6 tag at the N-terminal end of rMSMEG_0220 was removed by TEV digestion, and the cleaved recombinant enzyme was purified by exclusion from a Ni2+-NTA agarose column. Proteolytic digestion of rMSMEG_0220 by TEV protease was performed overnight at 4°C, using TEV and rMSMEG_0220 at a ratio of 1:15. After the purification procedure, rMSMEG_0220 activity was measured, and the protein concentration was estimated from the OD280 value using 0.867 M−1 cm−1 as the molar absorption coefficient. The enzyme was then concentrated to 0.6 mg ml−1 and stored at −80°C.
pH-Stat kinetic assays of rMSMEG_0220 and rRv0183.
The enzymatic activities of rMSMEG_0220 and rRv0183 were monitored potentiometrically using a pH-Stat instrument (718 Stat Titrino, Swiss; Metrohm) adjusted to a constant endpoint value. Each assay was performed at pH 8.0 in a thermostated vessel containing substrates at various concentrations (Table 1) and 2.5 mM Tris-HCl buffer containing 150 mM NaCl and 3 mM NaTDC in a final volume of 15 ml. When required, the final concentration of NaTDC was varied from 0 to 10 mM. When activities were assayed at pH values below 8.0, back titration experiments were performed following the protocol described by Fernandez et al. (12).
TABLE 1.
Specific activities of rRv0183 and rMSMEG_0220 on several lipidsa
| Substrate (concn, mM) | Sp act (U mg−1) |
|
|---|---|---|
| rRv0183 | rMSMEG_0220 | |
| Triacylglycerols | ||
| Triacetin (153) | 0 | 0 |
| Tributyrin (110) | 0 | 0 |
| Trioctanoin (71) | 0 | 0 |
| Triolein (7.5) | 0 | 0 |
| Diacylglycerols | ||
| Dioctanoin (7.7) | 43 ± 2 | 0 |
| Dimyristin (7.8) | 0 | 0 |
| Diolein (10.7) | 0 | 0 |
| Monoacylglycerols | ||
| Monooctanoin (6.1) | 431 ± 1 | 0 |
| Monomyristin (5.5) | 395 ± 14 | 80 ± 7.5 |
| Monoolein (9.2) | 300 ± 7 | 143 ± 6 |
| Tween 80 (5.1) | 0 | 0 |
Activities of rRv0183 and rMSMEG_0220 were measured using the pH-Stat technique. Experiments were performed at 37°C and at pH 8.0 in 15 ml of reaction mixture containing 2.5 mM Tris-HCl buffer, 150 mM NaCl, and 3 mM sodium taurodeoxycholate (NaTDC) with stirring, using mono-, di-, and triacylglycerol substrates. Before use, all substrates, except triacetin and tributyrin, were emulsified in 10% (wt/vol) gum arabic by sonication. Values are the means from 3 independent assays.
For the comparison of rMSMEG_0220 and rRv0183 biochemical properties, a sample of purified rRv0183 was diluted up to 0.6 mg ml−1 and dialyzed against 10 mM Tris-HCl (pH 8.5) buffer containing 150 mM NaCl and 10% (vol/vol) MPD. Specific activities are expressed in units per milligram of pure enzyme. One unit corresponds to the release of 1 μmol of fatty acid per minute.
pH stability of rMSMEG_0220 and rRv0183.
The pH stability profiles of rMSMEG_0220 and rRv0183 were determined after incubation of the enzymes (60 μg) for 1 h at 25°C in 0.2 ml of several buffers at 200 mM containing 150 mM NaCl and 10% (vol/vol) MPD, set at various pH levels: sodium acetate for pH 4.0 and 5.0, MES (morpholinoethanesulfonic acid) for pH 6.0 and 6.5, HEPES for pH 7.0 and 7.5, Tris-HCl for pH 8.0 and 8.5, and glycine for pH 9.0. The residual activity was determined potentiometrically at 37°C and at pH 8.0 using monoolein (9.2 mM) as the substrate in the presence of 3 mM NaTDC.
Western blotting.
The production of MSMEG_0220 protein in the M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains was checked by performing immunoblotting assays, using purified polyclonal antibodies previously raised against the homologue rRv0183 (7). M. smegmatis cells from 50 ml of saturated culture (final OD600 of ≈4) were harvested by centrifugation at 3,000 × g for 10 min and resuspended in 25 ml of buffer B (50 mM Tris-HCl [pH 8.0], 150 mM NaCl). Cells were broken twice in a French press at high pressure (1,100 lb/in2), and the cell lysates were subjected to centrifugation at 5,000 × g for 15 min. The supernatant (SN1) was recovered. The pellet was resuspended in buffer B containing 1% N-lauroylsarcosine sodium salt (vol/vol) and subjected to sonication two times for 30 s each. Unbroken cells and bacterial debris were removed by centrifugation at 5,000 × g for 15 min, and the supernatant (SN2) was recovered. Proteins from SN1 and SN2 fractions were separated by SDS-PAGE (12% polyacrylamide gel) before being transferred onto a nitrocellulose membrane (15). The immunoblotting experiments were carried out using a Snap i.d. system (Millipore) and horseradish anti-rabbit immunoglobulins conjugated to peroxidase as secondary antibodies, in line with the manufacturer's instructions. The immunoreactive proteins were revealed after the membrane was covered with ECL (enhanced chemiluminescence) reagent (Pierce) and exposed to Hyperfilm ECL film (Amersham).
Mycobacterial lipid extraction and analysis.
M. smegmatis wild-type, MsΔ0220, and ComMsΔ0220 strains were grown in 50 ml of 7H9 broth not supplemented with Tween 80 until saturation (final OD600 of ≈4). Cells recovered by centrifugation at 4,000 × g for 10 min were subjected to lipid extraction as described previously (36). Mycobacterial wet pellets were extracted at room temperature, once with 50 ml chloroform-methanol at a ratio of 1:2 (vol/vol) and twice with 50 ml chloroform-methanol at a ratio of 2:1 (vol/vol), for 24 h for each solvent mixture. The organic phases containing the whole-cell lipid extracts were pooled, washed twice with distilled water, and evaporated to dryness. Lipids were resuspended in a minimal volume of chloroform and precipitated by trickling methanol. After the mixtures were left to stand for 2 h at 4°C, methanol-soluble lipids were recovered by centrifugation at 4°C for 20 min (8,000 × g). Lipids were identified by thin-layer chromatography (TLC) on Silica Gel 60 plates (Macherey-Nagel) run in chloroform-methanol (90:10, vol/vol) or chloroform-methanol-water (65:25:4, vol/vol/vol). Sugar-containing compounds were visualized by spraying plates with 0.2% anthrone in concentrated sulfuric acid, followed by heating.
Bacterial residues obtained after lipid extraction with organic solvents were saponified by a mixture of 40% KOH and methoxyethanol (1:7, vol/vol) at 110°C for 3 h in a screw cap tube. After acidification, fatty acids were extracted with diethyl ether and methylated with an ethereal solution of diazomethane (26). The mycolate patterns of M. smegmatis wild-type, MsΔ0220, and ComMsΔ0220 strains were determined by analytical TLC on Silica Gel 60 plates with dichloromethane. Revelation of lipid spots was performed by spraying the plates with molybdophosphoric acid (10% in ethanol), followed by heating.
Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry detection of purified samples in reflectron mode was performed on an Applied Biosystems 4700 mass spectrometer (Voyager DE-STR; Applied Biosystems, Framingham, MA) as described previously (26). Lipid samples dissolved in chloroform at the concentration of 1 mg ml−1 were directly spotted onto the target plate as 0.5-μl droplets, followed by the addition of 0.5 μl of matrix solution (10 mg ml−1 of 2,5-dihydroxybenzoic acid in chloroform-methanol [1:1, vol/vol]), and allowed to crystallize at room temperature.
Antimicrobial susceptibilities of M. smegmatis strains.
The susceptibilities of M. smegmatis strains to various antibiotics were measured in Middlebrook 7H11 plates that contained increasing concentrations of the following drugs: novobiocin (5, 10, 15, 25, and 35 μg ml−1), rifampin (25, 50, 100, and 150 μg ml−1), isoniazid (2, 5, 10, 15, and 20 μg ml−1), and chloramphenicol (5, 15, 25, and 50 μg ml−1). Fifteen microliters (1 × 108 cells per ml) of serial 10-fold dilutions of actively growing cultures was plated and incubated at 37°C for 3 days. The MIC of each antibiotic, defined as the minimum concentration required to inhibit 99% of growth, was estimated.
RESULTS
Selection of MSMEG_0220.
In a previous study (7), Rv0183 was the first monoacylglycerol lipase from M. tuberculosis H37Rv to be identified. This enzyme has homologues in M. leprae (ML2603), which is believed to contain only enzymes necessary for its growth in host cells, and in the saprophytic M. smegmatis strain mc2155 (MSMEG_0220). An amino acid sequence identity of 68% was observed between Rv0183 and MSMEG_0220. Two common lipase motifs, the GXSXG pentapeptide and the His-Gly dipeptide, and the secondary structure analysis of MSMEG_0220 indicate that, like Rv0183, this protein belongs to the α/β fold family. These bioinformatic data suggest that these two proteins may have similar enzymatic activities. In addition, immunolocalization studies showed that Rv0183 and MSMEG_0220 are located in the same cell fractions (see “Complementation of the MSMEG_0220 disrupted mutant” below). All of these findings suggest that Rv0183 and MSMEG_0220 may be involved in the same metabolic pathway and have similar physiological functions.
Production and purification of rMSMEG_0220.
To determine the putative role of MSMEG_0220, the recombinant protein (rMSMEG_0220) was expressed in the form of a His6-tagged fusion protein in E. coli Rosetta(DE3)/pLysS under induction at 25°C for 20 h with IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM). After its purification, this protein was found unstable in 10 mM Tris-HCl buffer (pH 8.5) containing 150 mM NaCl (i.e., under the conditions previously used for the purification of rRv0183). This instability consisted of a decrease in the enzyme activity during the purification process and precipitation probably due to protein aggregation. In fact, rMSMEG_0220 precipitated at concentrations above 0.1 mg ml−1. To improve the stability of rMSMEG_0220, various pH values ranging from 7.0 to 9.0 were tested during the purification procedure, with little success. Solvents such as isopropanol, glycerol, and MPD were then added in various proportions (5, 10, and 20% [vol/vol]) to the purification buffer to stabilize rMSMEG_0220. This protein showed maximum stability in buffer containing 10% (vol/vol) MPD. However, addition of MPD to the buffer used for the Ni2+-NTA agarose column step resulted in a very low recovery yield (10%) of active protein. MPD was therefore added only to the tubes used for collecting fractions eluted from the column to prevent the proteins from precipitating. Under these conditions, between 4 and 5 mg of pure rMSMEG_0220 was purified from the 7 mg obtained per liter of culture (giving a yield of 57 to 71%). The His6 tag at the N-terminal end was removed, with a yield of about 75%, by performing TEV cleavage overnight at 4°C, followed by exclusion from a Ni2+-NTA agarose column. A molecular mass of about 30 kDa was estimated from the migration of purified rMSMEG_0220 without the His6 tail upon performance of SDS-PAGE (Fig. 1, lane 2). The His6 tag cleavage was confirmed by the N-terminal sequencing of the recombinant protein. Lastly, rMSMEG_0220 was concentrated and stored at −80°C.
FIG. 1.
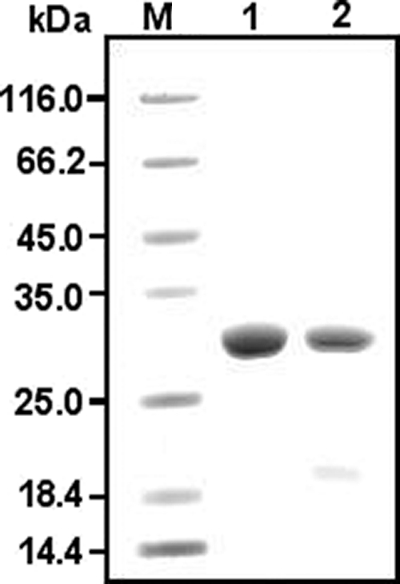
SDS-PAGE analysis of purified rRv0183 and rMSMEG_0220 after TEV digestion. Protein samples were loaded onto a 12% SDS-polyacrylamide gel under reducing conditions. Lane 1, 20 μg of rRv0183; lane 2, 15 μg of rMSMEG_0220; lane M, molecular mass markers (Fermentas).
Biochemical properties of rMSMEG_0220 and comparison with those of its homologue rRv0183.
Biochemical characterization studies were performed with rMSMEG_0220, and the results were compared with those obtained for rRv0183. Since rMSMEG_0220 was purified in a buffer containing 10% (vol/vol) MPD, this solvent was also added to the purified rRv0183 (Fig. 1, lane 1) in order to work under the same conditions; since MPD might have affected the enzyme activity, we checked that this solvent did not change the enzyme activity of rRv0183 (Table 1). We also checked that the recombinant MSMEG_0220 enzyme with its catalytic serine (S111) mutated into an alanine residue was inactive whatever the substrate used.
Substrate specificity.
In order to study the substrate specificity of rMSMEG_0220, measurements of activity on several substrates were performed using the pH-Stat technique. Like rRv0183, rMSMEG_0220 was inactive on emulsified TAG and phospholipids, whatever the substrate chain length (C2, C4, C8, or C18). In contrast to rRv0183, which shows activity on dioctanoin but not on long-chain diacylglycerol (DAG), rMSMEG_0220 does not hydrolyze any DAG (Table 1), even in the presence of gum arabic and NaTDC. rMSMEG_0220 was found to be active on monoacylglycerols (MAG), like rRv0183. However, rMSMEG_0220 hydrolyzes only MAG with fatty acid chains ranging from C14 to C18, whereas rRv0183 showed its maximum specific activity on monooctanoin (Table 1). When monoolein was used as the substrate, the specific activity recorded was 143 ± 6 U mg−1 with rMSMEG_0220, compared to 300 ± 7 U mg−1 with rRv0183. The hydrolysis of Tween 80, a detergent added systematically to the culture medium and used for detecting lipase/esterase activity, was investigated. Neither rRv0183 nor rMSMEG_0220 showed any activity toward this substrate. Based on these results, it was concluded that rMSMEG_0220 is a monoacylglycerol lipase, like rRv0183. In the subsequent experiments, monoolein was used as the standard substrate for the enzyme activity measurements.
Effects of pH and NaTDC on rMSMEG_0220 and rRv0183 activities.
As observed in the case of rRv0183, the specific activity of rMSMEG_0220 does not exceed 35 U mg−1 on monoolein in the absence of detergent in the reaction mixture. This specific activity was enhanced by supplementing the reaction mixture with NaTDC. Indeed, the specific activity of rMSMEG_0220 determined at 3 mM NaTDC was 4 times higher than that measured in the absence of this detergent. The pH dependency profile showed that, like rRv0183, rMSMEG_0220 is a basic enzyme, showing maximum activity at pH 8.0 (Fig. 2 A).
FIG. 2.

pH dependence activity (A) and pH stability (B) of rMSMEG_0220 (▵) and rRv0183 (⋄) when monoolein was used as the substrate. (A) Activity was measured at various pH values using the back titration method. (B) Activity recorded after 1 h of incubation at several pH values versus the activity measured under the optimum conditions. Values are the average results from 3 independent assays, with error bars indicating standard deviations.
Effect of pH on rMSMEG_0220 and rRv0183 stability.
The optimum pH for rMSMEG_0220 and rRv0183 stability was determined by measuring the residual activities of these enzymes after incubation for 1 h at various pH values ranging from 4.0 to 9.0 (Fig. 2B). rMSMEG_0220 and rRv0183 are highly stable from pH 6.5 to 9.0. rRv0183 is largely inactivated at pH 5 and below, in the presence or absence of MPD (7), whereas rMSMEG_0220 still shows a residual activity of 50% after 1 h of incubation at pH 4.0 (Fig. 2B). These results indicate that rMSMEG_0220 and rRv0183 exhibit similar biochemical behaviors, especially in regard to activity and specificity for MAG. This finding was therefore in line with the hypothesis that these enzymes may play similar physiological roles.
Disruption of the MSMEG_0220 gene in the M. smegmatis mc2155 strain.
In order to identify the physiological role of the monoacylglycerol lipase MSMEG_0220, an M. smegmatis mc2155 mutant strain was constructed by disrupting the MSMEG_0220 gene. The MSMEG_0220 mutant was obtained by exchanging the wild-type allele of MSMEG_0220 with the MSMEG_0220 gene disrupted by inserting a Km resistance cassette allele provided by the thermosensitive pRD222 plasmid (Fig. 3 A). After electroporation of the M. smegmatis wild-type strain and a selection procedure, more than 200 colonies were grown on Km-sucrose plates, and 42 putative mutants were further analyzed by PCR. The genomic DNA of these clones could contain either the inactivated MSMEG_0220 allele introduced after a double crossover or the entire plasmid pRD222, which would result from a single recombination event and a spontaneous resistance to sucrose. To address the question of whether the mutant strains carried the inactivated MSMEG_0220 allele, genomic DNA from putative mutant and wild-type strains was extracted and amplified by PCR. As expected with the wild-type DNA, a fragment of about 1 kb and two other fragments of about 1.3 kb were obtained when P5/P6, P3/P6, and P5/P4, respectively, were used as primer pairs (Fig. 3B). Genomic DNA from two putative mutants out of 42 yielded 2-kb and two 2.4-kb PCR products when the same respective primer pairs were used (Fig. 3B). These fragment sizes corresponded to those calculated for the disrupted MSMEG_0220 gene. The last two clones were therefore both mutants with the MSMEG_0220 gene disruption and were selected for further analysis; the results obtained with a single positive clone, named MsΔ0220, are presented throughout this paper.
FIG. 3.

Generation of an MSMEG_0220 disrupted mutant of M. smegmatis. (A) Schematic diagram of the MSMEG_0220 disrupted gene construct. The black boxes and those with horizontal dashes depict the gene sequence of MSMEG_0220 and the flanking regions used to obtain the disrupted construct, respectively. The part of the MSMEG_0220 gene replaced by the Km gene cassette (gray box) is shown as a hatched box. The primer pairs P3/P6, P5/P4, and P5/P6 were used to confirm the MSMEG_0220 gene disruption. The predicted sizes of the PCR products are indicated in each case. (B) PCR analysis of the wild-type mc2155 (lanes 1, 3, and 5) and recombinant MsΔ0220 (lanes 2, 4, and 6) strains, carried out using various combinations of specific primers (see Table S1 in the supplemental material). M, 1-kb DNA ladder (Promega).
To confirm the disruption of MSMEG_0220 gene expression, protein extracts from the MsΔ0220 strain were analyzed by performing immunoblotting assays using purified polyclonal antibodies directed against rRv0183 (7). These antibodies cross-reacted with the purified rMSMEG_0220, as expected for these two enzymes, which share 68% amino acid sequence identity (Fig. 4, lanes 1 and 6), allowing their direct use to screen for the MSMEG_0220 protein. With protein extracts from the MsΔ0220 strain, no recognition was observed, in contrast to what occurred with the protein extracts from the wild-type strain (Fig. 4, lanes 2 and 3), establishing the disruption of MSMEG_0220 gene expression.
FIG. 4.

Immunoblotting analysis of MSMEG_0220 production. The cytosol containing intracellular nonsoluble proteins (SN2) that was obtained after cell fractionation was subjected to immunoblotting procedures using the purified polyclonal antibodies raised against rRv0183. Except for the purified proteins, the same amount of protein (120 μg) was loaded for each sample. Lane 1, Rv0183 (100 ng); lane 2, SN2 from wild-type mc2155 cells; lane 3, SN2 from MsΔ0220 cells; lane 4, SN2 from ComMsΔ0220 cells; lane 5, SN2 from ComMsΔ0220S111A cells; lane 6, rMSMEG_0220 purified protein (100 ng); lane M, molecular mass markers (Fermentas).
Complementation of the MSMEG_0220 disrupted mutant.
The E. coli/Mycobacterium shuttle vectors pRD223 and pRD224 were used to restore MSMEG_0220 gene expression and protein production in MsΔ0220. Two clones showing Km and Hyg resistance conferred by these vectors were selected and called, respectively, ComMsΔ0220 and ComMsΔ0220S111A. Production of recombinant proteins in the complemented mutants was checked by performance of immunoblotting assays using the purified polyclonal antibodies raised against rRv0183. Cell cultures of M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains were harvested, and after the cells were broken, two fractions, SN1 (the cytosol that contains intracellular soluble proteins) and SN2 (the cytosol that contains intracellular nonsoluble and cell envelope proteins), were obtained (see Materials and Methods). Aliquots (≈120 μg of total protein) of these two fractions were loaded onto 12% SDS-polyacrylamide gels to screen for the expression of MSMEG_0220. As mentioned above, no MSMEG_0220 production was detected in the SN1 and SN2 protein fractions of MsΔ0220 cells. In contrast, MSMEG_0220 protein was detected in the SN2 fractions obtained from M. smegmatis wild-type, ComMsΔ0220, and ComMsΔ0220S111A cells (Fig. 4, lanes 2, 4, and 5). Comparable levels of MSMEG_0220 expression seem to occur in the ComMsΔ0220, ComMsΔ0220S111A, and wild-type strains. The MSMEG_0220 protein was also recognized in the SN1 fractions from the strains (data not shown). These findings clearly suggest that MSMEG_0220 was expressed in the complemented mutant, as observed in the wild-type strain. Furthermore, MSMEG_0220 protein was not detected in the culture medium of M. smegmatis wild-type, ComMsΔ0220, and ComMsΔ0220S111A strains, which suggests that MSMEG_0220 was likely an intracellular protein with a putative predilection for the cell envelope. This result is in agreement with those obtained previously in the case of Rv0183 (7).
Study of the physiological role of MSMEG_0220. (i) MSMEG_0220 disruption causes major phenotype alterations.
Intriguingly, the disruption of the MSMEG_0220 gene affected the morphology of the colony (Fig. 5 A and B). When grown on 7H11 agar plates, MsΔ0220 colonies had a very smooth, wet, and round appearance (Fig. 5B). This phenotype is in stark contrast to the dry, rough, and irregular morphology of the colonies of the wild-type mc2155 strain (Fig. 5A). Like those of the wild-type strain, the ComMsΔ0220 and ComMsΔ0220S111A colonies were irregular and rough (Fig. 5C and D). When the four strains were grown in 7H9 broth that did not contain Tween 80, the MsΔ0220 strain showed a homogeneous suspension culture, with individualized cells (Fig. 5F), whereas the wild-type mc2155, the ComMsΔ0220, and the ComMsΔ0220S111A strains showed a strong pattern of cell aggregation (Fig. 5E, G, and H). All of these changes suggested that the mycobacterial cell envelope could be modified and that cell aggregation was impaired upon MSMEG_0220 gene disruption. To test this hypothesis, cell wall lipids from the wild-type, MsΔ0220, and ComMsΔ0220 strains were extracted and analyzed both by TLC and by MALDI-TOF mass spectrometry (see Fig. S1 in the supplemental material). TLC analysis showed that the glycopeptidolipid (GPL) compositions in the wild-type, MsΔ0220, and ComMsΔ0220 strains were very similar (see Fig. S1A in the supplemental material). MALDI-TOF mass spectrometry analysis confirmed and extended these similarities by showing that these strains produced the same types of GPLs (see Fig. S1B in the supplemental material). Furthermore, TLC and MALDI-TOF mass spectrometry analyses of cell wall-bound mycolic acids and polar and apolar lipids of these three strains were strictly similar (data not shown). Therefore, the morphological changes observed in MsΔ0220 were not due to a change in the lipid composition and contents of the cell wall.
FIG. 5.

Growth characteristics of M. smegmatis wild-type (A and E), MsΔ0220 (B and F), ComMsΔ0220 (C and G), and ComMsΔ0220S111A (D and H) strains. (A to D) Five-day-old colonies of M. smegmatis strains on 7H11 solid medium, viewed from the top of the colony, showing rough (A, C, and D) and smooth (B) morphologies. (E to H) Optical microscopy (magnification, ×63) observation of M. smegmatis strains grown in Tween 80-free 7H9 broth. M. smegmatis wild-type (E), ComMsΔ0220 (G), and ComMsΔ0220S111A (H) strains developed cell aggregates, while MsΔ0220 bacteria (F) showed a homogeneous cell suspension with no aggregation.
(ii) Growth experiments in broth culture and solid medium.
Growth of M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains was monitored by measuring the absorbance at 600 nm at least every 3 h for 3 days. In a 7H9 broth containing ADC enrichment, these four strains showed similar patterns of growth. This finding suggests that the disruption of MSMEG_0220 did not affect cell growth under these conditions.
Since biochemical data proved that MSMEG_0220 is a monoacylglycerol lipase located in the cell wall, growth of M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains was studied first on Middlebrook 7H9 broth medium containing 0.02% monoolein and 0.05% Tween 80 to test the ability of these strains to hydrolyze this exogenous lipid. With stirring, the culture medium became a lipid emulsion in the presence of Tween 80. Under these conditions, it was difficult to assess the cellular growth and to observe a difference among all strains. Moreover, when strains were cultured in the absence of Tween 80, an inhomogeneous medium was obtained and the numbers of CFU were irreproducible. Consequently, M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains were grown on Middlebrook 7H9 agar supplemented or not supplemented with 0.02% (or 1%) monoolein and 0.05% Tween 80. The log10 CFU was calculated after 3 to 5 days of incubation at 37°C. When these four strains were grown on 7H9 agar plus monoolein, differences in the growth patterns were observed. The CFU value calculated in the case of the MsΔ0220 strain was about 2 log lower than that obtained with the M. smegmatis wild-type strain under the same conditions (Fig. 6). The mutant complemented with an active MSMEG_0220 protein recovered the ability to grow on monoolein to the same magnitude as the wild type, contrary to the mutant complemented with an inactive protein, which behaves like the MsΔ0220 strain (Fig. 6). Therefore, the MSMEG_0220 protein seems to be important to ensure M. smegmatis cell growth in the presence of monoolein. In contrast, all strains showed similar log10 CFU values (9 to 9.5) when grown on Middlebrook 7H9 agar supplemented with monoolein and Tween 80 (Fig. 6), as well as on 7H9 agar supplemented by either triacylglycerols (tributyrin and olive oil) or oleic acid (data not shown).
FIG. 6.

Assessment of growth of M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains on Middlebrook 7H9 agar supplemented with monoolein (MO) or with MO and Tween 80 (T 80). The log10 CFU were calculated for all four strains growing in different agar plates. Error bars indicate standard deviations.
Antimicrobial susceptibility.
Antimicrobial susceptibility was tested to determine whether the mycobacterial envelope permeability was modified and could support the change in the architecture of the cell envelope. Accordingly, M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains were grown on solid media with or without antibiotics. When novobiocin or chloramphenicol was used, the strains showed the same MIC value, 25 μg ml−1 or 15 μg ml−1, respectively. Interestingly, the MsΔ0220 strain was more sensitive to rifampin than the wild type (Fig. 7). The MIC value determined for this mutant was about 50 μg ml−1, versus 100 μg ml−1 for the wild type. In contrast, when isoniazid was used as the drug, the MsΔ0220 strain exhibited a slight resistance in comparison to the wild-type strain. In fact, the determined MIC values were 10 μg ml−1 and 15 μg ml−1 for the wild-type and the MsΔ0220 strains, respectively. In each case, complementation of the mutant with an active or inactive MSMEG_0220 protein restored the drug susceptibility to the same MIC values as for the WT.
FIG. 7.

Antimycobacterial effect of rifampin against M. smegmatis wild-type and MsΔ0220 strains. The susceptibilities of these strains were determined on Middlebrook 7H11 containing increasing inhibitor concentrations. Serial 10-fold dilutions of 1 × 108 cells per ml (indicated on the plates) were plated.
DISCUSSION
Expression and purification of rMSMEG_0220.
Despite a high amino acid sequence identity between MSMEG_0220 and Rv0183, their expression levels in E. coli Rosetta (DE3)/pLysS differed. Three times more rRv0183 protein was produced per liter of cell culture than rMSMEG_0220. This low production level of active rMSMEG_0220 might be due to the instability observed during the purification process and the storage of the pure protein. To overcome the instability of rMSMEG_0220, which appeared to be due mainly to protein aggregation and precipitation, the effects of some alcohols, such as glycerol, MPD, and isopropanol, on enzyme stability were assessed. Organic solvents are known to minimize the interactions between protein molecules and thus prevent protein precipitation (1, 5). In our case, MPD at a final concentration of 10% (vol/vol) improved the stability of the protein rMSMEG_0220 in terms of enzyme activity.
Biochemical characterization and immunolocalization.
rMSMEG_0220 showed activity only on monoacylglycerols, with a maximum activity on monoolein, which was twice as high as that on monomyristin. In comparison, the rRv0183 enzyme showed its maximum activity on monoacylglycerol but was also active on medium-chain diacylglycerol and vinyl esters. rMSMEG_0220, therefore, shows a greater substrate specificity than rRv0183 and can be considered a monoacylglycerol lipase. This conclusion is in line with predictions about the MSMEG_0220 protein, which was annotated as a monoacylglycerol lipase from sequence analysis (http://cmr.jcvi.org/tigr-scripts/CMR/shared/GenePage.cgi?locus=MSMEG_0220). Furthermore, rMSMEG_0220 was more stable at acidic pH values than rRv0183, in that rMSMEG_0220 still showed 50% of its initial activity after being incubated for 1 h at pH 4.0, whereas rRv0183 was entirely inactivated at this pH value. The MPD was not responsible for the enzyme stability observed in this experiment, since rRv0183 was also prepared in a buffer containing this solvent. The stability of rMSMEG_0220 observed at acidic pH levels may therefore be an intrinsic property of this enzyme. In addition, with antibodies directed against rRv0183, which cross-react with rMSMEG_0220, MSMEG_0220 was found to be located in the cell envelope of M. smegmatis, like Rv0183 in M. tuberculosis. As MSMEG_0220 and Rv0183 show similar patterns of biochemical behavior and cell localization, these two enzymes may play similar physiological roles. Therefore, MSMEG_0220 is clearly a good surrogate for exploring the functional role of Rv0183, based on the use of an MSMEG_0220 disrupted mutant.
Disruption of the MSMEG_0220 gene in M. smegmatis and complementation.
The frequency of recombination between the MSMEG_0220 allele and the Km resistance cassette disrupted allele was increased when a thermosensitive vector was used (31). By use of this procedure, a single or double crossover could occur. Only two clones out of the 42 screened, which were denoted MsΔ0220, showed the disrupted MSMEG_0220::Km phenotype, giving a very low yield of mutagenesis in comparison with the results obtained by Pelicic et al., who obtained 100% positive allelic exchange mutants of the M. tuberculosis purC gene (31). Immunoblotting analysis unambiguously confirmed that the two clones selected were generated from an allelic exchange event. The pVV16 expression vector used to complement the MSMEG_0220 mutation allowed restoration of an MSMEG_0220 expression level in the complemented mutant similar to that of the wild-type strain. Moreover, immunolocalization studies showed that the MSMEG_0220 protein produced by the complemented mutants (ComMsΔ0220 and ComMsΔ0220S111A) was properly localized like the native protein produced by the M. smegmatis wild-type strain.
Study of the physiological role of MSMEG_0220.
The disruption of the MSMEG_0220 gene led to a shift from a rough to a smooth morphology. The complementation with either an active or an inactive MSMEG_0220 protein almost completely restored the wild-type phenotype, which confirms that this alteration was directly due to the MSMEG_0220 disruption and suggests a structural role of the protein in the morphological change. In addition, the MsΔ0220 cell culture showed a homogenous cell suspension in Tween 80-free liquid medium, unlike M. smegmatis wild-type, ComMsΔ0220, and ComMsΔ0220S111A cell cultures, in which cell aggregates were observed. Previous authors have attributed this kind of change to an alteration in the lipid composition of the mycobacterium cell envelope having an impact on cell hydrophobicity (2, 11, 18, 30, 34). The conversion from a rough to a smooth appearance may be due to an overproduction of glycopeptidolipids on the cell surface (22) or a lack of trehalose dimycolate (30). Contrary to our expectations, the MsΔ0220 cell envelope exhibited the same GPL, mycolic acid, and polar and apolar lipid contents as the M. smegmatis wild-type and ComMsΔ0220 cell envelopes. However, the observed changes in colony morphology may be a consequence of an organizational change in this envelope. Indeed, the presence of the MSMEG_0220 protein, in an active or inactive form, is sufficient to restore a colony morphology comparable to that of the wild-type strain. Therefore, the hypothesis of a change in organization of the envelope was checked by testing cell envelope permeability (27). The mycobacterial cell envelope forms an effective barrier, preventing the passage of both hydrophilic and hydrophobic compounds, and any change in its architecture will impact its permeability (14). Hydrophilic (isoniazid and chloramphenicol) and hydrophobic (novobiocin and rifampin) antimicrobial compounds were therefore used to test the drug sensitivity profiles of M. smegmatis strains. It has been proposed that hydrophobic antibiotics may cross the cell wall by diffusion through the hydrophobic bilayer composed of mycolic acids and glycolipids, while hydrophilic antibiotics are thought to use porin channels for penetrating the cells (25). The MsΔ0220 strain did not show the same behavior toward hydrophilic and/or hydrophobic compounds as the complemented and wild-type strains. In fact, the MsΔ0220 strain was slightly more sensitive to rifampin and more resistant to isoniazid than the other strains. However, all strains responded in the same way to chloramphenicol and novobiocin. The absence of MSMEG_0220 in the cell envelope may have led to the alteration in the cell envelope architecture. This hypothesis was strengthened by results obtained with the complemented ComMsΔ0220S111A mutant strain suggesting that the envelope modification is due mainly to the presence or absence of the protein and not related to the enzyme activity. The same observation has been reported in the case of the Erp protein in M. smegmatis, which is anchored to the cell wall and might play an important role in the cell wall structure (21).
The analysis of growth of the M. smegmatis wild-type, MsΔ0220, ComMsΔ0220, and ComMsΔ0220S111A strains on medium containing monoolein as the sole carbon source indicated that MSMEG_0220 contributes to cell growth, probably by hydrolyzing exogenous lipids. Moreover, the growth of the MsΔ0220 strain on monoolein was not completely abolished, suggesting that this protein may be important but not essential. Finally, the ability of MsΔ0220 to grow on monoolein could be explained by the production of one or several lipases able to hydrolyze not only monoacylglycerols but also Tween 80, since the addition of this substrate restored the default growth of MsΔ0220 and ComMsΔ0220S111A. When agar plates were supplemented with triacylglycerols, whatever the chain length of the fatty acids, the growth patterns of all mutant strains as well as the wild type were similar. Once again, these data are in agreement with the biochemical data showing that MSMEG_0220 is a specific monoacylglycerol lipase and suggest the presence in the cell wall and/or the excretion of another lipolytic enzyme able to use TAG as the carbon source.
Rv0183 may show activity similar to that of MSMEG_0220 and be involved in the hydrolysis of exogenous host lipids during chronic infection in association with other lipolytic enzymes that generate monoacylglycerols. Since mycobacteria might encounter triacylglycerol mainly under physiological conditions (29, 32), one hypothesis can be made that, like some mammalian cells (e.g., adipocytes), mycobacteria can produce several enzymes whose activities occur sequentially on triacylglycerol, with the monoacylglycerol lipase being the last enzyme involved. Monoacylglycerol degradation provides free fatty acids that can be used as a carbon source by M. tuberculosis. Finally, the morphology change observed in the disrupted mutant MsΔ0220 is encouraging, as the phenotype change may affect the stability of the strain inside macrophages. As M. smegmatis is not a virulent mycobacterial species, being degradable by macrophages, the stability of the M. tuberculosis Rv0183 and M. bovis BCG BCG_0220 gene mutants in immune cells is under investigation.
Supplementary Material
Acknowledgments
This research was funded by GIP ANR 06-JCJC-0067-01.
We thank Karen Côtes for her contribution in the early stages of the work and Régine Lebrun (IMM, Marseille) for performing the N-terminal sequencing. We are indebted to Jessica Blanc for revising the English manuscript.
R. Dhouib was the recipient of a Ph.D. fellowship from CNRS (BDI-PED, CNRS).
Footnotes
Published ahead of print on 2 July 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Asakura, T., K. Adachi, and E. Schwartz. 1978. Stabilizing effect of various organic solvents on protein. J. Biol. Chem. 253:6423-6425. [PubMed] [Google Scholar]
- 2.Bhatt, A., N. Fujiwara, K. Bhatt, S. S. Gurcha, L. Kremer, B. Chen, J. Chan, S. A. Porcelli, K. Kobayashi, G. S. Besra, and W. R. Jacobs, Jr. 2007. Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc. Natl. Acad. Sci. U. S. A. 104:5157-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boshoff, H. I., and C. E. Barry. 2005. A low-carb diet for a high-octane pathogen. Nat. Med. 11:599-600. [DOI] [PubMed] [Google Scholar]
- 4.Brennan, P., and H. Nikaido. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29-63. [DOI] [PubMed] [Google Scholar]
- 5.Canaan, S., L. Dupuis, M. Rivière, K. Faessel, J. L. Romette, R. Verger, and C. Wicker-Planquart. 1998. Purification and interfacial behavior of recombinant human gastric lipase produced from insect cell in a bioreactor. Protein Expr. Purif. 14:23-30. [DOI] [PubMed] [Google Scholar]
- 6.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry, III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, B. G. Barrell, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 7.Côtes, K., R. Dhouib, I. Douchet, H. Chahinian, A. de Caro, F. Carriere, and S. Canaan. 2007. Characterization of an exported monoglyceride lipase from Mycobacterium tuberculosis possibly involved in the metabolism of host cell membrane lipids. Biochem. J. 408:417-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daffe, M., and P. Draper. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131-203. [DOI] [PubMed] [Google Scholar]
- 9.Daniel, J., C. Deb, V. S. Dubey, T. D. Sirakova, B. Abomoelak, H. R. Morbidoni, and P. E. Kolattukudy. 2004. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 186:5017-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deb, C., J. Daniel, T. D. Sirakova, B. Abomoelak, V. S. Dubey, and P. E. Kolattukudy. 2006. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J. Biol. Chem. 281:3866-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Etienne, G., C. Villeneuve, H. Billman-Jacobe, C. Astarie-Dequeker, M. A. Dupont, and M. Daffe. 2002. The impact of the absence of glycopeptidolipids on the ultrastructure, cell surface and cell wall properties, and phagocytosis of Mycobacterium smegmatis. Microbiology 148:3089-3100. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez, S., V. Jannin, J. D. Rodier, N. Ritter, B. Mahler, and F. Carriere. 2007. Comparative study on digestive lipase activities on the self emulsifying excipient Labrasol, medium chain glycerides and PEG esters. Biochim. Biophys. Acta 1771:633-640. [DOI] [PubMed] [Google Scholar]
- 13.Garton, N., H. Christensen, D. Minnikin, R. Adegbola, and M. Barer. 2002. Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148:2951-2958. [DOI] [PubMed] [Google Scholar]
- 14.Gebhardt, H., X. Meniche, M. Tropis, R. Kramer, M. Daffe, and S. Morbach. 2007. The key role of the mycolic acid content in the functionality of the cell wall permeability barrier in Corynebacterineae. Microbiology 153:1424-1434. [DOI] [PubMed] [Google Scholar]
- 15.Gershoni, J. M., and G. E. Palade. 1982. Electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to a positively charged membrane filter. Anal. Biochem. 124:396-405. [DOI] [PubMed] [Google Scholar]
- 16.Goldman, R. C., K. V. Plumley, and B. E. Laughon. 2007. The evolution of extensively drug resistant tuberculosis (XDR-TB): history, status and issues for global control. Infect. Disord. Drug Targets 7:73-91. [DOI] [PubMed] [Google Scholar]
- 17.Honer zu Bentrup, K., and D. G. Russell. 2001. Mycobacterial persistence: adaptation to a changing environment. Trends Microbiol. 9:597-605. [DOI] [PubMed] [Google Scholar]
- 18.Howard, S. T., E. Rhoades, J. Recht, X. Pang, A. Alsup, R. Kolter, C. R. Lyons, and T. F. Byrd. 2006. Spontaneous reversion of Mycobacterium abscessus from a smooth to a rough morphotype is associated with reduced expression of glycopeptidolipid and reacquisition of an invasive phenotype. Microbiology 152:1581-1590. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs, W. R. 2000. Mycobacterium tuberculosis: a once genetically intractable organism, p. 1-16. In G. F. Hatfull and W. R. Jacobs (ed.), Molecular genetics of mycobacteria. ASM Press, Washington, DC.
- 20.Kaufmann, S. H. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1:20-30. [DOI] [PubMed] [Google Scholar]
- 21.Kocincova, D., B. Sonden, L. de Mendonca-Lima, B. Gicquel, and J. M. Reyrat. 2004. The Erp protein is anchored at the surface by a carboxy-terminal hydrophobic domain and is important for cell-wall structure in Mycobacterium smegmatis. FEMS Microbiol. Lett. 231:191-196. [DOI] [PubMed] [Google Scholar]
- 22.Kocincova, D., N. Winter, D. Euphrasie, M. Daffe, J. M. Reyrat, and G. Etienne. 2009. The cell surface-exposed glycopeptidolipids confer a selective advantage to the smooth variants of Mycobacterium smegmatis in vitro. FEMS Microbiol. Lett. 290:39-44. [DOI] [PubMed] [Google Scholar]
- 23.Kremer, L., C. de Chastellier, G. Dobson, K. J. Gibson, P. Bifani, S. Balor, J. P. Gorvel, C. Locht, D. E. Minnikin, and G. S. Besra. 2005. Identification and structural characterization of an unusual mycobacterial monomeromycolyl-diacylglycerol. Mol. Microbiol. 57:1113-1126. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 25.Lambert, P. A. 2002. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J. Appl. Microbiol. 92(Suppl.):46S-54S. [PubMed] [Google Scholar]
- 26.Laval, F., R. Haites, F. Movahedzadeh, A. Lemassu, C. Y. Wong, N. Stoker, H. Billman-Jacobe, and M. Daffe. 2008. Investigating the function of the putative mycolic acid methyltransferase UmaA: divergence between the Mycobacterium smegmatis and Mycobacterium tuberculosis proteins. J. Biol. Chem. 283:1419-1427. [DOI] [PubMed] [Google Scholar]
- 27.McDonnell, G., and A. D. Russell. 1999. Antiseptics and disinfectants: activity, action, and resistance. Clin. Microbiol. Rev. 12:147-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra, K. C., C. de Chastellier, Y. Narayana, P. Bifani, A. K. Brown, G. S. Besra, V. M. Katoch, B. Joshi, K. N. Balaji, and L. Kremer. 2008. Functional role of the PE domain and immunogenicity of the Mycobacterium tuberculosis triacylglycerol hydrolase LipY. Infect. Immun. 76:127-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neyrolles, O., R. Hernandez-Pando, F. Pietri-Rouxel, P. Fornes, L. Tailleux, J. A. Payan, E. Pivert, Y. Bordat, D. Aguilar, M. C. Prevost, C. Petit, and B. Gicquel. 2006. Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLoS One 1:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen, L., S. Chinnapapagari, and C. J. Thompson. 2005. FbpA-dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J. Bacteriol. 187:6603-6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelicic, V., M. Jackson, J. M. Reyrat, W. R. Jacobs, Jr., B. Gicquel, and C. Guilhot. 1997. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:10955-10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peyron, P., J. Vaubourgeix, Y. Poquet, F. Levillain, C. Botanch, F. Bardou, M. Daffe, J. F. Emile, B. Marchou, P. J. Cardona, C. de Chastellier, and F. Altare. 2008. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4:e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portevin, D., W. Malaga, and C. Guilhot. 2009. The use of temperature-sensitive plasmids in mycobacteria. Methods Mol. Biol. 465:229-242. [DOI] [PubMed] [Google Scholar]
- 34.Recht, J., and R. Kolter. 2001. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J. Bacteriol. 183:5718-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simeone, R., P. Constant, W. Malaga, C. Guilhot, M. Daffe, and C. Chalut. 2007. Molecular dissection of the biosynthetic relationship between phthiocerol and phthiodiolone dimycocerosates and their critical role in the virulence and permeability of Mycobacterium tuberculosis. FEBS J. 274:1957-1969. [DOI] [PubMed] [Google Scholar]
- 36.Villeneuve, C., G. Etienne, V. Abadie, H. Montrozier, C. Bordier, F. Laval, M. Daffe, I. Maridonneau-Parini, and C. Astarie-Dequeker. 2003. Surface-exposed glycopeptidolipids of Mycobacterium smegmatis specifically inhibit the phagocytosis of mycobacteria by human macrophages. Identification of a novel family of glycopeptidolipids. J. Biol. Chem. 278:51291-51300. [DOI] [PubMed] [Google Scholar]
- 37.Zechner, R., P. C. Kienesberger, G. Haemmerle, R. Zimmermann, and A. Lass. 2009. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 50:3-21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.