

Summary
Retroviruses are well known for their ability to incorporate envelope proteins from other retroviral strains and genera and even from other virus families. This characteristic has been widely exploited for the generation of replication-defective retroviral vectors, including those derived from murine leukemia virus (MLV), bearing heterologous envelope proteins. We desired to investigate the possibility of “genetically” pseudotyping replication-competent MLV by replacing the native env gene in a full-length viral genome with that of another gammaretrovirus. We previously developed replication-competent versions of MLV that stably transmit and express transgenes inserted in the 3′ untranslated region of the viral genome. In one such tagged MLV expressing green fluorescent protein, we replaced the native env sequence with that of gibbon ape leukemia virus (GALV). Although the GALV Env protein is commonly used to make high titer pseudotypes of MLV vectors, we found that the env replacement greatly attenuated viral replication. However, passage of cells exposed to the chimeric virus resulted in selection of mutants exhibiting rapid replication kinetics and different variants arose in different infections. Two of these variants had acquired mutations at or adjacent to the splice acceptor site and three others had acquired dual mutations within the long terminal repeat. Analysis of the levels of unspliced and spliced viral RNA produced by the parental and adapted viruses showed that the mutations gained by each of these variants functioned to reverse an imbalance in splicing caused by the env gene substitution. Our results reveal the presence of previously unknown cis-acting sequences in MLV that modulate splicing of the viral transcript and demonstrate that tagging of the retroviral genome with an easily assayed transgene can be combined with in vitro evolution to efficiently generate and screen for replicating mutants of replication-impaired recombinant viruses.
Keywords: retrovirus, murine leukemia virus, envelope, evolution, splicing
Introduction
The genomic RNA of simple retroviruses such as murine leukemia virus (MLV) encodes three genes - gag, pol, and env - which are each indispensable for replication. The Gag and Pol proteins are translated from the full-length, unspliced transcript, which also serves as the packaged and transmitted genome. A fraction of the transcript undergoes a splicing event that places the env open reading frame near the 5′ cap, permitting translation of the surface and transmembrane subunits of the envelope (Env) protein, which mediates the binding and entry of the virus into the host cell through interaction with cognate receptors on the cell surface. Retroviruses have evolved Env proteins that utilize a variety of cellular receptors 1, and retrovirus particles can functionally incorporate envelope proteins from other retroviral strains and genera, and even from other virus families, when expressed in the same cell. These properties have been extensively exploited to generate pseudotyped replication-defective retroviral vectors that exhibit redirected or broadened tropism and greater biophysical stability 2. Furthermore, “genetically pseudotyped” infectious retroviruses can arise during infection by natural recombination events that result in replacement of env sequences with those of a co-infecting or endogenous virus 3; 4 and such env gene alterations can play an important role in virulence or pathogenicity and cross-species transmissibility.
The construction of chimeras between different strains, species or genera of retroviruses represents a useful strategy for investigating retrovirus biology, and a large number of such chimeras have been reported previously 5; 6; 7; 8; 9; 10. However, the production of replication-competent chimeric viruses requires that not only the proteins encoded by the heterologous sequence functionally substitute for any replaced proteins, but also that the sequence within the viral genome not itself impair viral replication. In fact, artificially constructed chimeric retroviruses are very often partially or completely replication-impaired 7; 8; 10; 11; 12; 13. The availability of fully replication-competent forms of such viruses would permit more thorough characterization and provide new tools for probing retroviral biology, and the identification of mutations that impart or improve replication competence may itself provide novel insights into retroviral replication.
One elegant approach to circumventing a block to replication in an impaired virus, and which does not require knowledge of the mechanism responsible for the block, is to employ adaptive evolution. Retroviruses are particularly well-suited to this approach due their high mutation rates. The methodology used in this study combines adaptive evolution with genetic tagging of the virus to simplify identification of efficiently replicating mutants. We previously developed replication-competent variants of MLV containing an internal ribosome entry site (IRES)-transgene insert in the virus's 3′ untranslated region 14; 15; 16. These viruses replicate to high titer and can efficiently transmit and express exogenous genes in mammalian cells. Additionally, the use of a fluorescent reporter transgene, such as GFP, in these viruses allows spread to be easily tracked and quantitated by flow cytometry or fluorescence microscopy. The IRES-GFP cassette is very well tolerated in the MLV genome, is retained over a large number of cell-free passages in culture, and its presence is compatible with replication kinetics comparable to wild type virus.
Here, we employed this tagged retrovirus system to monitor the consequences of experimentally introduced genetic alterations to the viral genome. We describe a GFP-tagged chimeric virus in which the env gene in an MLV genome was replaced with the env of another gammaretrovirus, gibbon ape leukemia virus (GALV). While the amphotropic MLV Env protein utilizes the Pit-2 phosphate transporter for viral entry, the GALV Env protein utilizes the Pit-1 phosphate transporter 1. Surprisingly, although the GALV Env protein is commonly used to make high titer pseudotypes of standard MLV vectors 17; 18; 19, the chimeric virus exhibited greatly reduced replication kinetics.
However, retroviruses display a high inherent rate of mutation, thought to be in large part a consequence of the error-prone nature of reverse transcriptase 20. This property allows these viruses to evolve rapidly to overcome obstacles to their propagation and has proven experimentally useful for the generation of recombinant retroviruses with desirable qualities and in the analysis of retrovirus proteins 21; 22; 23. In this study, we took advantage of this property to select for variants of the MLV-GALV chimera that had acquired replication-enhancing mutations. We found that prolonged passage of cells exposed to the chimeric virus allowed the natural selection of rapidly replicating variants and that repeated attempts to generate such variants resulted in the selection of viruses with mutations in different locations. Our results reveal previously unidentified elements in MLV that control the extent to which the viral transcript undergoes splicing, and illustrate a novel approach to efficiently identifying and isolating replication-competent variants of replication-impaired recombinant retroviruses.
Results
Construction and adaptive evolution of a tagged MLV-GALV chimera
To evaluate the possibility of altering the receptor specificity of MLV by incorporation of a heterologous env gene, we replaced the env gene of a GFP-tagged, replication-competent proviral clone of MLV 15 with that of GALV. In the resulting construct, designated GZAP-GFP (Figure 1(a)), a short portion of the signal peptide-coding sequence of the MLV env was retained in order to avoid alteration of the MLV pol sequence that overlaps with env. The retroviral Env signal peptide serves to target the Env protein for translation in the endoplasmic reticulum. The Env of GZAP-GFP thus contains a hybrid signal peptide, but is expected to be of fully GALV origin when mature, according to analysis by the SignalP program 24. Whereas the signal peptide of MLV is 33 amino acids 25, that of GZAP-GFP and wild type GALV Env were predicted by SignalP to be 35 and 42 amino acids, respectively. The length of the hybrid signal peptide is anticipated to be intermediate between MLV and GALV and is therefore not expected to compromise proper processing of the Env protein.
Figure 1.
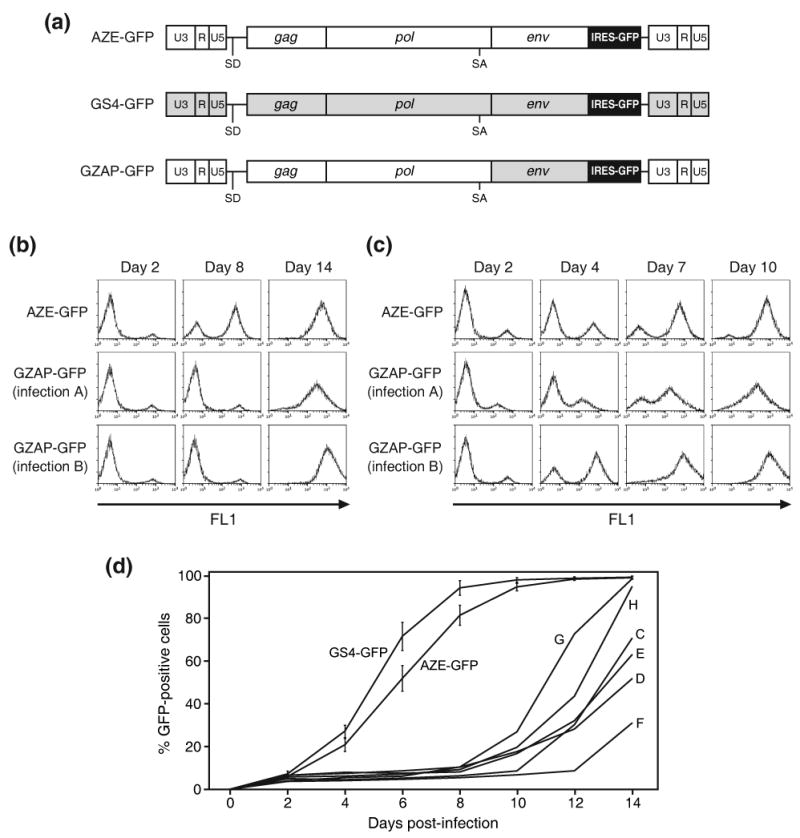
Structure and in vitro adaptation of an MLV-GALV chimera. (a) Schematic of the viruses used in this study. AZE-GFP and GS4-GFP are replication-competent clones of MLV and GALV, respectively, containing an IRES-GFP cassette between the env gene and 3′ UTR. GZAP-GFP is an MLV-GALV chimera identical to AZE-GFP except that it contains the env gene of GALV. Unshaded regions represent sequences derived from MLV and grey shaded regions represent those derived from GALV. (b) Histograms from flow cytometric analysis of primary infections with AZE-GFP or GZAP-GFP at equal MOI. LNCaP cells were exposed to supernatant from cells transfected with the provirus-containing plasmids and viral spread was monitored by GFP expression on the indicated days post-exposure. (c) Histograms from secondary infections of LNCaP cells carried out using supernatant from Day 14 of the infections shown in (b). (d) Results of additional primary infections of LNCaP cells with GZAP-GFP, AZE-GFP and GS4-GFP. The values for AZE-GFP and GS4-GFP are the averages obtained from three infections carried out in parallel and error bars represent standard deviations. Letters C through H denote single, independent infections with GZAP-GFP.
Virus was generated by transfection of 293T cells with plasmid encoding GZAP-GFP or AZE-GFP (Figure 1(a)), an efficiently replicating, GFP-tagged amphotropic clone of MLV 15. The two viruses differ only in their env sequence. After LNCaP cells were exposed to the viruses, viral spread was monitored by flow cytometry for GFP expression over the subsequent two weeks. Figure 1(b) shows the results of two independent infections with GZAP-GFP, designated A and B, and one with AZE-GFP, carried out using the same number of GFP-transducing units of virus. While the transduction levels at day 2 were comparable for all three infections, only infection with AZE-GFP resulted in progressive transmission of GFP during the first week, indicating that the switching of the env gene had greatly impaired replication of GZAP-GFP. However, at day 14 both cultures infected with GZAP-GFP had progressed to transduction levels near 100%, suggesting that the virus might have undergone adaptation and acquired the ability to replicate rapidly. Interestingly, the mean fluorescence intensity (MFI) of these two fully-transduced GZAP-GFP cultures differed significantly, by approximately 5-fold (Figure 1(b)). To determine if the chimeric virus had undergone changes that allowed efficient replication, we used supernatant from the three initial infections for secondary infections of fresh cultures. In these secondary infections, carried out using equal doses of virus, both passaged populations of GZAP-GFP, like the amphotropic virus, mediated rapid transmission of GFP with no lag phase (Figure 1(c)), providing further evidence that the virus had undergone adaptation. Furthermore, the large difference in MFI of the two GZAP-GFP-infected cultures was retained, suggesting that distinct alterations had arisen in the virus during the two initial infections.
To determine whether additional putative mutants might arise during independent infections with GZAP-GFP, six more infections were carried out using virus produced by transfection. In each case, the spread of GFP exhibited a pattern similar to that seen earlier, with a lag phase of 8-12 days after an initial transduction level of 5-10%, followed by rapid spread (Figure 1(d)). Infections with AZE-GFP and GS4-GFP at equivalent multiplicity of infection (MOI) demonstrated that MLV and GALV replicated in these cells at a comparable rate, and that the delayed kinetics of GZAP-GFP was due to the combining of the MLV and GALV sequences rather than to an inability of either MLV or GALV to replicate efficiently in these cells. Each of the eight GZAP-GFP populations, designated A-H, was then subjected to three cell-free passages at low MOI to enrich for rapidly replicating virus, and cellular DNA was isolated after the final passage for screening for sequence changes in the proviruses.
Identification of replication-enhancing splice acceptor mutations in adapted GZAP-GFP
To identify mutations that might have been acquired during the eight GZAP-GFP infections, we first performed a functional screen of the right half of the provirus of each of the eight passaged virus populations. The 4.1-kb SalI-MluI region of the viral genome from the middle of pol to the 3′ end of env was PCR amplified from the cellular DNA and cloned back into pGZAP-GFP (Figure 2(a)). Virus was generated from the resulting plasmids by transfection and used to again infect LNCaP cells. GFP expression was monitored over a short (6-day) time course to screen for replication-competent clones. Five of the eight recloned fragments, from infections A, B, D, F, and G, conferred upon GZAP-GFP the ability to replicate rapidly and without delay (Figure 2(b)). For each of these, the kinetics of spread was similar to that of the corresponding passaged virus from which the insert was amplified. The fragments from infections C, E and H were not found to enhance the replication-competence of GZAP-GFP, despite the screening of multiple independently reconstructed plasmid clones. To further localize the mutations in infections A, B, D, F and G, we analyzed the left (SalI-NdeI) and right (NdeI-MluI) halves of the 4.1-kb fragment for their ability to confer replication competence on GZAP-GFP by cloning them separately back into pGZAP-GFP. In each case, the NdeI-MluI fragment alone enhanced transmission of GFP through the cultures to occur (data not shown), demonstrating that the mutation was contained within this fragment.
Figure 2.

Screening of the eight passaged populations of GZAP-GFP for replication-enabling mutations in the 4.1-kb SalI-MluI pol-env region of the virus. (a) Schematic of the provirus showing the region that was amplified by PCR from genomic DNA and reintroduced into pGZAP-GFP for functional analysis. (b) Results from infections initiated with either passaged virus from the original eight infections or with virus produced by transfection of pGZAP-GFP reconstructed with the corresponding amplified fragments. LNCaP cells were exposed to an equal number of GFP-transducing units of virus from the original infections (“passaged GZAP-GFP”) or from transfections with reconstructed pGZAP-GFP, and spread was quantitated by flow cytometry at the indicated days post-infection. Infections with parental GZAP-GFP and AZE-GFP, produced by transfection, were included as controls. Values shown are the average obtained from triplicate infections and error bars indicate standard deviations. (c) The adaptive splice acceptor mutations that arose in GZAP-GFP during infections A, B, D, F and G. The mutant and wild type sequences around the acceptor site are shown aligned. Above each nucleotide sequence is the corresponding translation for integrase. The mutations and the amino acids that are altered are in bold. The locations of the MLV splice acceptor (SA) site, polypyrimidine tract (PPT), and putative branch point sequence (BPS) are indicated.
Sequencing of the NdeI-MluI region in each of the eight reconstructed plasmids revealed that the variants that arose in infections A, B, D, F and G all contained one of two mutations at the MLV splice acceptor site 26, and that those from infections C, E and H contained no changes in this region. The variants from infections A, D and F all possessed the same G→A transition at the 3′ terminal nucleotide of the viral intron (Figure 2(c)). This mutation, 5490G>A, converted the AG splice acceptor dinucleotide, which is nearly invariant among major-class introns, to AA and would therefore be expected to abolish splicing at this site 27. (All MLV nucleotide numbering is in accordance with Shinnick et al. 28, GenBank accession no. J02255). This mutation also lies within the sequence coding for integrase, and converts the alanine at amino acid 293 of the protein to threonine.
The variants that appeared in infections B and G both exhibited an insertion of TCC just upstream of the splice acceptor. This insertion lies within the polypyrimidine tract of the splice acceptor, extending the tract from 11 nucleotides to 14, and creates a duplication of Ser 290 of integrase. No alterations other than these two mutations were observed. Thus, none of the eight adapted viruses contained any alteration of the GALV env gene itself, demonstrating that the GALV envelope protein containing the hybrid signal peptide functioned effectively for replication of the chimeric virus.
Identification of replication-enhancing LTR mutations in adapted GZAP-GFP
To screen for mutations that arose during infections C, E and H, we directly sequenced PCR products amplified from the corresponding cellular DNA from the 5′ terminus of the provirus to the middle of pol. The viruses from all three of these infections were found to each possess two mutations in the LTR, which are diagrammed in Figure 3(a). The variants from infections C, E and H each contained a mutation between the CAAT and TATA boxes of the viral promoter in the U3 region along with a point mutation in either the R or U5 region. The U3 mutation of infection C was a G→A transition while those of infections E and H were deletions of 19 and 10 base pairs, respectively. The two deletions were found to have occurred between short direct repeats in the MLV LTR (Figure 3(b)). In a previous study, we observed similar repeats flanking deletions that appeared within the IRES-GFP cassette over long-term viral propagation 15. PCR products amplified from the 5′ LTRs of the other five mutant viruses containing splice acceptor mutations were also amplified and sequenced and were found to contain no changes relative to pGZAP-GFP, as expected.
Figure 3.

Replication-enabling mutations that arose in GZAP-GFP during infections C, E and H. (a) Schematic of the LTRs of the mutants showing the locations of deletions and point mutations identified by sequencing of proviral DNA. Hatched segments indicate deletions. (b) Alignment of the U3 region sequences between the CAAT and TATA boxes of each mutant with the corresponding wild type MLV sequence. The CAAT and TATA boxes are in bold, the 8217C>A mutation is marked by an asterisk, deleted nucleotides are represented by dashes and the short direct repeats that flanked the regions deleted in infections E and H are indicated below the wild type MLV sequence. (c) Comparison of the replication of virus from the passaged GZAP-GFP populations to that of GZAP-GFP reconstructed to contain the corresponding dual LTR mutations shown in (a). Values shown are the average obtained from triplicate infections and error bars indicate standard deviations.
In order to assess the influence of the LTR mutations on the replication of these mutants, we mutated pGZAP-GFP to contain each of these three pairs of mutations. The resulting plasmids were constructed so that the LTR alterations of the variants from infections C, E or H were present in both the 5′ and 3′ LTR. Infection of LNCaP cells with these mutants showed that each mutation pair imparted to GZAP-GFP the ability to replicate with kinetics similar to that of the corresponding passaged GZAP-GFP population (Figure 3(c)), demonstrating that these mutations enabled the mutant viruses that arose in these infections to replicate efficiently.
To determine whether or not both mutations of each mutation pair were necessary for replication of these variants, we constructed a series of additional versions pGZAP-GFP in which each mutation was present in isolation. Again, each virus was constructed so that both the 5′ and 3′ LTR contained the mutation in question. Figure 4 shows the results of infections with these variants. For each of the three mutation pairs, both mutations were found to be required together for efficient replication. Only the 79G>A mutation had any effect on its own on replication of GZAP-GFP, allowing the virus to spread, but only with very slow kinetics. None of the other mutations on their own had a discernable effect on replication. Thus, both mutations in each LTR mutation pair acted in concert enhance spread of the variants that arose in infections C, E and H.
Figure 4.

Evaluation of the importance of each of the LTR mutations from infections C, E and H for viral replication. For each of these three GZAP-GFP mutants, two variants were constructed, each possessing one of the two mutations in isolation. Infections with these variants or the original mutants were carried out using equal doses of virus, and viral spread was assessed by flow cytometry at the indicated days post-infection. Values shown are the average obtained from triplicate infections and error bars indicate standard deviations.
Transmission of GFP through cultures infected with the adapted variants corresponds with replication as measured by a standard retrovirus assay
To determine if the spread of GFP expression by the GZAP-GFP mutants as measured by flow cytometry reliably reflects viral spread as measured by a more conventional assay for retroviral replication, we examined the RT activity in cell cultures following exposure to each virus (Figure 5). While cells infected with AZE-GFP or any of the mutants of GZAP-GFP exhibited a progressive increase in RT activity over the subsequent 9 days, cells exposed to parental GZAP-GFP exhibited only a low RT level through the course of the experiment. Furthermore, the relative rate of replication of each virus as measured by RT activity was consistent with that measured by transmission of GFP. Variant B of GZAP-GFP exhibited in both assays the most rapid kinetics, while variant A exhibited the slowest. GZAP-GFP variants C, E and H and AZE-GFP replicated at intermediate rates by both measurements. Hence, the transmission of GFP served as a convenient and reliable indicator of replication for these viruses.
Figure 5.

Replication of viruses as measured by reverse transcriptase activity. LNCaP cells were exposed to each virus at an MOI of 0.1 with GZAP-GFP, the indicated GZAP-GFP variants, or AZE-GFP, and RT activity in the cultures was monitored over the following nine days. RT activities are expressed in arbitrary units and are the average obtained from triplicate infections. Error bars represent standard deviations. Asterisks denote RT activities that were significantly different (p < 0.05 by two-tailed, paired t-test) from those of AZE-GFP at the same time point. The values for AZE-GFP are shown by a dashed line for reference. The RT value for Day 0 represents the activity in cultures immediately before infection.
The adapted variants of GZAP-GFP retain the receptor specificity of GALV env
To confirm that the GZAP-GFP variants utilized the GALV receptor for cell entry, we performed infections on additional cell lines. Due to species-specific differences in the Pit-1 protein, GALV Env can mediate infection of human but not murine cells 29. By contrast, amphotropic MLV Env allows infection of cells from both species. We therefore exposed human PC-3 and murine NIH3T3 cells to AZE-GFP or the variants of GZAP-GFP at equal doses. Both cell lines were readily infected with AZE-GFP, but the GZAP-GFP variants were only capable of infecting the human cell line (Figure 6). Exposure of additional murine cell lines to the viruses likewise resulted in rapid spread of AZE-GFP, but no infection by any of the GZAP-GFP variants (data not shown), confirming that the chimeric viruses utilize Pit-1 as expected.
Figure 6.

Comparison of the infectivity of the adapted chimeric viruses on human and mouse cells. PC-3 human prostate carcinoma and NIH3T3 mouse fibroblast cells were infected at a MOI of 0.1 and were analyzed by flow cytometry at 5 days post-infection. Vertical axis: cell number, horizontal axis: GFP fluorescence measured using the FL1 channel
The mutations of the adapted GZAP-GFP variants reversed an imbalance in the unspliced:spliced viral RNA ratio
The occurrence of mutations at the splice acceptor site in two of the adapted GZAP-GFP variants suggested that the replication defect in the parental chimeric virus was the result of a flaw involving splicing of the viral RNA. As the unspliced RNA of simple retroviruses serves for expression of the gag and pol genes and functions as the transmitted genome, while the unspliced transcript is required for expression of the env gene, a balance of spliced and unspliced RNA must be maintained for efficient replication to occur. We therefore tested the possibility that the defect involved a disruption of the relative levels of the two RNAs.
First, we needed to determine if GZAP-GFP and its variants utilized the native splice donor and acceptor sites of MLV. Because we also desired to evaluate the levels of spliced and unspliced RNA produced by GALV, it was necessary identity the splice junction of this virus, which has not yet been reported. We therefore performed RT-PCR on RNA from cells transfected with the parental and mutant GZAP-GFP plasmids and wild type and GFP-tagged GALV plasmids and sequenced the resulting amplification products. The forward primers used for amplification were specific for the U5 region and the reverse primers were specific for the sequence just upstream of env. Sequencing of the amplified product from the RNA of the chimeric viruses showed that each one, except for that possessing the 5490G>A splice acceptor mutation, utilized the native MLV splice donor and acceptor (data not shown). The 5490G>A mutant of GZAP-GFP was found to use the native MLV splice donor and a cryptic acceptor located 11 bp downstream of the native acceptor. Sequencing of the GALV RT-PCR products (Figure 7), revealed that the 5′ and 3′ ends of the GALV intron are at nucleotides 204 and 5445 of the viral RNA, respectively, in both the wild type and tagged virus (numbering is in accordance with Delassus et al.30, GenBank accession no. M26927).
Figure 7.
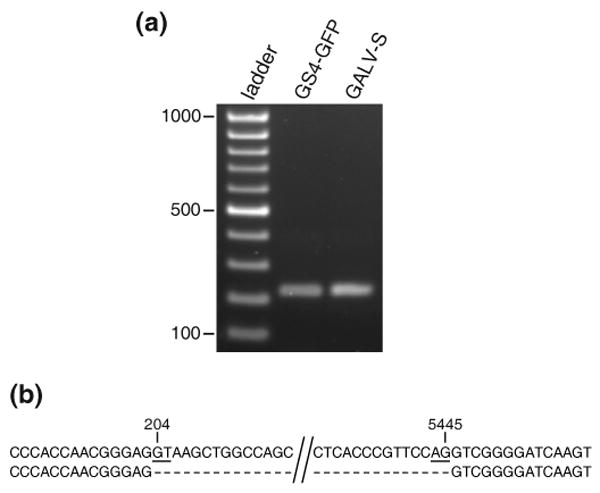
Identification of the splice junction of GALV. RNA was isolated from cells transfected with either pGALV-S, which contains a wild type proviral clone of GALV, or pGS4-GFP and used for RT-PCR. A forward primer in U5 and a reverse primer immediately upstream of the env start codon were used to amplify across the splice junction. The amplified products were gel purified and directly sequenced. (a) Agarose gel of the RT-PCR reactions. (b) Alignment of the amplified sequences (top strand) with that of GALV genomic sequence (bottom strand). The GALV RNA genome coordinates at the 5′ and 3′ ends of the identified intron are shown. The GT dinucleotide of the splice donor and the AG of the splice acceptor are underlined.
We then performed quantitative RT-PCR on the RNA of GZAP-GFP and its variants and several control RNAs from wild type and GFP-tagged MLV and GALV. To measure the level of unspliced transcript, we used primer-probe sets specific for pol (Figure 8(a) and Table 1). To measure the level of spliced transcript, we used 5′ primers specific for the sequence upstream of the splice donor and probes and 3′ primers specific for the region downstream of the splice acceptor. The quantitation revealed that introduction of the GALV env into the MLV genome did in fact affect viral splicing, increasing the unspliced:spliced transcript ratio by roughly three-fold (Figure 8(b)) compared to wild type or GFP-tagged MLV. Each of the single or dual mutations of the five variants of GZAP-GFP caused a strong reversal in this ratio, not merely restoring it to that of wild type or tagged MLV, but reducing it by 10- to 30-fold, depending on the variant. This suggests that the chimeric viruses required a higher fraction of spliced RNA than did MLV for optimal replication. Additionally, both wild type and GFP-tagged GALV exhibited a transcript ratio similar to those of the adapted GZAP-GFP variants. Additionally, three of the LTR mutations were tested in isolation and each one, when present alone, reduced the ratio to a lesser extent than did the dually mutated LTRs, indicating that the LTR mutations functioned cooperatively to restore replication-competence by an additive effect on splicing. This is consistent with our earlier observation that both LTR mutations were together required for correction of the replication defect of GZAP-GFP.
Figure 8.

Quantitation of the levels of spliced and unspliced viral RNA produced by MLV, GALV and chimeric viruses. RNA isolated from cells transfected with the provirus-containing plasmids was reverse transcribed and used in TaqMan PCR. (a) Schematic diagram of the gammaretroviral RNA genome showing locations of the primers (arrowheads) and probes (rectangles) used to measure the levels of unspliced and spliced transcript. The primer-probe sets above the RNA diagram represent the location of those used for wild type and GFP-tagged MLV and for GZAP-GFP and its mutants. The primer probe sets below the RNA diagram represent the location of those used for wild type and GFP-tagged GALV. Black circles indicate the locations of the splice donor and acceptor sites in the viral RNA. (b) Results of quantitation of the transcripts, expressed as the ratio of unspliced to spliced viral RNA. The ratio for wild type MLV (Moloney strain) was arbitrarily set to 1, and the ratios for all other viruses are normalized to this value. Each value is the mean ratio obtained from at least three amplifications, and error bars represent standard deviations. ZAPm-GFP is identical to AZE-GFP and GZAP-GFP but contains the ecotropic MLV env gene.
Table 1.
Primers and probes used in quantitative RT-PCR for unspliced and spliced viral RNA.
| A. MLV and hybrid viruses | |||
|---|---|---|---|
| Target transcript | Primer or probe | Sequencea | Coordinates in MLV genomeb |
| Unspliced | Forward primer | 5′-AACAAGCGGGTGGAAGACATC-3′ | 2952-2972 |
| Probe | 5′-FAM-CCCACCGTGCCCAACCCTTACAACC-TAMRA-3′ | 2976-3000 | |
| Reverse primer | 5′-CAAAGGCGAAGAGAGGCTGAC-3′ | 3088-3108 | |
| Spliced | Forward primer | 5′-CTCCTCTGAGTGATTGACTACCC-3′ | 103-125 |
| Probe | 5′-VIC-TGCCGCCAGAGGTCTCCAGACTTCG-TAMRA-3′ | 5522-5546 | |
| Reverse primer | 5′-CGGTCCAGTTGTTCTTGGTAGG-3′ | 5548-5569 | |
| B. GALV viruses | |||
|---|---|---|---|
| Target transcript | Primer or probe | Sequencea | Coordinates in GALV genomec |
| Unspliced | Forward primer | 5′-TGGTATACAGACGGTAGCAGTTTC-3′ | 4102-4125 |
| Probe | 5′-FAM-CCCTGCTCTCCGTTTACCTTCCGTGA-TAMRA-3′ | 4127-4152 | |
| Reverse primer | 5′-CCGTCCGCTTGCCATCTAC-3′ | 4162-4180 | |
| Spliced | Forward primer | 5′-ACCACCGACCCACCAACG-3′ | 182-199 |
| Probe | 5′-VIC-TTCCACCGAGGCTCAAGGCTGCTG-TAMRA-3′ | 5481-5504 | |
| Reverse primer | 5′-GGTAGTCAGCAACACCAGGTATG-3′ | 5510-5532 | |
Abbreviations: FAM, 5-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine. VIC is a proprietary fluorescent label of Applied Biosystems.
Numbered in accordance with the MLV RNA genome sequence of GenBank accession no. J02255. The MLV splice donor and acceptor sites are located at nucleotides 206 and 5490, respectively.
Numbered in accordance with the GALV RNA genome sequence of GenBank accession no. M26927. The GALV splice donor and acceptor sites are located at nucleotides 204 and 5445, respectively.
As in all retroviruses, the MLV LTR contains the sequences that govern viral transcription. Therefore, it might be expected that the LTR mutations in the adapted GZAP-GFP variants affected the rate of transcription of the viral RNA and may have thereby influenced the ability of the mutants to replicate. To evaluate the effects of the LTR mutations on transcription, we constructed reporter plasmids in which the LTRs of the GZAP-GFP variants from infections C, E and H drove expression of a luciferase reporter. Transfection of these constructs revealed that the LTR of infection E, containing the 8205-8223del and 5C>T LTR mutations, did exhibit a significantly higher transcriptional activity than the wild type LTR, mediating nearly three-fold higher expression of the reporter (Figure 9). The two other mutant LTRs, however, did not exhibit transcriptional activity significantly different from that of the wild type LTR. Thus the mutated LTR from infection E may have functioned to enhance replication of the chimeric virus not only by its effect on splicing but also through its greater transcriptional activity.
Figure 9.

Comparison of the transcriptional activity of the wild type and mutant LTRs. LNCaP cells were transiently transfected with reporter plasmids containing the wild type or mutant LTRs linked to Gaussia luciferase. Shown for each construct are the average relative light units (RLU) produced per cell, after normalization for transfection efficiency. Values were obtained from four replicates and error bars represent standard deviations. The asterisk indicates a statistically significant difference between the activity of the LTR of mutant E and that of wild type virus (p = 0.012 by two-tailed, paired t-test).
Discussion
In the present study, we employed a full-length, GFP-tagged MLV to evaluate the replication of a chimeric variant in which the natural env gene had been replaced by that of GALV, and to investigate the role of compensatory mutations that arose upon natural selection. The presence of the IRES-GFP insert in the viral genome made it possible to easily assess replication, to detect the outgrowth of mutant viruses, to localize their mutations, and to characterize the mutants after cloning. Conventional assays for retroviral replication, such as measurement of reverse transcriptase activity, are time and labor-intensive and often require the use of radioisotopes. By contrast, flow cytometric analysis for GFP expression to detect and quantitate viral spread is sensitive, comparatively simple and rapid and does not require the use of hazardous materials. This approach should be applicable generally to the efficient isolation of replicating variants of recombinant, full-length retroviruses whose replication is somehow impaired.
Interestingly, although high-titer preparations of GALV Env-pseudotyped MLV vectors can be readily made, and the two viruses belong to the same retroviral genus, the replacement of the native env gene in the MLV genome with that of GALV impaired replication of the virus. This result provides an illustration of the difficulty associated with predicting what changes to the retroviral genome can made without compromising or eliminating replication-competence.
In order for gag, pol, and env genes to be expressed at appropriate levels and for efficient replication to occur, an appropriate balance between spliced and unspliced RNA is required. By contrast, cellular mRNAs generally must undergo complete splicing prior to export from the nucleus. Examination of the levels of spliced and unspliced RNA produced by the parental chimeric virus showed that the replacement of the native env gene of MLV with the env of GALV resulted in a large decrease in spicing of the viral RNA.
However, prolonged passage of cells exposed to the virus allowed the selection and outgrowth of rapidly replicating mutants. Repeated attempts to generate such variants resulted in the selection of viruses with mutations clustering in one of two regions. One group of variants exhibited mutations at the splice acceptor and the other had acquired dual alterations in the LTR. For both groups, the perturbation in the unspliced:spliced transcript ratio caused by the env substitution had been reversed by the mutations. The variants containing alterations in the LTR all possessed a combination of two mutations and the presence of both mutations together was essential to each variant both for efficient replication and for complete rebalancing of the transcript ratio. Interestingly, the adapted chimeric viruses all exhibited a spliced:unspliced transcript ratio significantly lower than MLV but comparable to that of GALV. This may indicate that the level of GALV Env that is optimal for viral replication is higher than that of MLV Env and that viruses encoding this Env protein may therefore need to maintain higher levels of splicing than those encoding an MLV Env. Further investigation would be necessary to address this possibility.
How do these mutations shift the balance of viral RNA towards the spliced form? While the cis-acting sequences in the genomes of gammaretroviruses such as MLV that are responsible for the maintenance of the balance between spliced and unspliced RNA remain poorly defined, certain inferences can be made. In the case of the splice acceptor mutations, a direct effect on splicing efficiency is the most likely mechanism. The polypyrimidine tract TCC insertion most likely enhanced splicing efficiency by increasing the tract's length and uridine content. Polypyrimidine tract length 31; 32 and U-richness 33; 34 have been shown to correlate with splicing efficiency, presumably by influencing the affinity of the sequence for the splicing factor U2AF65 35. How the 5490G>A mutation caused an increase in the fraction of spliced RNA is less clear. This mutation, which abolished the natural MLV acceptor AG, shifted splicing to an AG several nucleotides downstream and caused an approximately 30-fold rise in the ratio of the spliced relative to the unspliced RNA, an effect stronger than any of the other mutations or mutation pairs we found. One possibility is that the new acceptor site is inherently more efficient than the natural acceptor, but the natural acceptor's proximity to the branch site makes it the preferred location for splicing. The first AG downstream of an intron's branch site is normally selected for exon ligation 36. Thus, mutation of the native acceptor AG would allow splicing to occur at the next AG downstream, whose context may be better suited for efficient splicing.
There are multiple potential mechanisms whereby the LTR mutations shifted the balance of RNA towards the spliced form. One possibility is that the mutations directly influence the efficiency of the splicing reaction itself by modulation of the activity of a splicing enhancer or silencer in the LTR. Such regulatory elements have been identified in other retroviruses 37; 38; 39, although none of these have been located in the LTR. Another possibility is that the mechanism involves the transport pathway used to export the unspliced viral RNA from the nucleus. In contrast to cellular mRNAs, which are in general fully spliced before nuclear export, a portion of retroviral RNA must be exported to the cytoplasm in unspliced form, and in the case of complex retroviruses, partially spliced form 40. In the simple retroviruses Mason Pfizer monkey virus (MPMV) and simian retrovirus-1 (SRV-1), export of the unspliced RNA is mediated by a cis-acting sequence termed the constitutive transport element (CTE). Deletion of the CTE in either of these viruses results in an increase in the overall fraction of spliced viral RNA in the cell 41; 42. Alteration of the rate of entry of the unspliced transcript into the pathway used for export of unspliced MLV RNA, which may sequester the RNA away from the splicing machinery, could change the relative levels of spliced versus unspliced RNA in the cell. For example, the env gene of human immunodeficiency virus (HIV) possesses important cis-acting sequences, including the Rev-responsive element 43, in addition to coding for the Env protein, and substitution of the env gene of this virus with that of another could result in impairment of replication due to loss of such functions. While, to our knowledge, no such sequences have been reported in the env genes of gammaretroviruses, their existence remains a possibility. Additionally, these same general mechanisms, or their inhibition, may be invoked to explain why replacement of the native MLV env with GALV env shifted the transcript ratio towards the unspliced form. Although the env substitution did not alter the ratio as dramatically as the mutations acquired by the mutants, our results provide evidence that the env itself may contain sequences that influence splicing.
A third mechanism by which the LTR mutations might affect splicing is via alterations in the RNA polymerase II complex or the associated transcriptional regulatory proteins that assemble on the proviral LTR. Evidence has accumulated in recent years for an important role of transcription in regulation of splicing 44. Promoter structure 45 and association with transcription factors 46 and coregulators 47 have been demonstrated to be capable of strongly influencing patterns of alternative splicing through as-yet undefined mechanisms that are independent of transcriptional efficiency. As the transcriptional control sequences in retroviruses are located primarily within the LTR, and most importantly in the U3 region, the LTR mutations we identified may alter the extent to which the viral transcript is spliced by modulating interactions with components of the transcription machinery. Further investigation of how these mutations act should provide additional insight into the regulation of retroviral RNA processing.
A previous study showed that the R region of MLV contains a short stem-loop structure that is required for the accumulation of the unspliced, but not the spliced, transcript in the cytoplasm 48. The R region mutation that we identified, 5C>T, is located within the stem portion of this structure and may therefore function by modulating its activity. The means by which the stem-loop operates has not yet been determined. While the mechanisms by which the LTR mutations function are uncertain, it is clear that none of them, when present alone, are sufficient to impart high-level replication-competence to the chimeric virus. The ability of a U3 mutation to complement a U5 mutation to restore replication is particularly interesting because these regions lie at opposite ends of the viral RNA. This raises the possibility that some functional interaction between the ends of the RNA molecule might occur during MLV replication.
The mutant viruses isolated in this study not only reveal a novel splice-regulatory mechanism in MLV, but should also prove useful specifically in pre-clinical safety evaluation of GALV-pseudotyped defective MLV vectors. The GZAP-GFP variants we isolated represent potential replication-competent viruses that might arise by recombination in oncoretroviral vector-producing cells expressing the GALV env gene, such as the widely used PG13 packaging cell line 49. Until now, no reference standards were available for the development and validation of appropriate detection assays for such replication-competent MLV-GALV recombinants. Furthermore, our results indicate that the likelihood of formation of replication-competent retrovirus from MLV vector packaging cells expressing the GALV env gene is lower than from packaging cells expressing an MLV envelope, due to a requirement in the former for additional mutations to correct the perturbation in splicing that a recombinant MLV containing the GALV env gene would likely exhibit.
Finally, our results demonstrate a methodology for the development and optimization of genetically pseudotyped retroviruses as vehicles for gene delivery. We and others have shown that MLV can be modified to transmit and express transgenes with high efficiency throughout solid tumors in vivo without detectable spread to normal tissues 16; 50; 51; 52; 53; 54; 55; 56. Such modified retroviruses show promise as agents for gene therapy of cancer. There remains cause for caution, however, in the development of such viruses, in view of previous reports of lymphoma developing in lethally irradiated macaques 57, and clonal T cell proliferation in pediatric patients with severe combined immunodeficiency after transplantation with retrovirus vector-transduced hematopoietic stem cells 58. However, based on reports from classical virology literature on MLV 59; 60 as well as more recent studies demonstrating lack of retrovirus-induced pathology following exposure to wild type amphotropic MLV by either direct systemic injection or transplantation of infected bone marrow cells in normal primates 61; 62, and lack of detection of any virus by quantitative PCR after intravenous injection of transgene-carrying MLV in immunocompetent mice 50, there is likely to be little biohazard risk to normal individuals, and a relatively favorable risk:benefit ratio associated with the use of such replicating retroviruses in the context of cancer gene therapy, particularly for poor-prognosis malignancies
Materials and Methods
Plasmids
Plasmid pGZAP-GFP was constructed by replacement of the env region of pZAPm-GFP 15, from the HpaI site near the 5′ end of env to the MluI site at the 3′ terminus of env, with GALV env sequence amplified by PCR from plasmid p386, which harbors a full-length molecular clone of the SEATO strain of GALV (generously provided by Maribeth Eiden, National Institute of Mental Health). The primers used for amplification of the env sequence were: AACCCGCGAGGCCCCCTAATCCTGATCATCCTCTTAAGCTGC, and GTTCTGTGACGCGTTTAAAGGTTACCTTCGTTCTCTAGG, which adds an MluI site (underlined) after the env stop codon. To construct plasmid pGS4-GFP, we first replaced the plasmid backbone of p386 with the replication origin and ampicillin resistance gene of pUC19, producing pGALV-S. To introduce MluI and NotI sites at the end of the env gene for insertion of transgene sequences, we PCR amplified the 3′ end of the env gene of p386 with primers TCCTCCTTCTGTTGCTCATCCTC and TGTGAAGCGGCCGCATCTTACGCGTAAATTAAAGGTTACCTTCGTTC, and the 3′UTR-3′LTR region with primers GAACGAAGGTAACCTTTGCGGCCGCTCTAAGATTAGAGCTAT and AAAACAGGCCGGCCATGTGAGACCCCCGAAGTG (NotI, MluI and FseI sites in the primers are underlined). The env PCR product was cut with NsiI and NotI and the 3′UTR-3′LTR product was cut with NotI and FseI. These two fragments were then ligated into pGALV-S at the NsiI site in env and an FseI site at the end of the 5′ LTR, producing pGALV-S-MCS. To produce pGS4-GFP, the IRES-GFP cassette of plasmid pZAPm-GFP was inserted into the MluI and NotI sites of pGALV-S MCS. Plasmids pAZE-GFP, which contains an amphotropic IRES-GFP-tagged clone of MLV, and pZAP2, which contains a full-length clone of wild type Mo- MLV, were described previously 15.
Cell culture and transfections
293T human embryonic kidney, LNCaP and PC-3 human prostate carcinoma and NIH3T3 mouse fibroblast cells were obtained from the American Type Culture Collection. 293T and NIH3T3 cells were cultivated in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. LNCaP and PC-3 cells were cultivated in RPMI 1640 with 10% FBS. All media was supplemented with penicillin/streptomycin. Lipofectamine 2000 (Invitrogen) was used for plasmid transfection of 293T cells to produce virus as described previously 63. Fugene 6 (Roche, Indianapolis, IN) was used for transfection of LNCaP cells for RNA isolation and reporter assays. All cell culture procedures were conducted under protocols approved by the UCLA Institutional Biosafety Committee at Biosafety Level 2 (BSL2), with adequate containment and decontamination precautions.
Infections and titration
For all infections, cells were at 20-30% confluence at the time of exposure to virus and were replated at a density of 20-30% at each passage. Polybrene was added to all cultures immediately prior to infection to a concentration of 5 μg/ml. Only infections that resulted in 5-15% transduction at two days post-infection were further propagated and analyzed. For enrichment of rapidly replicating mutants, after the percentage of GFP-positive cells in each culture reached 90-100%, the supernatant was collected, filtered through a 0.45 μm Acrodisc Supor syringe filter (Pall Gelman Sciences, Ann Arbor, MI), and used to infect fresh LNCaP cultures at a 100-fold dilution. Virus titers, in terms of GFP-transducing units, were determined by exposing 20 to 30% confluent LNCaP cultures in the presence of 4 μg of polybrene/ml to dilutions of virus produced by transfection of 293T cells. The following day, the medium on the cells was replaced with fresh medium containing 50 μM 3′-azido-3′-deoxythymidine to block secondary spread of virus. On the third day, the cells were analyzed by flow cytometry to determine the percentage GFP-positive cells. Titers were calculated according to the formula: transducing units per ml = (number of cells counted at the time of exposure to virus × percentage of GFP-positive cells determined by flow cytometry)/dilution factor of transfection supernatant.
Flow cytometry
Epics XL (Beckman Coulter, Fullerton, CA) and FACScan (Becton Dickinson, Franklin Lakes, NJ) flow cytometers were used for analysis of GFP expression in infected and transfected cells using a 525 or 530 nm bandpass emission filter. 5-10 thousand cells from each sample were analyzed after trypsinization and suspension in phosphate buffered saline.
PCR amplification and reconstruction of adapted viruses
High molecular weight DNA was isolated from infected LNCaP cells using the GenomicPrep kit (GE Healthcare, Piscataway, NJ). Proviral sequences were amplified from the LNCaP DNA using Pfu Ultra polymerase (Stratagene, La Jolla, CA). For screening of the SalI-MluI region from the middle of pol to the 5′ end of the IRES, we performed PCR using primers GGGGTTGCCAGATTTGACTAAG and AAAAGACGGCAATATGGTGGAA. The resulting products were digested with SalI and MluI and inserted into pGZAP-GFP at the same sites to replace the original sequence. For screening of the region of the virus from the 5′ LTR to the middle of pol, we amplified this region by PCR using primers AAATAAGAATTCAATGAAAGACCCCA and CGTTAGGACACCTTTGGCGTA. The resulting PCR products were gel purified using the Wizard SV kit (Promega, Madison, WI), and directly sequenced. To recreate the mutant proviruses exhibiting mutations in the LTR, we performed site-directed mutagenesis using the QuikChange II and QuikChange Multi kits (Stratagene, La Jolla, CA), according to the manufacturer's instructions, on versions of pGZAP-GFP that had either the 5′ or 3′ half of the viral genome deleted. The version of pGZAP-GFP lacking the 5′ half of the genome was produced by removal of the region from the SacI site in the 5′ LTR to the SacI site in pol, and the version of pGZAP-GFP lacking the 3′ half of the genome was produced by removal of the region from the SacI site in pol to the SacI site in the 3′ LTR. After the desired mutation(s) were introduced into these plasmids, full-length proviral clones containing the mutations in both LTRs were constructed by ligating the following three fragments: one from the EcoRI site 5′ of the 5′ LTR to the XhoI site in gag, one from the XhoI site in gag to the MluI site at the 3′ end of env, and one from the MluI site at the end of env to the EcoRI site 5′ of the 5′ LTR. Additional details on the mutagenesis procedures and mutagenic primer sequences used are available upon request. The integrity of all plasmids in the regions encompassing the mutations was verified by sequencing. Laragen, Inc. (Los Angeles, CA) performed all sequencing for this study.
Reverse transcriptase assays
The reverse transcriptase (RT) activity in supernatants of infected cell cultures was assayed as described previously 64. Quantitation of the reaction products was performed using an FX Pro Plus molecular imager (Bio-Rad, Hercules, CA).
Standard and quantitative RT-PCR
LNCaP cells were transfected with virus-encoding plasmid and total cellular RNA was isolated with the RNeasy Mini Kit with DNase I treatment (Qiagen, Valencia, CA). Cell homogenization for RNA isolation was carried out with Qiashredder columns (Qiagen). RNA yield was determined by absorbance at 260 nm and 2 μg of each RNA preparation was used for synthesis of cDNA with Superscript III reverse transcriptase and random hexamers (Invitrogen) according to the manufacturer's instructions. The primers used for identification of the splice junction of GZAP-GFP and its variants are the same as those used for quantitative RT-PCR and are shown in Table 1A. The primers used for identification of the splice junction of GALV were GAGCCGTGGTCTCGTTGTTC and GGTCGGGGTAGTCAGCAACA. The relative levels of the spliced and unspliced viral transcripts were determined by TaqMan amplification of the cDNAs in an ABI PRISM 7700 using the TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA). The primers and probes were synthesized by Invitrogen and Applied Biosystems, respectively. In each experiment, standard curves for both the unspliced and spliced amplicons were run in triplicate and used to determine the starting quantities of the experimental samples. Experimental samples were amplified in triplicate, and for each primer pair, control reactions using cDNA from untransfected cells were included as controls.
Luciferase reporter assays
Full-length wild type and mutant LTRs were PCR amplified from the corresponding provirus-containing plasmids and inserted into the MluI and HindIII sites of Gaussia luciferase expression plasmid pCMV-GLuc1 (Nanolight, Pinetop, AZ), replacing the CMV promoter. LNCaP cells were co-transfected with the luciferase plasmids and GFP expression plasmid pGFPemd-cmv[R]-control (Packard Biosciences, Meriden, CT) to normalize for transfection efficiency and at 36 hours post-transfection the culture supernatant was collected for luciferase assays and the cells were analyzed by flow cytometry to determine transfection efficiency, measured as the percentage of GFP-positive cells. A Gaussia Luciferase Assay Kit (New England Biolabs, Ipswich, MA) and a MicroLumatPlus luminometer (Berthold Technologies, Oak Ridge, TN) were used to quantitate luciferase activity in the supernatants.
Acknowledgments
This work was supported by NIH grants P01CA59318 and R01CA105171 (to N. K.) and NCI Cancer Education Grant R25 CA098010 (to C.L.). We thank Aki Logg for assistance during the early stages of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Overbaugh J, Miller AD, Eiden MV. Receptors and entry cofactors for retroviruses include single and multiple transmembrane-spanning proteins as well as newly described glycophosphatidylinositol-anchored and secreted proteins. Microbiol Mol Biol Rev. 2001;65:371–89. doi: 10.1128/MMBR.65.3.371-389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders DA. No false start for novel pseudotyped vectors. Curr Opin Biotechnol. 2002;13:437–42. doi: 10.1016/s0958-1669(02)00374-9. [DOI] [PubMed] [Google Scholar]
- 3.Mild M, Esbjornsson J, Fenyo EM, Medstrand P. Frequent Intrapatient Recombination between Human Immunodeficiency Virus Type 1 R5 and X4 Envelopes: Implications for Coreceptor Switch. J Virol. 2007;81:3369–76. doi: 10.1128/JVI.01295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy-Burman P. Endogenous env elements: partners in generation of pathogenic feline leukemia viruses. Virus Genes. 1995;11:147–61. doi: 10.1007/BF01728655. [DOI] [PubMed] [Google Scholar]
- 5.Chen BK, Rousso I, Shim S, Kim PS. Efficient assembly of an HIV-1/MLV Gag-chimeric virus in murine cells. Proc Natl Acad Sci U S A. 2001;98:15239–44. doi: 10.1073/pnas.261563198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray M, Prasad S, Dubay JW, Hunter E, Jeang KT, Rekosh D, Hammarskjold ML. A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. Proc Natl Acad Sci U S A. 1994;91:1256–60. doi: 10.1073/pnas.91.4.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deminie CA, Emerman M. Incorporation of human immunodeficiency virus type 1 Gag proteins into murine leukemia virus virions. J Virol. 1993;67:6499–506. doi: 10.1128/jvi.67.11.6499-6506.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nack U, Schnierle BS. Replacement of the murine leukemia virus (MLV) envelope gene with a truncated HIV envelope gene in MLV generates a virus with impaired replication capacity. Virology. 2003;315:209–16. doi: 10.1016/s0042-6822(03)00519-1. [DOI] [PubMed] [Google Scholar]
- 9.Owens CM, Yang PC, Gottlinger H, Sodroski J. Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J Virol. 2003;77:726–31. doi: 10.1128/JVI.77.1.726-731.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita M, Emerman M. The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog. 2005;1:e18. doi: 10.1371/journal.ppat.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ambrose Z, Boltz V, Palmer S, Coffin JM, Hughes SH, Kewalramani VN. In vitro characterization of a simian immunodeficiency virus-human immunodeficiency virus (HIV) chimera expressing HIV type 1 reverse transcriptase to study antiviral resistance in pigtail macaques. J Virol. 2004;78:13553–61. doi: 10.1128/JVI.78.24.13553-13561.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon DT, Li G, Aldovini A. Nucleocapsid and matrix protein contributions to selective human immunodeficiency virus type 1 genomic RNA packaging. J Virol. 1998;72:1983–93. doi: 10.1128/jvi.72.3.1983-1993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shikova-Lekova E, Lindemann D, Pietschmann T, Juretzek T, Rudolph W, Herchenroder O, Gelderblom HR, Rethwilm A. Replication-competent hybrids between murine leukemia virus and foamy virus. J Virol. 2003;77:7677–81. doi: 10.1128/JVI.77.13.7677-7681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logg CR, Logg A, Matusik RJ, Bochner BH, Kasahara N. Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J Virol. 2002;76:12783–91. doi: 10.1128/JVI.76.24.12783-12791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Logg CR, Logg A, Tai CK, Cannon PM, Kasahara N. Genomic stability of murine leukemia viruses containing insertions at the Env-3′ untranslated region boundary. J Virol. 2001;75:6989–98. doi: 10.1128/JVI.75.15.6989-6998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Logg CR, Tai CK, Logg A, Anderson WF, Kasahara N. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 2001;12:921–32. doi: 10.1089/104303401750195881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunnell BA, Kluge KA, Lee-Lin SQ, Byrne ER, Orlic D, Metzger ME, Agricola BA, Wersto RP, Bodine DM, Morgan RA, Donahue RE. Transplantation of transduced nonhuman primate CD34+ cells using a gibbon ape leukemia virus vector: restricted expression of the gibbon ape leukemia virus receptor to a subset of CD34+ cells. Gene Ther. 1999;6:48–56. doi: 10.1038/sj.gt.3300808. [DOI] [PubMed] [Google Scholar]
- 18.Horn PA, Topp MS, Morris JC, Riddell SR, Kiem HP. Highly efficient gene transfer into baboon marrow repopulating cells using GALV-pseudotype oncoretroviral vectors produced by human packaging cells. Blood. 2002;100:3960–7. doi: 10.1182/blood-2002-05-1359. [DOI] [PubMed] [Google Scholar]
- 19.Movassagh M, Boyer O, Burland MC, Leclercq V, Klatzmann D, Lemoine FM. Retrovirus-mediated gene transfer into T cells: 95% transduction efficiency without further in vitro selection. Hum Gene Ther. 2000;11:1189–200. doi: 10.1089/10430340050015239. [DOI] [PubMed] [Google Scholar]
- 20.Svarovskaia ES, Cheslock SR, Zhang WH, Hu WS, Pathak VK. Retroviral mutation rates and reverse transcriptase fidelity. Front Biosci. 2003;8:d117–34. doi: 10.2741/957. [DOI] [PubMed] [Google Scholar]
- 21.Barsov EV, Payne WS, Hughes SH. Adaptation of chimeric retroviruses in vitro and in vivo: isolation of avian retroviral vectors with extended host range. J Virol. 2001;75:4973–83. doi: 10.1128/JVI.75.11.4973-4983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marzio G, Verhoef K, Vink M, Berkhout B. In vitro evolution of a highly replicating, doxycycline-dependent HIV for applications in vaccine studies. Proc Natl Acad Sci U S A. 2001;98:6342–7. doi: 10.1073/pnas.111031498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Reilly L, Roth MJ. Second-site changes affect viability of amphotropic/ecotropic chimeric enveloped murine leukemia viruses. J Virol. 2000;74:899–913. doi: 10.1128/jvi.74.2.899-913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Swanstrom R, Wills JW. Synthesis, Assembly, and Processing of Viral Proteins. In: Coffin JM, Hughes SH, Varmus H, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Plainview, N.Y.: 1997. pp. 263–334. [PubMed] [Google Scholar]
- 26.Lazo PA, Prasad V, Tsichlis PN. Splice acceptor site for the env message of Moloney murine leukemia virus. J Virol. 1987;61:2038–41. doi: 10.1128/jvi.61.6.2038-2041.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green MR. Pre-mRNA splicing. Annu Rev Genet. 1986;20:671–708. doi: 10.1146/annurev.ge.20.120186.003323. [DOI] [PubMed] [Google Scholar]
- 28.Shinnick TM, Lerner RA, Sutcliffe JG. Nucleotide sequence of Moloney murine leukaemia virus. Nature. 1981;293:543–8. doi: 10.1038/293543a0. [DOI] [PubMed] [Google Scholar]
- 29.Johann SV, van Zeijl M, Cekleniak J, O'Hara B. Definition of a domain of GLVR1 which is necessary for infection by gibbon ape leukemia virus and which is highly polymorphic between species. J Virol. 1993;67:6733–6. doi: 10.1128/jvi.67.11.6733-6736.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delassus S, Sonigo P, Wain-Hobson S. Genetic organization of gibbon ape leukemia virus. Virology. 1989;173:205–13. doi: 10.1016/0042-6822(89)90236-5. [DOI] [PubMed] [Google Scholar]
- 31.Bouck J, Fu XD, Skalka AM, Katz RA. Genetic selection for balanced retroviral splicing: novel regulation involving the second step can be mediated by transitions in the polypyrimidine tract. Mol Cell Biol. 1995;15:2663–71. doi: 10.1128/mcb.15.5.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Santen VL, Spritz RA. mRNA precursor splicing in vivo: sequence requirements determined by deletion analysis of an intervening sequence. Proc Natl Acad Sci U S A. 1985;82:2885–9. doi: 10.1073/pnas.82.9.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coolidge CJ, Seely RJ, Patton JG. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 1997;25:888–96. doi: 10.1093/nar/25.4.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roscigno RF, Weiner M, Garcia-Blanco MA. A mutational analysis of the polypyrimidine tract of introns. Effects of sequence differences in pyrimidine tracts on splicing. J Biol Chem. 1993;268:11222–9. [PubMed] [Google Scholar]
- 35.Zamore PD, Patton JG, Green MR. Cloning and domain structure of the mammalian splicing factor U2AF. Nature. 1992;355:609–14. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 36.Adams MD, Rudner DZ, Rio DC. Biochemistry and regulation of pre-mRNA splicing. Curr Opin Cell Biol. 1996;8:331–9. doi: 10.1016/s0955-0674(96)80006-8. [DOI] [PubMed] [Google Scholar]
- 37.Amendt BA, Si ZH, Stoltzfus CM. Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: evidence for inhibition mediated by cellular factors. Mol Cell Biol. 1995;15:6480. doi: 10.1128/mcb.15.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arrigo S, Beemon K. Regulation of Rous sarcoma virus RNA splicing and stability. Mol Cell Biol. 1988;8:4858–67. doi: 10.1128/mcb.8.11.4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staffa A, Cochrane A. Identification of positive and negative splicing regulatory elements within the terminal tat-rev exon of human immunodeficiency virus type 1. Mol Cell Biol. 1995;15:4597–605. doi: 10.1128/mcb.15.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cullen BR. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 2003;28:419–24. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 41.Ernst RK, Bray M, Rekosh D, Hammarskjold ML. A structured retroviral RNA element that mediates nucleocytoplasmic export of intron-containing RNA. Mol Cell Biol. 1997;17:135–44. doi: 10.1128/mcb.17.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saavedra C, Felber B, Izaurralde E. The simian retrovirus-1 constitutive transport element, unlike the HIV-1 RRE, uses factors required for cellular mRNA export. Curr Biol. 1997;7:619–28. doi: 10.1016/s0960-9822(06)00288-0. [DOI] [PubMed] [Google Scholar]
- 43.Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–7. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- 44.Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. Rna. 2004;10:1489–98. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cramer P, Pesce CG, Baralle FE, Kornblihtt AR. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci U S A. 1997;94:11456–60. doi: 10.1073/pnas.94.21.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. J Biol Chem. 2002;277:43110–4. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]
- 47.Auboeuf D, Dowhan DH, Dutertre M, Martin N, Berget SM, O'Malley BW. A subset of nuclear receptor coregulators act as coupling proteins during synthesis and maturation of RNA transcripts. Mol Cell Biol. 2005;25:5307–16. doi: 10.1128/MCB.25.13.5307-5316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trubetskoy AM, Okenquist SA, Lenz J. R region sequences in the long terminal repeat of a murine retrovirus specifically increase expression of unspliced RNAs. J Virol. 1999;73:3477–83. doi: 10.1128/jvi.73.4.3477-3483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65:2220–4. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solly SK, Trajcevski S, Frisen C, Holzer GW, Nelson E, Clerc B, Abordo-Adesida E, Castro M, Lowenstein P, Klatzmann D. Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther. 2003;10:30–9. doi: 10.1038/sj.cgt.7700521. [DOI] [PubMed] [Google Scholar]
- 51.Tai CK, Wang WJ, Chen TC, Kasahara N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005;12:842–51. doi: 10.1016/j.ymthe.2005.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hiraoka K, Kimura T, Logg CR, Kasahara N. Tumor-selective gene expression in a hepatic metastasis model after locoregional delivery of a replication-competent retrovirus vector. Clin Cancer Res. 2006;12:7108–16. doi: 10.1158/1078-0432.CCR-06-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metzl C, Mischek D, Salmons B, Gunzburg WH, Renner M, Portsmouth D. Tissue- and tumor-specific targeting of murine leukemia virus-based replication-competent retroviral vectors. J Virol. 2006;80:7070–8. doi: 10.1128/JVI.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiao J, Moreno J, Sanchez-Perez L, Kottke T, Thompson J, Caruso M, Diaz RM, Vile R. VSV-G pseudotyped, MuLV-based, semi-replication-competent retrovirus for cancer treatment. Gene Ther. 2006;13:1457–70. doi: 10.1038/sj.gt.3302782. [DOI] [PubMed] [Google Scholar]
- 55.Wang WJ, Tai CK, Kasahara N, Chen TC. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 2003;14:117–27. doi: 10.1089/104303403321070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kikuchi E, Menendez S, Ozu C, Ohori M, Cordon-Cardo C, Logg CR, Kasahara N, Bochner BH. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007;14:279–86. doi: 10.1038/sj.cgt.7701013. [DOI] [PubMed] [Google Scholar]
- 57.Donahue RE, Kessler SW, Bodine D, McDonagh K, Dunbar C, Goodman S, Agricola B, Byrne E, Raffeld M, Moen R, et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J Exp Med. 1992;176:1125–35. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 59.Moloney JB. The Rodent Leukemias: Virus-Induced Murine Leukemias. Annu Rev Med. 1964;15:383–92. doi: 10.1146/annurev.me.15.020164.002123. [DOI] [PubMed] [Google Scholar]
- 60.Klein E, Klein G. Antibody response and leukemia development in mice inoculated neonatally with the Moloney virus. Cancer Res. 1965;25:851–4. [PubMed] [Google Scholar]
- 61.Cornetta K, Moen RC, Culver K, Morgan RA, McLachlin JR, Sturm S, Selegue J, London W, Blaese RM, Anderson WF. Amphotropic murine leukemia retrovirus is not an acute pathogen for primates. Hum Gene Ther. 1990;1:15–30. doi: 10.1089/hum.1990.1.1-15. [DOI] [PubMed] [Google Scholar]
- 62.Cornetta K, Morgan RA, Gillio A, Sturm S, Baltrucki L, O'Reilly R, Anderson WF. No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum Gene Ther. 1991;2:215–9. doi: 10.1089/hum.1991.2.3-215. [DOI] [PubMed] [Google Scholar]
- 63.Logg CR, Kasahara N. Retrovirus-mediated gene transfer to tumors: utilizing the replicative power of viruses to achieve highly efficient tumor transduction in vivo. Methods Mol Biol. 2004;246:499–525. doi: 10.1385/1-59259-650-9:499. [DOI] [PubMed] [Google Scholar]
- 64.Telesnitsky A, Blain S, Goff SP. Assays for retroviral reverse transcriptase. Methods Enzymol. 1995;262:347–62. doi: 10.1016/0076-6879(95)62029-x. [DOI] [PubMed] [Google Scholar]