

Abstract
Autoantibodies directed against the skeletal muscle acetylcholine receptor (AChR) play a critical role in the pathogenesis of the autoimmune disease, myasthenia gravis (MG). The pathogenic importance of anti-AChR antibodies is substantiated clinically by the often dramatic clinical improvement that follows removal of circulating antibodies utilizing extracorporeal plasma exchange (PE). Unfortunately, the effects of PE are non-specific as immunoglobulins (IgG) and other plasma proteins are removed in addition to anti-AChR IgG. In this study, we have successfully incorporated the AChR protein purified from Torpedo Californicus into a Nanodisc (ND) membrane scaffold protein/phospholipid structure. We go on to demonstrate the effectiveness of this ND-AChR complex, administered intravenously, in the in vivo down-modulation of anti-AChR antibodies and subsequent amelioration of clinical disease in the experimental murine model of MG. These results provide proof-of-principle for the in vivo antigen-specific reduction of pathogenic anti-AChR antibodies utilizing ND-AChR particles. Further development of this strategy may provide an effective, antigen-specific, and readily accessible acute therapy for exacerbating MG or myasthenic crisis.
Keywords: Acetylcholine receptor, nanodisc, myasthenia gravis, immunoadsorption
Introduction
Autoimmune myasthenia gravis (MG) is a T-cell dependent, antibody-mediated disease in which autoantibodies directed against the skeletal muscle nicotinic acetylcholine receptor (AChR) impair neuromuscular transmission resulting in a loss in the number of functional AChRs at the motor endplate (Hoedemaekers et al., 1997; Vincent et al., 2001; Meriggioli and Sanders, 2009). The AChR is a ligand-gated ion channel receptor transmembrane protein formed by five subunits (α2, β, γ (ε), and δ), with the α–subunit constituting the main immunogen involved in the majority of cases of MG (Lindstrom, 2000). The important role of the antibody-mediated attack on the AChR, resulting in destruction of the muscle endplate region is well established (Engel et al., 1977; Sahashi et al., 1978). An experimental animal model of MG, experimental autoimmune myasthenia gravis (EAMG), is induced in mice by immunization with AChR purified from the electric organs of the electric ray, Torpedo californicus (Berman and Patrick, 1980). In both MG and EAMG, anti-AChR antibodies bind to the AChR at the neuromuscular junction, activate complement, and accelerate AChR destruction, culminating in neuromuscular transmission failure and fatigable muscle weakness. The majority of pathogenic anti-AChR antibodies are directed against the main immunogenic region of the α subunit (core amino acids 67–76 and 125–147) separate from the acetylcholine binding sites, and the binding of anti-AChR autoantibodies is highly conformation-dependent (Luo et al., 2009).
An important intervention in treating MG, particularly when a quick therapeutic response is desirable, is extracorporeal plasmapheresis or plasma exchange (PE). PE has been successfully used to treat severe exacerbations of MG, and often produces rapid improvement in myasthenic weakness associated with reductions in the titer of anti-AChR-Abs and immunoglobulin (IgG) levels (Dau et al., 1977; Chiu et al., 2000; Gajdos et al., 1997). However, this method removes normal plasma components as well as IgG, and removes IgG non-specifically rather than anti-AChR IgG selectively. In addition to the removal of factors and immunoglobulins of potential pathogenic significance, nonspecific immunoglobulin depletion may have adverse affects on MG, possibly removing regulatory antibodies (Jambou et al., 2003), leading to increased synthesis of new pathogenic anti-AChR antibodies. Although very effective in inducing clinical improvement, the general usefulness of PE is also limited by its restriction to major medical centers and the frequent need for large-bore venous catheters. Infections and thrombotic complications related to venous access occur (Gajdos et al., 1997; Seybold, 1987). PE can also reduce coagulation factors, particularly with repeated treatments, leading to bleeding tendencies (Seybold, 1987).
Nanodiscs (ND) are soluble, nanoscale phospholipid bilayers which can self-assemble and incorporate membrane proteins for biophysical, enzymatic or structural investigations (Borch and Hamann, 2009; Nath et al., 2007). The ND consists of a non-covalent assembly of a phospholipid bilayer surrounded by an annulus composed of two copies of the amphipathic membrane scaffold protein (MSP) (Denisov et al., 2004). A trans- membrane protein inserted in a Nanodisc is thus surrounded by a lipid bilayer providing an environment that approximates its native state. The ND system provides a novel platform that has been utilized mainly for the purpose of understanding membrane protein function. Recently however Nanodisc incorporated Hemagglutinin vaccine has been shown to illicit a protective immune response in an animal model, demonstrating the potential of Nanodiscs as a vaccine platform (Bhattacharya et al., 2010). Membrane associated proteins, such as the AChR, are particularly suited for ND incorporation, potentially allowing for other in vivo delivery applications in addition to vaccines. We investigated a novel application of this technology, hypothesizing that AChR incorporated Nanodiscs (ND-AChR) could function as effective “autoantibody traps” for antigen-specific, adsorption of pathogenic anti-AChR antibodies in MG. Accordingly, we have successfully incorporated the AChR protein purified from Torpedo Californicus into the ND MSP/phospholipid structure, and report the effects of intravenous administration of ND-AChRs on disease severity and levels of anti-AChR antibodies in EAMG.
Materials and Methods
Purification of T. Californicus AChR
AChR was purified from the electric organs of Torpedo californicus by affinity chromatography using a conjugate of neurotoxin coupled to agarose, as previously described (Wu et al., 1997; Sheng et al., 2006). Purity of the isolated product was tested by SDS-PAGE. Intact AChR complex was obtained in mg quantities by extraction with Triton X-100 and subsequent chromatographic separation. The purified AChR was used for incorporation into Nanodiscs as well as to induce EAMG.
Protein isolation and purification and assembly of ND-AChR
Details of the production and purification of the membrane scaffold protein (MSP) component and the assembly of ND-AChR are available on-line. Briefly, assembly conditions for Nanodiscs were performed as described previously (Bayburt et al., 2007). Purified AChR (0.35 mg/ml) obtained from Torpedo californicus membranes (Wu et al., 1997) was incubated with sodium cholate solubilized phospholipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and membrane scaffold proteins (MSP1E3D1, 32.6 kDa) in the presence of 0.1% Triton X-100 while incubating at 4°C. Torpedo ND-AChR assemblies were formed by a self-assembly reaction initiated by the removal of detergent using hydrophobic Bio-Beads (Denisov et al., 2004; Bayburt et al., 2003).
Purification of ND-AChR
The ND-AChR assembly mixture was batch adsorbed with Ni-NTA resin equilibrated with 20 mM Tris-HCl (pH 7.4), 100 mM NaCl. The column was washed with 50 mM imidazole and ND and ND-AChR particles were eluted with 0.4M imidazole. Ni-column eluate was dialyzed overnight at 4°C against low-ionic strength 20mM Tris-HCl buffer (pH 7.4), 20 mM NaCl and the solution was passed over an affinity resin comprised of the acetylcholine analog 4,7,10-trioxa-1,13-carboxyethyl-2-trimethylaminetridecanediaamine (TDAC) (Tierney et al., 2004). The column was extensively washed with 10 column volumes of low-salt buffer to eliminate empty NDs, and ND-AChR was eluted with a step gradient of 50–150 mM NaCl to enrich the sample for ND-AChR. The 50 and 100 mM fractions containing AChR were concentrated (Amicon Ultra 15, 10,000 MWCO), passed through a 0.2 μm filter and separated by SEC.
ND-AChR quantification
AChR content and ND content (as determined by concentration of MSP) were measured by ELISA. Briefly, 96 well ELISA plates were coated with varying amounts of AChR and MSP in 0.1 M carbonate buffer (pH 9.6) by overnight incubation at 4° C. Plates were blocked (25° C for 2h) with PBS containing Tween 20 (0.05% v/v) and BSA (3% w/v). After a thorough washing with PBS-Tween 20 (0.05% v/v) the plates were further incubated with anti-AChR antibodies (1:5000, from EAMG mouse sera) or anti-His tag antibodies (1:5000, BD 6xHistag Ab). After further washing, samples were incubated (25° C for 1 h) separately with HRP anti-mouse IgG (1:5000) (for AChR), and HRP anti-mouse IgG2b (1:5000) (for MSP). Samples were then incubated with TMB substrate solution in the dark for 15 minutes. The reaction was stopped by adding 2M H2SO4 and the colorimetric change was measured as the optical density at 450 nm (OD450) using a microplate reader (Bio-Rad, Model 550). Concentration dependent standard curves were constructed for known quantities of AChR and MSP from the respective OD450 values, and AChR and MSP concentrations of the unknown ND-AChR mixtures were then calculated from the corresponding OD450 values by extrapolation. The concentration of AChR in the pooled sample used for treatment was determined to be 7.5 μg/100 μL; for MSP 15 μg/100 μL. The volume administered was 200 μL, based on an estimate of a safe volume for injection in the mouse.
Induction and Clinical Scoring of EAMG
Animal experiments were carried out under a protocol approved by the Animal Care and Use Committee of the University of Illinois at Chicago. Eight-week old female C57BL6/J mice were immunized with 40 μg of Torpedo AChR emulsified in CFA, 200ul subcutaneously, and received two “boosts” with 20μg of AChR in IFA in 200 μl volume injected in the flanks and tail base every 30 days. Mice were observed and scored every other day. For clinical examination, mice were evaluated for myasthenic weakness and assigned clinical scores as previously described (Sheng et al., 2006; Sheng et al., 2008). Briefly, mice were observed on a flat platform for a total of two minutes. They were then exercised by gently dragging them suspended by the base of the tail across a cage top grid repeatedly (20–30 times) as they attempted to grip the grid. They were then placed on a flat platform for two minutes and again observed for signs of EAMG. Clinical muscle weakness was graded as follows: grade 0, mouse with normal posture, muscle strength, and mobility at baseline and after exercise; grade 1, normal at rest but with muscle weakness characteristically shown by a hunchback posture, restricted mobility, and difficulty in raising the head after exercise; grade 2, grade 1 symptoms without exercise during observation period; grade 3, dehydrated and moribund with grade 2 weakness; and grade 4, dead. The evaluator was blinded to treatment status for all clinical evaluations.
Administration of ND-AChR
After initial priming and two booster immunizations, the mice were divided into four groups of 10 mice per group, consisting of equal numbers of mice with various disease severities in each group. Mice received 1) ND-AChR, 2) “bare AChR” (7.5 μg), 3) empty ND (15 μg), or 4) PBS in a total volume of 200 μL via tail vein injection on five consecutive days, followed by 2 days of “rest”, and then five additional days of treatment. The day of treatment initiation was designated “day 0” and the mice were evaluated for myasthenic weakness and assigned clinical scores as described above.
ELISA for anti-mouse AChR antibody isotypes and serum C3 levels
Mice were bled prior to initiation of treatment and 8 days following completion of therapy. Affinity-purified mouse AChR (0.5ug/ml) was used to coat 96-well microtiter plates (Corning Costar 96 wells plate, eBioscience, San Diego, CA) with 0.1 M carbonate bicarbonate buffer (pH 9.6) overnight at 4 °C. Serum samples diluted 1:2000 to 1:10000 in blocking buffer were added and incubated at 37 °C for 90 min. After four washes, horseradish peroxidase-conjugated (HRPO) goat anti-mouse IgG and IgG2b were diluted in 1:2000 (Caltag Laboratories, Burlingame, MA) in blocking buffer were added and incubated at 37 °C for 90 min. Subsequently, TMB substrate solution (eBioscience) was added, and color was allowed to develop at room temperature in the dark for 15 minutes. The reaction was stopped by adding 2M H2SO4 and absorbance values were measured at a wavelength of 450 nm using a microplate reader (Bio-Rad, model 550) and the results were expressed as OD values.
For complement C3 determination, ninety-six-well microtiter plates were covered with goat Abs to mouse C3 (ICN Biomedicals/Cappel, Aurora, OH) in 0.1 M sodium carbonate buffer (pH 8.2) overnight at 4°C. The plates were then blocked with 2% BSA in PBS at room temperature for 30 min. Diluted serum samples (30 μl) were added and incubated at 37°C for 90 min. After four washes, HRP-conjugated goat anti-mouse C3 complement (ICN Biomedicals/Cappel), diluted 1/500 in dilution buffer, was added and incubated at 37°C for 90 min.
Negative Stain Electron Microscopy
Copper grids (TAAB, 400 mesh) on a formvar film were coated with a thin layer of carbon and glow discharged for 1–2 minutes prior to use. Protein samples (2–5μl) were added to the carbon-coated grids and stained with 1% uranyl acetate. Samples were viewed using a Phillips CM100 electron microscope operating at 100 kV. Specimens were viewed at 46,000x nominal magnification and digital images were recorded using an AMT-V600 CCD.
Histological Studies
At the end of the study (18 days after completion of treatment), mice were euthanized and the vital organs (liver, kidney, spleen and lungs) were collected and fixed with a 10% formalin neutral buffer solution, embedded in paraffin, and cut into 5-μm-thick sections. Sections were stained with hematoxylin and eosin (H and E) staining for general histology according to published literature (Tarnopolsky MA et al., 2003).
Statistics
Mean, SD, and statistical significance were calculated using SPSS software applications. Student’s t-test was utilized for determining the significance of autoantibody changes in ND-AChR-treated animals compared to controls (PBS). A value of p ≤0.05 was considered significant.
3. Results
Incorporation of AChR into NDs
Self-assembled Nanodiscs incorporate membrane proteins into a lipid bilayer contained within stable discoidal structures of controlled size (Denisov et al., 2004; Bayburt et al., 2003). Once assembled, the structures remain soluble and retain their membrane protein payload in the lipid membrane plug without the need for surfactants. The principle of the self-assembly process has been well studied (Denisov et al., 2004; Bayburt et al., 2003), and pioneering studies for proteins containing various transmembrane segments, membrane associations, and multimeric complexes have been reported (Bayburt et al., 2003; Bayburt et al., 2006; Bayburt et al., 2007; Boldog et al., 2006; Leitz et al., 2006). We used this system to incorporate purified AChR, consisting of five subunits (α2, β, γ (ε), and δ), from the electric organ of Torpedo Californicus. ND-AChR complexes were formed by a self-assembly reaction using a ratio of 200 ND : 1 AChR, and initiated by the removal of detergent using hydrophobic beads as described [29] (Fig. 1). Because ND-AChR particles are prepared in the presence of an excess of MSP and phospholipid, the self-assembly process yields a mixture of ND-AChR and empty ND.
Figure 1.

Schematic figure illustrating protocol utilized for incorporation of T. Californicus AChR into the Nanodisc structure. Purified AChR obtained form Torpedo californicus membranes was incubated with sodium cholate solubilized phospholipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and membrane scaffold proteins (MSP1E3D1, 32.6 kDa). ND-AChR assemblies were formed by a self-assembly reaction initiated by the removal of detergent using hydrophobic beads. A dual affinity chromatography process was used to purify ND/nAChR particles. First, the assembly mixture was batch adsorbed with Ni-NTA resin (Ni column) and the His-tag present in the MSPs was allowed to bind to the resin. The Ni-column eluate, containing a mixture of excess empty NDs and ND-AChR, was dialyzed against low-ionic strength Tris buffer, passed over an affinity resin comprised of the acetylcholine analog 4,7,10-trioxa-1,13-carboxyethyl-2-trimethylaminetridecanediaamine (TDAC), and ND/AChR was eluted.
Confirmation of successful assembly and purification of ND-AChR
Size-exclusion chromatography (SEC) was used to assess the efficiency of the self-assembly reaction by comparison to the Stokes radius of protein standards and empty ND controls. First, this analysis indicated no gross formation of aggregated material in the column void (15–16 min). Second the primary protein peak, which eluted at ~26 min was identical to that of empty ND control and represents particles of ~12.5 nm in size, the expected size of MSP1E3D1 based ND. This is consistent with the 200-fold excess of ND used in the self-assembly mixture. Following TDAC affinity purification, which eliminates empty ND but retains ND-AChR particles, the primary peak remains consistent with MSP1E3D1 derived particles, though it reflects a shorter SEC retention time due to the increased hydrodynamic that incorporated AChR contributes to the ND particle (Fig. 2A). Western blot analysis of SEC fractions indicated that the bulk of AChR had eluted between 19–26min (data not shown), based on the calibrated standards, this elution profile is consistent with the predicted 16 nm length of an AChR pore. Finally, fractionation of AChR alone resulted in aggregation of AChR and elution in the column void volume.
Figure 2.

Purification of ND-AChR. A. Size-exclusion chromatography (SEC) assessing the efficiency of the self-assembly reaction (post-assembly) with comparison to TDAC purified ND-AChR particles. The primary protein peak eluted at ~26 min which was identical to that of empty ND control and represents particles of ~12.5 nm in size, the expected size of MSP1E3D1 based ND (Boldog et al., 2007). B. The eluted sample following purification (containing a mixture of assembled ND-AChR and empty NDs) was further purified by TDAC affinity chromatography and analyzed by SDS-PAGE. The sample was loaded (FT= flow through) and column was processed with a step gradient of NaCl (20mM, 50mM, 100mM). Increasing NaCl concentration enriches sample for ND-AChR as demonstrated on SDS-PAGE gel (lower panel). C. Western blot (using mouse anti-AChR sera) of ND-AChR self-assembly mixture (Mix) following purification and SDS-PAGE separation. Mixture was loaded (FT), washed with 50mM imidazole buffer (W1, W2) and eluted with 0.4M imidazole (El). Four bands corresponding to the four AChR subunits are apparent.
A dual affinity chromatography process was used to purify ND-AChR particles as described in Materials and Methods. First, the assembly mixture was batch adsorbed with Ni-NTA resin and the 6xHis-tag present on the MSP was allowed to bind to the resin. The column was subsequently washed to remove any unincorporated AChR and ND-AChR particles were eluted. The eluted solution, containing a mixture of excess empty NDs and ND-AChR, was dialyzed against low-ionic strength Tris buffer (20 mM NaCl) and the solution was passed over TDAC affinity resin which contains an acetylcholine analog that binds only to intact and properly folded AChR (Tierney et al., 2004). Empty NDs were then removed by extensive washing in low-ionic strength buffer. The ligand binding site of AChR is located between each α subunit and the face of its respective γ or δ subunit neighbor (Wang, 1996). ND-AChRs bind tightly to TDAC-sepharose under low ionic strength conditions and this binding strongly suggests that the receptor complex is intact. In contrast, snake venom toxins which are frequently used to purify AChR (Wu et al., 1997), bind only to the α-subunit regardless of whether the complex is intact. Accordingly, ND-AChR binds at 20 mM NaCl and elutes between 50–100 mM NaCl, so ND-AChR particles were eluted by increasing ionic strength (50–100 mM NaCl) (Fig. 2B). The purified mixture was then concentrated (Fig 2C) and separated into fractions by SEC. After correcting for the dilution ratios used in the respective ELISA assays, the pooled fractions were found to contain 75 μg/mL AChR and 150 μg/mL MSP. The ND-AChR mixture was then concentrated, exchanged with TBS and stored for the in vivo adsorption of anti-AChR antibodies.
Structural assessment of ND-AChR
We further assessed the ND-AChR mixture from a structural/morphologic perspectives by subjecting the purified sample to negative stain electron microscopy. Visualized individual particles showed an apparent five-fold symmetry, consistent with the known five subunit molecular structure of the AChR (Fig. 3).
Figure 3.
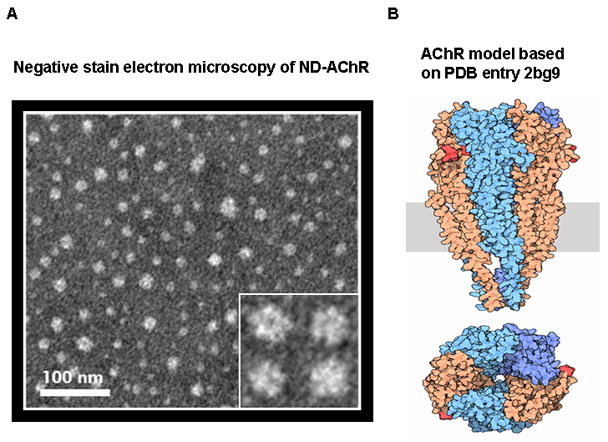
Negative stain electron microscopy of ND-AChR. A. Inset depicts individual particles at 4x magnification which show apparent five-fold symmetry consistent with the known structure of the AChR (B). Diagram of the AChR receptor (Unwin, 2005; reproduced with permission from RCSB PDB: http://dx.doi.org/10.2210/rcsb_pdb/mom_2005_11) for comparison, showing extracellular (above rectangle), transmembrane (rectangle) and intracellular (below rectangle) domains. Bottom figure shows view down the pore axis.
Intravenous administration of ND-AChR complex ameliorates clinical EAMG
We then tested the efficacy of intravenously administered ND-AChR in the treatment of EAMG. Disease severity scores were monitored on an every other day basis, and are plotted for the 4 experimental groups in Fig. 4. In the PBS and ND treatment groups, there was no change in the average clinical disease score over the length of the study. As shown in Fig. 4, the average disease severity in the ND-AChR- treated animals gradually lessened, while in the “bare AChR” treatment group, a gradual worsening of disease severity was noted, coinciding with changes seen in autoantibody levels (see below).
Figure 4.

Effects of ND-AChR on clinical disease severity. Mice received a priming immunization with AChR and two booster immunizations administered 30 days apart. This resulted in clinical myasthenic weakness with an average clinical score of approximately 1.5. Clinical disease severity in the four experimental groups (AChR (n=10) : “bare” AChR; PBS (n=7): phosphate buffered saline; ND (n=7): unincorporated nanodisc; ND-AChR (n=10): nanodisc incorporated AChR) is plotted for 30 days after initiation of treatment. Mice received treatment on days 1–5 and days 8–12. Results shown are representative of two separate experiments.
Intravenous administration of ND-AChR reduces circulating levels of anti-AChR antibodies
We measured the serum concentration of anti-mouse AChR antibodies and circulating levels of C3 by ELISA prior to and after treatment (Fig. 5). Although all 4 experimental groups had comparable clinical disease severity, the group of animals treated with “bare AChR” protein had lower baseline autoantibody titers, on average. Total anti-AChR IgG levels showed no significant post-treatment change in mice treated with empty ND and PBS; as expected, mice given “bare AChR” protein actually had increased circulating autoantibody levels (Fig. 5A). In mice receiving ND-AChR, the anti-AChR antibody levels decreased after treatment, and this observed decrease included a reduction in the complement-fixing (and likely pathogenically relevant) IgG2b isotype (Fig 5B). Furthermore, circulating levels of the complement component C3 was significantly reduced in the ND-AChR treatment group (Fig. 5C). Examination of the time course of anti-AChR reduction in the ND-AChR treatment group indicates that the major reduction in autoantibody levels occurred on or before day 3 (following completion of therapy), and this reduction persisted until day 17 (Fig. 6).
Figure 5.

Anti-AChR antibody levels, IgG (A) and IgG2b (B) and serum C3 levels (C) before and after treatment in the four experimental groups. Significant reductions in anti-AChR antibody levels and serum C3 occurred only in mice treated with ND-AChR. Results are from one representative experiment, of two performed (n=7 per experimental group for ND and PBS; n=10 per group for ND-AChR and AChR; total number of mice used = 46).
Figure 6.
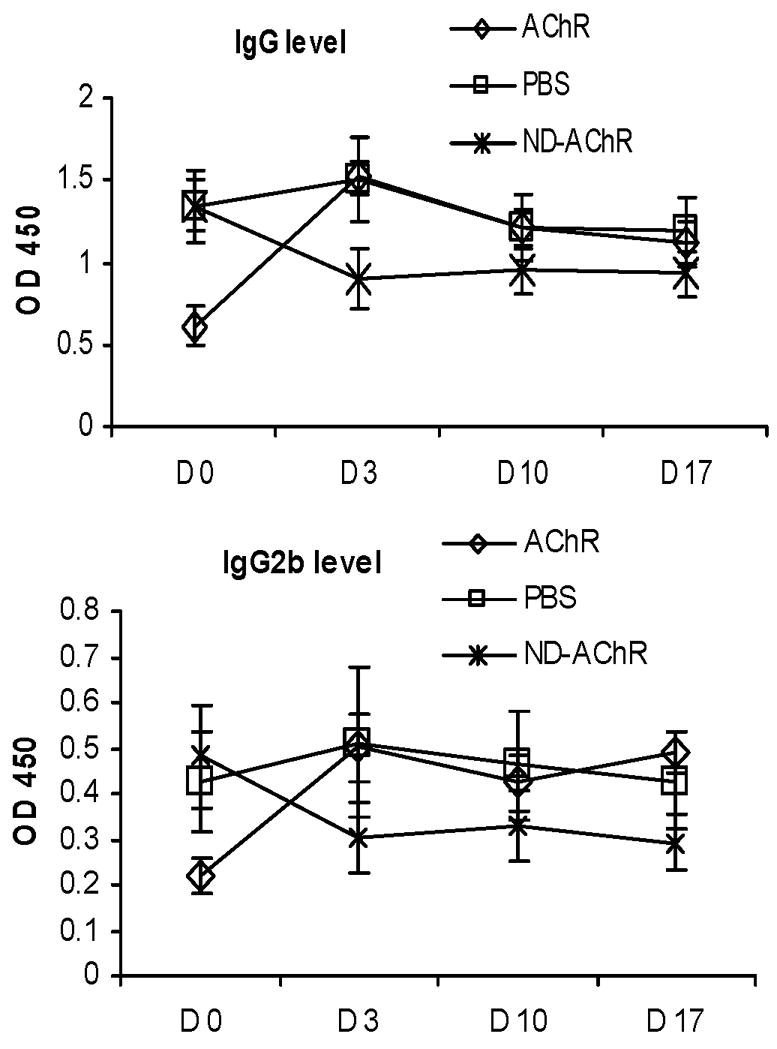
Time course of antibody decrease for IgG (A) and IGg2b (B) showing onset of decrease in ND-AChR-treated animals on or before day 3 and persisting until day 17 after completion of treatment course (n=5 per experimental group).
Intravenous administration of ND-AChR causes no obvious organ toxicity
There was no mortality during the course of the performed studies in any of the experimental groups. At the end of the treatment, weight gain in the treated as well as control groups was similar. Gross changes were not observed in any of the tissues/organs obtained from mice in the four treatment groups. Histopathological sampling of the lungs, liver, spleen, and kidneys revealed no discernable differences amongst the four groups. Representative sections from an ND-AChR-treated animal compared to the PBS control are shown in supplemental figure 1.
Discussion
Autoantibodies against the AChR play a critical role in the pathogenesis of MG mainly by causing a complement-mediated loss of functional receptors on the postsynaptic membrane (Vincent, 2006; Meriggioli and Sanders, 2009). As noted, the removal of anti-AChR antibodies utilizing PE results in clinical improvement in human MG, but has limitations. Recently, immunoadsorption (IA) columns have been developed using recombinant AChR fragments for selective, antigen-specific, ex vivo removal of anti-AChR antibodies (Psaridi-Linardki et al., 2005; Guo et al., 2005; Grob et al., 1995). While this strategy may represent a feasible therapeutic option to remove only the pathogenic autoantibodies from MG patient’s sera, the other limitations of extracorporeal plasma exchange (e.g., requirement for large bore venous access) are not addressed. Furthermore, the adsorption capacity of these columns and the incubation time needed for efficient adsorption of antibodies are also potential therapeutic limitations.
We have therefore investigated a novel technique for the in vivo, antigen-specific down-modulation of anti-AChR antibodies via intravenous administration of ND-AChR, and demonstrate herein the effectiveness and safety of this strategy in the murine model of MG. Since the majority of antibodies in MG patients are highly conformation-dependent (Luo et al., 2009), efficient adsorption of anti-AChR antibodies would require AChR with a conformation close to that of its native state, and ND incorporation of AChR provides an ideal method to achieve this.
We therefore first demonstrated the successful incorporation of AChR into the ND complex, and present evidence suggesting that a single AChR is not only incorporated in each ND, but also has an incorporated structure consistent with its native conformation, as demonstrated by negative stain electron microscopy showing particles with an apparent five subunit structure (Fig. 4). Having confirmed successful incorporation of ND-AChR, we proceed to to demonstrate the effectiveness of intravenous administration of ND-AChR in the in vivo reduction of anti-AChR antibodies and amelioration of signs of myasthenic weakness in EAMG. We limited our studies to the assessment of antibody concentrations, clinical disease scores, and assessment of toxicity in an effort to demonstrate efficacy and safety of this approach and to provide proof-of-principle for future studies. Importantly, the treatment was timed to coincide with expected rising antibody levels (eight days after the third immunization with AChR). Both serum anti-AChR antibody levels and clinical disease scores were reduced after treatment only in the group of mice treated with ND-AChR; while the control groups showed no significant change or actually worsened. The associated reduction in circulating C3 levels in mice receiving ND-AChR may be explained by the removal of circulating immune complexes or may indicate a concomitant inhibition in complement system reactivity. A similar effect has been reported in MG patients receiving extracorporeal immunoadsorption (Ptak and Lochman, 2005).
Interestingly, anti-AChR antibody levels increased (and disease severity worsened) in animals receiving “bare” AChR protein. This raises the obvious concern that administration of autoantigen intravenously may actually exacerbate, worsen, or even induce autoimmunity. It is important to note that in induced autoimmune animal models, induction of the anti-self response absolutely requires immunization with self antigen in an immunogenic form (e.g., in complete Freund’s adjuvant (CFA)) (Billiau and Mathys, 2001). Immunization with self-antigen alone is not sufficient. Notably, intravenous administration of soluble, self-antigen in a non-immunogenic form (without CFA) prior to immunization with an immunogenic form of the same antigen can actually induce peripheral tolerance and prevent induction of autoimmune disease (Gregorian et al., 1993). Administration of “bare” AChR in our studies may have activated and broadened the immune response since both extracellular and intracellular portions of the AChR were exposed. Additionally, a membrane protein such as AChR, when not incorporated into a membrane, will likely acquire a non-native conformation, and will likely be present in an aggregated, and much more immunogenic form. These features most likely rendered the protein “foreign” to the immune system. In contrast, embedding the AChR in ND may have prevented exposure of “hidden” epitopes, while the extracellular portions of the AChR were in a conformation close to the native state, thereby reducing immunogenicity, and possibly promoting tolerance. This speculation is consistent with the notion that most autoimmune responses are initiated by “altered self proteins” (Notkins et al., 1984).
Thus, our studies indicate that administration of bare “AChR” appears to exacerbate disease, “empty” ND has no effect, and ND-AChR ameliorates disease and lowers anti-AChR antibody levels. In mice, induction of experimental autoimmune thyroiditis and mouse thyroglobulin (MTg)-specific antibody production have been shown to be suppressed by intravenous administration of de-aggregated MTg in a nonimmunogenic manner, and CD4+ T suppressor cells were required for this suppression (Tang and Braley-Mullen, 1997). This observation raises the question of a possible alternative mechanism for the therapeutic effects of ND-AChR in our studies, via mobilization of tolerogenic suppressor cells, which may be explored in future studies. However, the induction of tolerance is unlikely to explain the changes in anti-AChR levels which occurred relatively rapidly (3 days).
For treatment of human MG, there are significant and obvious limitations associated with the use of a purified, non-human protein (Torpedo AChR), as was utilized in our investigations. The use of purified human receptor is probably not practical since native human AChR is available only in small amounts. Alternatively, large recombinant AChR fragments have been produced in several laboratories, and specifically, the N-terminal extracellular domain of the α-subunit (amino acids 1–210) has recently been expressed in P. pastoris with a native-like conformation, as shown by the high-affinity binding of conformation-dependent anti-AChR antibodies [32]. The use of this or a similar ND-incorporated recombinant AChR protein could potentially be tested for its ability to adsorb MG patients’ anti-AChR antibodies in ex vivo experiments initially.
In addition, the reduction in anti-AChR antibody levels and clinical severity appear to be modest in our studies, and a relatively large amount of ND-AChR was utilized. Nevertheless, the reduction in anti-AChR antibody levels was statistically significant and was associated with an observable clinical effect. As this is a proof-of-principle study, dose-ranging experiments were not performed, but it is possible that lower doses/concentrations may be as effective.
We observed no evidence for significant systemic or organ toxicity in our studies. The precise mechanism of clearance of nanoparticles is not completely understood, and there are very few studies in terms of in vivo biodistribution. Organs that take up nanostructures include the spleen, lymph node, and bone marrow, all of which contain large concentrations of macrophages (Fischer and Chan, 2007). It is therefore hypothesized that ND complexes are taken up by phagocytic cells of the reticuloendothelial system, but once sequestered, their ultimate fate is not known. Therefore, the use of ND-AChR for the selective, in vivo down-modulation of anti-AChR antibodies in patients will require careful in vivo toxicity studies in animal models to confirm that repeated intravenous administration of ND-AChR is safe, as well as effective. In summary, we have demonstrated proof-of-principle for the in vivo, antigen-specific down-modulation of anti-AChR antibodies utilizing ND-AChR administered intravenously in the murine model of MG. Reduction of circulating anti-AChR antibody levels was accompanied by amelioration of myasthenic weakness, without significant systemic or organ toxicity. These results suggest that further development of this strategy for in vivo modulation of circulating autoantibodies may provide a safe and accessible alternative to PE or other related extracorporeal techniques.
Supplementary Material
Representative histopathological studies (clockwise from upper left) of kidney, liver, spleen and lung from mice receiving ND-AChR compared to PBS control group. Magnification is 200X.
Acknowledgments
This work was supported by the NIH (National Institute of Neurologic Disorders and Stroke, K08NS058800, MNM; and National Institute of Allergy and Infectious Diseases, RO1 AI 058190, BSP); and by the Myasthenia Gravis Foundation of America –Postdoctoral Fellowship Award to JRS.
This work was partially funded by Nanodisc Inc., Enterprise Works Room 207, 60 Hazelwood, Champaign, IL 61820 USA.
The authors gratefully acknowledge Wancai Yang, M.D. for assistance with the histological studies for organ toxicity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bayburt TH, Sligar SG. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 2003;12:2476–2481. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Grinkova YV, Sligar SG. Assembly of single bacteriorhodopsin trimers in bilayer Nanodiscs. Arch Biochem Biophys. 2006;450:215–222. doi: 10.1016/j.abb.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- Berman PW, Patrick J. Experimental myasthenia gravis: a murine system. J Exp Med. 1980;151:204–223. doi: 10.1084/jem.151.1.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P, Grimme S, Ganesh B, Gopisetty A, Sheng JR, Martinez O, Jayarama S, Artinger M, Meriggioli M, Prabhakar BS. Nanodisc-incorporated hemagglutinin provides protective immunity against influenza virus infection. J Virol. 2010;84:361–371. doi: 10.1128/JVI.01355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukocyte Biol. 2001;70:849–860. [PubMed] [Google Scholar]
- Boldog T, Grimme S, Li M, Sligar SG, Hazelbauer GL. Nanodiscs separate chemoreceptor oligomeric states and reveal their signaling properties. Proc Natl Acad Sci U SA. 2006;103:11509–11514. doi: 10.1073/pnas.0604988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldog T, Li M, Hazelbauer GL. Using Nanodiscs to create water-soluble transmembrane chemoreceptors inserted in lipid bilayers. Methods Enzymol. 2007;423:317–335. doi: 10.1016/S0076-6879(07)23014-9. [DOI] [PubMed] [Google Scholar]
- Borch J, Hamann T. The Nanodisc: a novel tool for membrane protein studies. Biol Chem. 2009;390:805–814. doi: 10.1515/BC.2009.091. [DOI] [PubMed] [Google Scholar]
- Chen PS, Toribara TY, Warner H. Microdetermination of Phosphorous. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- Chiu HC, Chen WH, Yeh JH. The six year experience of plasmapheresis in patients with myasthenia gravis. Ther Apher. 2000;4:291–295. doi: 10.1046/j.1526-0968.2000.004004291.x. [DOI] [PubMed] [Google Scholar]
- Dau PC, Lindstrom JM, Cassel CK, Denys EH, Shev EE, Spitler LE. Plasmaphereses and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134–1140. doi: 10.1056/NEJM197711242972102. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Engel AG, Lambert EH, Howard FM. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc. 1977;52:267–280. [PubMed] [Google Scholar]
- Fischer HC, Chan WCW. Nanotoxicity: the growing need for in vivo study. Curr Opin Biotechnol. 2007;18:565–571. doi: 10.1016/j.copbio.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group. Ann Neurol. 1997;41:789–796. doi: 10.1002/ana.410410615. [DOI] [PubMed] [Google Scholar]
- Gregorian SK, Clark L, Heber-Katz E, Amento EP, Rostami A. Induction of peripheral tolerance with peptide-specific anergy in experimental autoimmune neuritis. Cell Immunol. 1993;150:298–310. doi: 10.1006/cimm.1993.1198. [DOI] [PubMed] [Google Scholar]
- Grob D, Simpson D, Mitsumoto H, Hoch B, Mokhtarian F, Bender A, Greenberg M, Koo A, Nakayama S. Treatment of myasthenia gravis by immunoadsorption of plasma. Neurology. 1995;45:338–344. doi: 10.1212/wnl.45.2.338. [DOI] [PubMed] [Google Scholar]
- Guo CY, Li ZY, Xu MQ, Yuan JM. Preparation of an immunoadsorbent coupled with a recombinant antigen to remove anti-acetylcholine receptor antibodies in abnormal serum. J Immunol Meth. 2005;303:142–147. doi: 10.1016/j.jim.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Hoedemaekers AC, van Breda Vriesman PJ, De Baets MG. Myasthenia gravis as a prototype autoimmune receptor disease. Immunol Res. 1997;16:341–354. doi: 10.1007/BF02786398. [DOI] [PubMed] [Google Scholar]
- Jambou F, Zhang W, Menestrier M, Klingel-Schmitt I, Michel O, Caillat-Zucman S, Aissaoui A, Landemarre L, Berrih-Aknin S, Cohen-Kaminsky S. Circulating regulatory anti-T cell receptor antibodies in patients with myasthenia gravis. J Clin Invest. 2003;112:265–274. doi: 10.1172/JCI16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of Beta2-adrenergic receptors utilizing self-assembling Nanodisc technology. Biotechniques. 2006;40:601–602. doi: 10.2144/000112169. [DOI] [PubMed] [Google Scholar]
- Lindstrom JM. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453–477. doi: 10.1002/(sici)1097-4598(200004)23:4<453::aid-mus3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Luo J, Taylor P, Losen M, de Baets MH, Shelton GD, Lindstrom J. Main immunogenic region structure promotes binding of conformation-dependent myasthenia gravis autoantibodies, nicotinic acetylcholine receptor conformation maturation, and agonist sensitivity. J Neurosci. 2009;29:13898–13908. doi: 10.1523/JNEUROSCI.2833-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriggioli MN, Sanders DB. Myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–490. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Atkins WM, Sligar SG. Application of phospholipid bilayer Nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- Notkins AL, Onodera T, Prabhakar BS. Virus-induced autoimmunity. In: Notkins AL, Oldstone MBA, editors. Concepts in Viral Pathogenesis. Springer-Verlag; New York: 1984. pp. 210–215. [Google Scholar]
- Psaridi-Linardki L, Trakas N, Mamlaki A, Tzartos J. Specific immunoadsorption of the autoantibodies from myasthenic patients using the extracellular domain of the human muscle acetylcholine receptor α-subunit. J Neuroimmunol. 2005;159:183–91. doi: 10.1016/j.jneuroim.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Ptak J, Lochman J. Imunoadsorption therapy and complement activation. Transfus Apher Sci. 2005;32:263–267. doi: 10.1016/j.transci.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Sahashi K, Engel AG, Linstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of immune complexes (IgG and C3) at the end-plate in experimental autoimmune myasthenia gravis. J Neuropathol Exp Neurol. 1978;37:212–223. doi: 10.1097/00005072-197803000-00008. [DOI] [PubMed] [Google Scholar]
- Seybold ME. Plasmapheresis in myasthenia gravis. Ann NY Acad Sci. 1987;505:584–587. doi: 10.1111/j.1749-6632.1987.tb51326.x. [DOI] [PubMed] [Google Scholar]
- Sheng JR, Li LC, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of experimental autoimmune myasthenia gravis (EAMG) by Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) is associated with an expansion of FoxP3+ regulatory T cells. J Immunol. 2006;177:5296–52306. doi: 10.4049/jimmunol.177.8.5296. [DOI] [PubMed] [Google Scholar]
- Sheng JR, Li L, Ganesh BB, Prabhakar BS, Meriggioli MN. Regulatory T cells induced by GM-CSF suppress ongoing experimental myasthenia. Clin Immunol. 2008;128:172–180. doi: 10.1016/j.clim.2008.03.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Braley-Mullen H. Intravenous administration of deaggregated mouse thyroglobulin suppresses induction of experimental autoimmune thyroiditis and expression of both Th1 and Th2 cytokines. Int Immunol. 1997;9:679–687. doi: 10.1093/intimm/9.5.679. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Bourgeois JM, Snow R, Keys S, Roy BD, Kwiecien JM, Turnbull J. Histological assessment of intermediate and long-term creatine monohydrate supplementation in mice and rats. Am J Physiol Regul Integr Comp Physiol. 2003;285:R762–769. doi: 10.1152/ajpregu.00270.2003. [DOI] [PubMed] [Google Scholar]
- Tierney ML, Osborn KE, Milburn PJ, Stowell MH, Howitt SM. Phylogenetic conservation of disulfide-linked, dimeric acetylcholine receptor pentamers in southern ocean electric rays. J Exp Biol. 2004;207:3581–3590. doi: 10.1242/jeb.01204. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet. 2001;357:2122–2128. doi: 10.1016/S0140-6736(00)05186-2. [DOI] [PubMed] [Google Scholar]
- Vincent A. Immunology of disorders of neuromuscular transmission. Acta Neurol Scand Suppl. 2006;183:1–7. doi: 10.1111/j.1600-0404.2006.00605.x. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Hardy SF, Hall ZW. Assembly of the nicotinic acetylcholine receptor. J Biol Chem. 1996;271:27575–27584. doi: 10.1074/jbc.271.44.27575. [DOI] [PubMed] [Google Scholar]
- Wu B, Goluszko E, Christadoss P. Experimental Autoimmune Myasthenia Gravis in the Mouse. Curr Protoc Immunol. 2001;15(Unit 15.8) doi: 10.1002/0471142735.im1508s21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative histopathological studies (clockwise from upper left) of kidney, liver, spleen and lung from mice receiving ND-AChR compared to PBS control group. Magnification is 200X.