

Abstract
Gastrointestinal inflammation significantly affects the electrical excitability of smooth muscle cells. Considerable progress over the last few years have been made to establish the mechanisms by which ion channel function is altered in the setting of gastrointestinal inflammation. Details have begun to emerge on the molecular basis by which ion channel function may be regulated in smooth muscle following inflammation. These include changes in protein and gene expression of the smooth muscle isoform of L-type Ca2+ channels and ATP-sensitive K+ channels. Recent attention has also focused on post-translational modifications as a primary means of altering ion channel function in the absence of changes in protein/gene expression. Protein phosphorylation of serine/theronine or tyrosine residues, cysteine thiol modifications, and tyrosine nitration are potential mechanisms affected by oxidative/nitrosative stress that alter the gating kinetics of ion channels. Collectively, these findings suggest that inflammation results in electrical remodeling of smooth muscle cells in addition to structural remodeling. The purpose of this review is to synthesize our current understanding regarding molecular mechanisms that result in altered ion channel function during gastrointestinal inflammation and to address potential areas that can lead to targeted new therapies
Ion channels play an essential role in the physiology of excitable cells and therefore, not surprisingly, defects in channel function have been implicated in several diseases. Altered channel function can either directly contribute to the symptoms of the underlying disease or be indirectly affected by the disease process. Studies over the last twenty years have demonstrated both inherited gene mutations as well as altered gene transcription of ion channels in disease states or following tissue insult, which have collectively been termed as “channelopathies”. An increasing list of ion channel encoding genes with inherited mutations have been linked to both ligand- and voltage-gated Na+, K+, Ca2+ and Cl− channels such as those resulting in long QT syndrome, Bartter’s syndrome, cystic fibrosis, etc. In addition, abnormal expression of channels may occur following tissue injury/inflammation that is associated with transcriptional regulation of normal ion channels. Altered channel function also occurs without concomitant changes in either mRNA or protein expression of the ion channels. In this case, post-translational modifications lead to altered function of the channel protein.
In Inflammatory or functional bowel diseases, increased peripheral sensitization of sensory neurons and altered gastrointestinal motility are two common occurrences. Enhanced pain perception can be related to the sensitization of sensory neurons due to changes in excitability at the spinal, supraspinal and intrinsic primary afferents of the gastrointestinal tract. Mawe et al. (1) recently reviewed the many significant changes in enteric neuron excitability in models of gastrointestinal inflammation. The pattern that emerges from this and other studies strongly implicates the sensitization process to alterations in ion channel function. Similarly, changes in gastrointestinal motility may be attributed to altered ion channel activity of smooth muscle cells following gastrointestinal inflammation (2–6). In the context of the changes in ion channel function, two main concepts have evolved from the various studies, 1) changes in the protein expression of channels, and 2) post-translational modifications in channel function. The goal of this review is to provide an overview of the mechanisms by which voltage-gated ion channels may be modulated during gastrointestinal inflammation. We focus on the smooth muscle ion channels, particularly calcium channels, as the primary example where recent studies implicate that colonic inflammation induces changes in protein expression and post-translational modifications of the channel. We provide examples of modulation of other ion channels specifically where information from gastrointestinal smooth muscle is lacking but pertinent to our perception of molecular mechanisms of aberrant channel function.
Ion channel structure and function
Our understanding of ion channel function has been greatly aided by development and combination of techniques in electrophysiology, molecular biology, and structural biology. Ion channels are composed of one or more pore-forming subunits, often in association with accessory proteins. Based on the crystal structures of bacterial ion channels, a comprehensive view of how ions permeate through the pore regions has been established (7). These structural studies have provided a mechanistic insight into how ions dock within the pore of the ion channels, the basis for ligand binding, regulation by accessory subunits and mechanisms associated with sensing of voltage changes. Further biochemical and molecular analysis have revealed that the regulation of cell excitability is dependent not only on the variability in the classes of expressed channels but also in diversity of the subtypes in individual cells. This is particularly relevant for the coordinated function of the gastrointestinal tract that is dependent on the excitability of various cell types e.g. nerves, muscle, interstitial cells of Cajal, etc. For each individual ion channel, the plethora of genes encoding the pore-forming α subunits is compounded by the additional involvement of accessory subunits, alternative splicing, heteromeric assembly of the subunits, and mRNA editing. Thus, an in-depth understanding of how each of these processes may be affected during insult/injury is therefore necessary to gain insight into the possible therapeutic modalities.
Most voltage-gated ion channels have a common structural feature consisting of domains of transmembrane α-helices that come together to form the α subunit. Functional ion channels, particularly the K+ channels are formed as tetramers of these cores of α subunits. Such tetramers may be monomeric or heteromultimeric thus increasing the diversity of the functional channels properties. For Na+ and Ca2+ channels, four domains each consisting of 6 transmembrane segments are linked by intracellular loops as a single α subunit. In addition to the α subunit, almost all channels are associated with accessory subunits which may regulate several channel properties including voltage-dependence, kinetics of activation, inactivation and deactivation, and membrane trafficking.
Calcium channels
Calcium influx in gastrointestinal smooth muscle is largely mediated by the voltage-gated L-type Ca2+ channel that is responsible for the upstroke of the action potential. The Ca2+ channel is a multimeric complex comprising of a central pore-forming and voltage-sensing α subunit with auxillary β and α2δ subunits. The mammalian α subunits are encoded by at least 10 different genes. Initially the α subunits were given alphabetical subscripts with the smooth muscle L-type channel being designated as α1c. This has now been superseded by numerical identifiers with α1c denoted as Cav1.2 (8). The Cav1.2 gene is encoded by at least 53 exons with alternative splicing resulting in tissue-specific variants. Alternative splicing of the first exon that encodes for the amino-terminal region of the α subunit distinguishes the major cardiac (Cav1.2a) and smooth muscle (Cav1.2b) isoforms (9). Exons 1a and 1b are the two 1st exons of Cav1.2 that encode for the longer and shorter isoforms, respectively. The Cav1.2a was first cloned from the rabbit heart (10) and later also identified in the rat aorta (11). Cav1.2b which differs by 46 amino acids in the N-terminal from Cav1.2a has been cloned from human fibroblasts, human jejunum and resembles the neuronal class of L-type Ca channels(12–14). These exons are independently controlled by separate promoters (15) with the major expression of Cav1.2b in smooth muscle.
The critical role of calcium channels in mediating gastrointestinal contractions is illustrated by selective inactivation of Cav1.2 in smooth muscle. Global knock-out of Cav1.2 is embryonic lethal. Wegener et al (16) utilized a tamoxifen-inducible Cre-lox based strategy in adult mice in which the Cre recombinase was placed under the control of the smooth muscle specific promoter. Excision of loxP-flanked exons 14 and 15 of Cav1.2 by Cre recombinase resulted in an inactivated calcium channel in vascular, urinary and gastrointestinal smooth muscle. Knock-out of smooth muscle Cav1.2 resulted in death with symptoms of paralytic ileus in ~ 4 wks after initiation of gene inactivation with tamoxifen. Consistent with the loss of Cav1.2 protein expression after 14 days, the amplitude of spontaneous contractions and that to carbachol in the colon were largely abolished. These direct studies provide strong evidence of the importance of the voltage-dependent calcium channel in colonic motility.
Calcium channel expression in experimental colitis
Partial loss of calcium channel function has been demonstrated in colonic inflammation. Direct measurements of calcium currents in single smooth muscle cells by whole-cell voltage clamp techniques in the dog, rat and mouse colon have shown a 50—70% decrease in calcium currents following inflammation(2, 4, 6). In accordance with this, colonic inflammation results in decreased contractions of colonic muscle strips in both animal models of colitis as well as in humans to depolarizing K+ solutions.
The decrease in the calcium currents raises the issue of whether this can be attributed to a downregulation of the ion channel protein. The amplitude of a macroscopic current is governed by the product of the number of channels (N), its open probability (Po) and the single channel current (I =NPoi). Like any other functional protein, ion channel expression is dependent on the relative amounts of mRNA, the environmental conditions that regulate the expression eg. mRNA stability, and on the trafficking to the membrane. Open probability measures the time spent in the open state and measures the inherent characteristics of the single channels kinetics, while the single channel current (i) reflects the amplitude of the current at a particular voltage. In the ethanol/acetic acid model of colonic inflammation in the dog, Liu et al. (4) initially demonstrated a decrease in the expression of the α subunit of the calcium channel in colonic smooth muscle cells. In these studies, the authors showed significant decrease in the protein expression after 48 hours of ethanol/acetic acid treatment by Western blots concomitant with 70% decrease in the peak amplitude of calcium currents measured in single smooth muscle cells by patch clamp recordings. Interestingly, the calcium channel agonist, BayK8644 enhanced the calcium currents to a similar extent in both controls and inflamed cells suggesting that the ability of the channel to respond to the dihydropyridine agonist was not functionally altered with inflammation. Kinoshita et al.(6) showed decreased calcium currents in single smooth muscle cells from the colon of the rat trinitrobenzene sulfonic acid (TNBS) (100 mg/kg) induced colonic inflammation, however, this did not correlate with changes in protein expression or mRNA of the Cav1.2. In the mouse dextran sulfate sodium (DSS) model of colonic inflammation, calcium currents decrease by 70% however no changes in mRNA expression or membrane protein expression was observed (2, 17). At first glance, differences in protein expression or mRNA in the different studies could be attributed to either species or methods of inflammation. However, in the same model of rat TNBS (68 mg/kg), Shi and Sarna (18) observed significant decreases in Cav1.2 expression. Determining protein expression by western blots remains a semi-quantitative approach (19, 20) and additional approaches including radiolabelled binding may be useful to establish changes in Cav1.2 protein. Another possible explanation for the differences between the different laboratories with regard to Cav1.2 expression may relate to the extent of inflammation. Although significant changes were observed with respect to the expression of cytokines such as IL-1β in both rat and mouse colon with inflammation (6, 17) a more thorough analysis of correlating ion channel expression with the complement of cytokines and extent of inflammation may be necessary. It is noteworthy that these models are mostly representative of an acute phase of inflammation and much less is known of the changes in a chronic model of inflammation. The protein expression of Cav1.2 was markedly decreased with high TNBS (68 mg/kg) but not when lower concentrations were used (17 mg/kg) whereby significant inflammation does not occur (18). In cDNA microarray analysis from patients with ulcerative colitis or Crohn’s disease (21) or in gene expression studies of rat TNBS model, evidence of changes in the pore-forming subunit of Cav1.2 were not reported (22, 23). The absence of changes in protein or mRNA expression of the alpha subunit of Cav1.2b in some colitis models is suggestive of post-translational modifications as an additional potential mechanism of altered channel function (see below).
Shi et al (24) further demonstrated that treatment of human colon smooth muscle cell cultures with TNF-α resulted in decreased protein and mRNA expression of Cav1.2b within 6–24 hours. This correlated with the activation of the transcription factor, NF-kB, that binds to the promoter region of the α subunit resulting in the suppression of Cav1.2b gene expression. The decrease in α subunit expression of almost 50% within 6 hrs by TNF-α suggests that protein turnover may be relatively rapid for the calcium channel. However, conditional gene inactivation of the intestinal calcium channel suggests a much slower time course for channel protein turnover (half-life ~ 14 days) (16) raising the possibility that TNF-α may additionally affect mRNA stability. In the rat TNBS colitis model, the downregulation of calcium currents was reversed by treatment with sulphasalazine (6). This could be due to either inhibition of NF-κB mediated repression of Cav1.2b gene expression or by scavenging of reactive oxygen species (25) thus affecting post-translational modifications of the calcium channel. The decrease in protein expression can also occur through various other pathways, including enhanced protein degradation and altered calcium channel trafficking to the membrane. Ion channels, like many membrane proteins, may undergo ubiquitylation and be targeted to the proteasome for degradation. It is now becoming increasingly clear that the ubiquitylation-proteasome pathway (UPS) underlies a major mechanism for degradation of intracellular proteins. Several ion channel subunits have been found to be ubiquitylated. Ubiquitin is a 76-amino acid protein that covalently attaches to lysines of the target protein. Ubiquitylation involves the successive action of a ubiquitin-activating enzyme E1, a ubiquitin conjugating enzyme E2, and a ubiquitin-protein ligase E3 that tags the substrate. In a second step, the tagged substrate is degraded by the 26S proteasome complex to small peptides. There is now a growing list of ion channels known to be ubiquitylated. Probably the best studied example is the epithelial sodium channel, ENaC. The carboxy terminus of ENaC β and γ subunits contains a PY motif (PPXYXXΦ), where P is a proline, X any amino acid, and Φ is a hydrophobic amino acid, that are binding sites for ubiquitin protein ligases of the Nedd4 family (an E3 ligase). Mutation in the PY motif leads to the stabilization of the complex resulting in the accumulation of the ENaC subunits enhancing reabsorption of Na+ and H2O that leads to Liddle’s syndrome, a severe form of hypertension. Other ion channels known to be ubiquitylated are the voltage-gated Na+ channels (Nav1.5), ClC-5 (a chloride/proton exchanger), Kir 1.1, Aquaporin-, Kv11.1 (see (26) for review). Ubiquitinylation processes may be regulated by several mechanisms including hormones and stress. It is not known how gastrointestinal inflammation affects the UPS although mRNA expression of a ubiquitin-conjugating enzyme and proteosome subunits are enhanced in colon biopsies from IBD patients (27). Studies to determine ion channel degradation through the UPS pathway in gastrointestinal inflammation merit further investigation.
ATP-sensitive K+ channels
In the DSS colitis mouse model, Akbarali et al (2) demonstrated that in addition to the down-regulation of Ca2+ currents, the response to the ATP-sensitive K+ channel agonist, lemakalim were significantly enhanced in inflamed smooth muscle cells. Further studies established an enhanced bursting activity of single K(ATP) channels from inflamed mice (3). K(ATP) channels are heterooctomers comprised of two subunits, an inwardly rectifying poreforming K+ channel (Kir 6.x) and the sulphonylurea receptor (SURx). In mouse colonic smooth muscle, the major isoforms constituting the K(ATP) channel were identified as Kir 6.1 and SUR2B. Gene expression showed that the mRNA for Kir 6.1 was up-regulated with inflammation while that of of SUR2B was down-regulated (3). However, the protein expression of the channel complex was not determined and therefore it is unclear if these changes in mRNA translate to altered protein expression. The role of K(ATP) channels in colitis was recently addressed in studies by Wallace et al (28) who showed that in colitis rats, treatment with the K(ATP) channel blocker, glibenclamide, resulted in significant mortality of inflamed rats, but not in healthy controls suggesting that upregulation of K(ATP) is an important protective mechanism in inflammation. There is emerging evidence that the K(ATP) channel is modified by hydrogen sulphide (29) (see below) which appears to be upregulated during colitis (28) and underscores the potential for post-translational modifications of ion channels during inflammation.
Post-translational modification of ion channels in inflammation
As stated above, features of gastrointestinal inflammation include changes in a) the electrical excitability of nociceptive neurons, particularly hyperexcitability of DRG neurons (30) resulting in increased peripheral sensitization, and b) motility of circular smooth muscle (5). Post-translational modifications of ion channel proteins present an alternate pathway to modify functional activity in both acute as well as in chronic inflammatory conditions (31). Many of the transcriptional and translational mechanisms are likely to have slower kinetics which may become predominant at later stages of inflammation, provided a continuous stimulus is present. Post-translational modifications of proteins on the other hand may be construed to occur with a faster onset, although significant overlap between these processes is likely to exist. In this case, the channel protein is directly modified affecting its biophysical properties. Changes in the gating kinetics of the individual single channel are reflected in the macroscopic current due to changes in the open probability. Considerable progress has been made in delineating many of the molecular details of how these modifications arise, including those due to phosphorylation of serine and tyrosine residues, oxidation, nitrosylation and sulfhydration of cysteine residues and nitration of tyrosine residues. Examples presented below, although by no means exhaustive, illustrate effects on ion channels pertinent to gastrointestinal inflammation.
Phosphorylation of calcium channels
Ion channels are the target of many intracellular signaling pathways, including protein phosphorylation and dephosphorylation. In fact, nearly every type of ion channel is regulated by phosphorylation, most notably by serine/thereonine kinases such as protein kinases A, C and CAMKII and by tyrosine kinases such as c-src kinase.
One of the best examples of how phosphorylation alters channel function comes from the extensive studies of the effects of β-adrenergic stimulation of the cardiac L-type Ca2+ channel(32). These studies have shown that β adrenergic stimulation of protein kinase A results in the phosphorylation of Ser1928 in the c-terminus of the Ca2+ channel. A combination of electrophysiological single channel, whole cell and molecular studies over the last 20 years have revealed that phosphorylation results in a shift in the modal gating of the channel such that long open events are favored while closed states are decreased by PKA-dependent phosphorylation (32). Ca2+ channels exist in three modes, 0, 1 and 2. Mode 0 constitutes the closed state, mode 1 as brief openings and mode 2 as long-lasting openings. Mode 2 is rarely observed but can be induced by dihydropyridine agonists, such as BayK8644 and by phosphorylation, including that by CAMKII (33). Increased mode 2 gating results in increased open probability and hence enhances ionic currents. Transition from one mode to the other is regulated by specific phosphatases (34). Thus, mutations within the phosphorylation sites or inhibition of phosphatases can result in altered biophysical properties of the channel. An example of this is seen in Timothy syndrome, a disease associated with neurological and cardiovascular effects where mutation of serine 439 in the cytoplasmic end of the S6 helix results in a “gain of function” due to enhancement of mode 2 gating. Similarly increased calcium channel mode 2 gating is observed due to cyclosporin neurotoxicity. Cyclosporin inhibits calcineurin, a phosphatase necessary for dephosphorylating CAMKII- mediated phosphorylation of the calcium channel. Enhanced tyrosine phosphatase activity has also been suggested to promote down-regulation of calcium currents in patients with atrial fibrillation (35).
In colonic inflammation, the phosphorylation of the smooth muscle isoform of the calcium channel by the tyrosine kinase, c-src kinase is attenuated (17) resulting in decreased calcium currents. Src-kinase mediated regulation of Cav1.2b has been demonstrated in vascular, gastrointestinal and urinary smooth muscle cells (36, 37) whereby tyrosine phosphorylation enhances calcium currents. Evidence that calcium channels are under basal regulation of c-src kinase comes from a) the ability of src inhibitors to decrease currents in the absence of receptor activation, b) tyrosine phosphatase inhibitors enhance calcium currents, and 3) co-transfection of hCav1.2b with active src results in enhanced currents(9). Furthermore, src co-precipitates with the calcium channel in rabbit colonic smooth muscle (36). Src kinase contains two protein binding domains, SH2 and SH3,that recognize specific consensus sequences within the target proteins. The SH3 binds to proline-rich regions, while the SH2 binds to phosphorylated tyrosine residues. Both these domains were mapped to the carboxy terminus of hCav1.2b (38). The mechanism by which tyrosine kinase(s) affect calcium channel was addressed by Nakayama and colleagues (39). These authors showed that Cav1.2b undergoes state transition to a second open state, termed O2, upon depolarization (40). In the O2 state, that is distinct from the mode 2 gating, the channels do not, or only very slowly, inactivate during depolarization with the functional consequence of sustained calcium influx thus promoting tonic contractions. The shift to O2 state is promoted by tyrosine kinase – mediated phosphorylation. Ross et al. (41) recently identified that colonic inflammation decreased the shift to O2 state and in transfected cells, mutations of c-terminal tyrosine residues of Cav1.2b prevents tyrosine kinase phosphorylation and the shift to O2 state. The decrease in the availability of the channels in the O2 state was shown to arise as a result of nitration of tyrosine residues (see below).
Phosphorylation of TRPV1
Gastrointestinal inflammation also alters the excitability of spinal afferent neurons (1) and there has been renewed interest in several different ion channels including the role of the transient receptor potential (TRP) non-selective cation channels (42). One of the best studied in this family are the TRPV1, a polymodal Ca2+ permeable cation channel that is activated by vanilloid compounds such as capsaicin, resinferotoxin and other stimuli that include moderate heat, low pH, and several endogenous activators e.g. eicosanoids, anandamide, etc. (43). Post-translational modification of the TRPV1 reduces its thermal threshold and enhances its response to stimuli thus potentiating its activity. This post-translational modification includes acute activation of protein kinases following tissue injury/inflammation that directly phosphorylate the channel. A large body of evidence has now accumulated establishing TRPV1 as a substrate for protein kinase A, protein kinase C, CAMK II and c-src kinase (31, 44). Many inflammatory mediators acutely sensitize the channel due to phosphorylation which results in shifting the threshold for channel activation by protons, pH, and thermal sensitivity. The specific amino acids that are affected by these kinases have been mapped by various methodologies, including site-directed mutagenesis, to lie largely within the N- and C-termini. These examples illustrate the potential alteration of ion channel function due to aberrant phosphorylation/dephosphorylation in disease states.
Oxidative stress
“Oxidative stress” in inflammatory bowel disease has been examined quite extensively and the overall chemical biology of oxidative and nitrosative stress studied in detail (45, 46). Oxidative stress was initially considered as mainly due to the production of superoxide (.O2−) and its conversion to oxidants such as hydrogen peroxide (H2O2) by superoxide dismutase. Hydrogen peroxide, although deactivated by glutathione peroxidase and catalase (Figure 1), in the presence of trace redox metals (Iron and copper) leads to the very reactive hydroxyl radicals (.OH) via Fenton chemistry that further enhances tissue damage. The preponderance of reactive oxygen species (ROS) in the inflamed intestine has been documented in clinical and experimental models, and interestingly altered transcription of endogenous antioxidants such as glutathione peroxidase were reported in these models (47). Hydrogen peroxide covalently modifies cysteine residues forming disulphide bridges between approximating thiols. Prasad and Goyal (48) found that H2O2 partially decreased the transient outward K+ current in mouse colonic smooth muscle cells. This effect was mimicked by the cysteine-modifying membrane impermeable DTNB and reversed by the intracellular dialysis with the reducing agent, dithiothreitol (DTT). The effects of H2O2 were specific for the transient outward K+ currents and did not affect the delayed rectifier K+ currents. Hydrogen peroxide can also be transformed to a more reactive oxidant through interaction with chloride and myeloperoxidase to form hypochlorous acid (HOCl) which can react with ammonia to form monochloramine. Under physiological conditions, bacteria ferment nitrogen-containing compounds in the lumen of the colon resulting in large amounts of ammonia in the local environment. Monochloramine is a highly potent lipophilic oxidant produced during colitis when the destruction of the epithelial barrier results in accumulation of ammonia within the submucosa. The presence of hydrogen peroxide and myeloperoxidase induce production of monochloramine. In the rabbit colonic muscularis mucosae, monochloramine enhanced the large-conductance BKCa channel activity and shifted the steady-state voltage dependence of activation to the left by -22 mV (49). This effect was blocked by the sulfhydryl alkylating agent, N-ethylamleimide (NEM). In the mouse colon, monochloramine abolished the transient outward K+ currents, and enhanced the delayed rectifier K+ currents through modification of cysteine and possibly methionine residues (48, 50). The question remains as to whether these effects on smooth muscle ion channels following inflammation are transient and can be reversed during the course of inflammation by intracellular enzymatic (glutathione peroxidase, superoxide dismutase, catalase) and non-enzymatic antioxidants (Vitamin C, E, glutathione). Alterations in the levels of anitoxidants in inflammatory bowel disease patients has been noted with conflicting data, however, correlation with total oxidative stress implicate an imbalance in both clinical and experimental colitis models (51). Covalently modified ion channels may accumulate and thus contribute to altered excitability in gastrointestinal inflammation analogous to the build-up of oxidized proteins in many other diseases. Determining the relative sensitivity of the various cysteine modifications to endogenous reducing agents will be important in developing therapeutic strategies in treating altered excitability.
Figure 1.
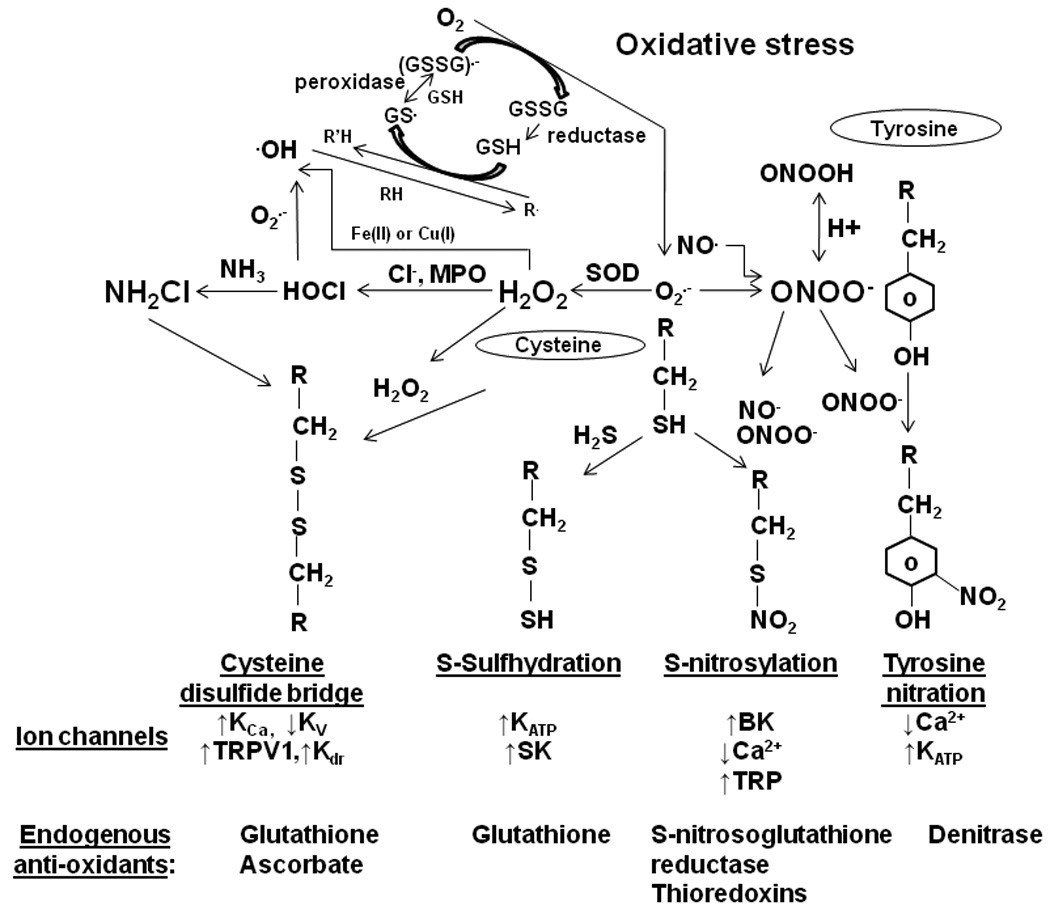
Schematic presentation of pathways associated with post-translational modification of amino acid residues of ion channels. The central components are the presence of reactive superoxide, nitric oxide and hydrogen sulfide. The dismutation of superoxide leads to the production of hydrogen peroxide (H2O2) that oxidizes cysteine residues resulting in disulfide bonds between two approximating thiols groups. Cysteine residues can also be modified by monochloramine (NH2Cl) that is produced from interaction of H2O2 with ammonia (NH3). Nitric oxide can directly s-nitrosylate the sulfhydryl group of the cysteine residues or combine with superoxide to generate peroxynitrite (ONOO−). Peroxynitrite interacts with tyrosine residues resulting in 3-nitrotyrosine that prevents tyrosine phosphorylation. Hydrogen sulfide interacts with cysteine residues to produce hydropersulfide. Ion channels are directly modified by phosphorylation and post-translational modification can also be altered/prevented by endogenous anti-oxidants
Oxidative stress on TRPV1
The molecular details of how oxidative challenge by hydrogen peroxide-induced cysteine modification alters ion channel function was recently addressed for TRPV1 (52). Hydrogen peroxide sensitized rTRPV1 expressing cells in excised patches resulting in enhanced responses to capsaicin, mild acid (pH 6.4) and receptor phosphorylation. Oxidized receptors were more resistant to capsaicin-induced desensitization while desensitized receptors could be primed following oxidation to respond to noxious stimuli. This interesting paradigm establishes that post-translational modification by oxidative stress during inflammation proceeds independently from other potential mechanisms such as phosphorylation/desensitization to alter the functional activity of ion channels. Oxidation induced sensitization remained until strong reducing agents were applied.
Nitrosative stress
Since the realization that nitric oxide (.NO) reacts with superoxide (.O2 −) to form the powerful oxidant peroxynitrite (ONOO−) (53), and other reactive nitrogen species (RNS), “nitrosative stress” has become intricately linked to oxidative stress. The prime biological action of .NO is its interaction with the heme centers of guanylate cyclase to activate cGMP. It was also recognized that NO induced post-translational modifications of various proteins by coupling to reactive cysteine thiols to form S-nitrosothiols (54) that may relate to the cGMP-independent signaling actions of nitric oxide. There are now a large number of proteins thought to be substrates for S-nitrosylation, including ion channels. Although most proteins possess cysteine residues, there is evidence of substrate specificity that include nucleophilicity, hydrophobic compartmentalization, and presence of allosteric regulators such as Ca2+, Mg2+, and H+ ions. S-nitrosylation is a reversible process with recent findings of denitrosylating enzymes, in particular S-nitrosoglutathione reductase and thioredoxins which may be dysregulated in disease states (55). The spatial and temporal specificity of S-nitrosylation for the S-NO bond formation potentially establishes it as an important cellular signaling process. Detection of S-nitrosylation has been possible by use of a biotin switch assay in which free sulfhydryl residues are initially protected by treatment with a sulfhydryl reactive compound (methyl methanethiosulfonate), S-nitrosylated proteins are then reduced with ascorbate followed by tagging with a biotinylated compound (56). Several ion channels that are regulated by S-nitrosylation have been demonstrated, including the L-type Ca2+ channel in heterologous expression systems whereby an inhibition has been suggested by direct nitrosylation(57). The molecular details, including the specific cysteine residues modified in calcium channels, are still unclear. An example of how s-nitrosylation can affect ion channel function was recently addressed by Yoshida et al. (58) who showed that s-nitrosylation of a family of TRP channels was linked to Ca2+ influx induced by ATP- mediated stimulation of eNOS. Nitrosylation of the cysteine residue and disulphide formation coupled the activation gating for the TRP channel in endothelial cells effectively making TRPC5 act as a NO sensor.
In contrast to the effects on cysteine residues and the regulation of cellular signaling by s-nitrosylation in physiological conditions, the formation of peroxynitrite becomes particularly relevant when the concentrations of .NO are enhanced in pathophysiological conditions to an extent that the reaction with superoxide outcompetes the disumation of .O2− by superoxide dismutase. The main outcome of producing peroxynitrite is the oxidation of tyrosine residues of proteins resulting in 3-nitrotyrosine. Tyrosine nitration has been detected in numerous diseases, including clinical and experimental inflammatory bowel disease (59–61) and has been considered as a surrogate marker for inflammation in several disease states. Enhanced production of nitric oxide during gastrointestinal inflammation via increased nitric oxide synthtases (NOS), particularly iNOS, has been well documented.
Ross et al (62) showed that treatment of colonic muscle tissues with peroxynitrite resulted in decreased calcium-influx dependent contractions and induced nitration of hCav1.2b in transfected cells. An increase in tyrosine nitrated calcium channel was observed in colonic smooth muscle from TNBS treated mice. Nitration of the c-terminal tyrosine residues of Cav1.2b prevented tyrosine phosphorylation and mutations of residues Y2134 and Y1837 resulted in decreased calcium currents. As described above, src-kinase promotes the transition of the smooth muscle calcium channel to a non-inactivating open state increasing availability at depolarized potentials. Tyrosine nitration prevents this shift and hence reduces the calcium influx (41). Calcium influx is also associated with excitation-transcription coupling and nitration of the calcium channel attenuated the phosphorylation of the transcription factor cyclic-AMP response element binding protein (CREB) in colonic inflammation (63). Several other ion channels have been reported to be nitrated including Kv1.2 in coronary arteries during diabetes which was reversed by treatment with the peroxynitrite scavenger, ebselen(64), the ATP-sensitive K+ channels in rabbit internal carotid artery (65) and the NMDA receptor channel during hypoxia (66).
Analogous to protein phosphorylation and dephosphorylation, denitration has been suggested as a mechanism for reversing nitrated proteins (67). Although the specific enzymes have not been identified, denitrase activity has been preferentially observed in some tissues such as spleen, lung and activated macrophages. Recently, Kang and Akbarali (68) demonstrated denitration of Cav1.2b by cell lysates from activated macrophages. Important questions remain with regard to the specific nature of the enzymes and if these are altered during inflammation.
S-Sulfhydration of K(ATP) by hydrogen sulfide
The criteria and role of hydrogen sulfide as a gaseous transmitter within the gastrointestinal tract has recently been addressed (69). Of particular interest have been the recent findings that the capacity for H2S synthesis is markedly enhanced in colitis and that inhibition of the synthesizing enzymes exacerbates colitis (28). Hydrogen sulfide inhibits gastrointestinal motility by direct actions on smooth muscle that are glybenclamide- and apamin-sensitive suggesting activation of ATP-sensitive K channels and small conductance apamin-sensitive channels(70). However, in other studies, including human colon, the effects of hydrogen sulfide were not blocked by glibenclamide (69) (71). Analogous to disulphide and s-nitrosylation of cysteine residues, the – SH from H2S is transferred to the sulfhydryl of cysteine to form hydropersulfide (-SSH) in the target protein. The effects of H2S on activation of the ATP-sensitive K channels have been demonstrated in vascular smooth muscle, cardiomyocytes, pancreatic beta-cells and neurons (72). Jiang et al (73) showed that H2S induced activation of K(ATP) was blocked when the extracellular cysteine residues were alkylated by NEM, but not by intracellular application of NEM or by intracellular oxidation of cysteine residues by chloramines-T. Mutation of extracellular cysteine residues of the sulphonylurea receptor, SUR1, also prevented activation of the K(ATP) channel by H2S suggesting that SUR but not the pore forming Kir6.1 is the potential target site for H2S. Using a biotin switch assay, Mustafa et al (29) recently showed S-sulfhydration of ATP-sensitive K channels (Kir 6.1/SUR2B) expressed in HEK cells although the specific subunit remains to be identified. In the DSS model of colitis, there is enhanced bursting activity of single ATP-sensitive K channels (3). The question of whether this occurs as a result of post-translational modification of the cysteine or tyrosine residues remains to be determined but merits investigation.
Conclusions and Future directions
Inflammation induced changes in electrical excitability of gastrointestinal smooth muscle cells were first established over twenty years ago by sharp microelectrode studies in whole tissue segments (74). We now know of specific changes in both protein expression and post-translational modifications of ion channels that results in electrical remodeling in pathophysiological settings. Important questions still remain with regard to identifying these changes in human GI smooth muscle cells, and what alterations occur in the acute vs. the chronic phases of inflammation. Studies to delineate the pathways for membrane trafficking and ion channel degradation and the influence of inflammation need to be established. It is important to note that each individual ion channel may be modulated at various sites by different “oxidative” elements. Although oxidative stress has been recognized as a key component in gastrointestinal inflammation and alterations in endogenous anti-oxidants have been reported in inflammatory bowel disease (75), antioxidant therapy still remains in its infancy (51). The focus of this review was to highlight the possible mechanisms involved in altered ion channel activity and the different facets of post-translational modifications. The latter also brings into question the role of various endogenous anti-oxidant mechanisms. For example, de-nitrosylation requires specific thioredoxins, oxidation of cysteine residues may be reduced by ascorbate and glutathione, while S-sulfhydration appears to be more stable. Recent studies have also addressed the potential of a “denitrase” which may allow for recovery of tyrosine nitrated proteins. A combination that takes into account the various antioxidant mechanisms could provide an important therapeutic approach in the treatment of gastrointestinal inflammatory disorders particularly towards restoring cellular excitability.
ACKNOWLEDGEMENT
The work in the authors laboratory is supported by NIH DK046367 and DA024009
Footnotes
CONFLICTS OF INTEREST: None
REFERENCES
- 1.Mawe GM, Strong DS, Sharkey KA. Plasticity of enteric nerve functions in the inflamed and postinflamed gut. Neurogastroenterol Motil. 2009;21:481–491. doi: 10.1111/j.1365-2982.2009.01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akbarali HI, Pothoulakis C, Castagliuolo I. Altered ion channel activity in murine colonic smooth muscle myocytes in an experimental colitis model. Biochem Biophys Res Commun. 2000;275:637–642. doi: 10.1006/bbrc.2000.3346. [DOI] [PubMed] [Google Scholar]
- 3.Jin X, Malykhina AP, Lupu F, Akbarali HI. Altered gene expression and increased bursting activity of colonic smooth muscle ATP-sensitive K+ channels in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G274–G285. doi: 10.1152/ajpgi.00472.2003. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Rusch NJ, Striessnig J, Sarna SK. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology. 2001;120:480–489. doi: 10.1053/gast.2001.21167. [DOI] [PubMed] [Google Scholar]
- 5.Malykhina AP, Akbarali HI. Inflammation-induced "channelopathies" in the gastrointestinal smooth muscle. Cell Biochem Biophys. 2004;41:319–330. doi: 10.1385/CBB:41:2:319. [DOI] [PubMed] [Google Scholar]
- 6.Kinoshita K, Sato K, Hori M, Ozaki H, Karaki H. Decrease in activity of smooth muscle L-type Ca2+ channels and its reversal by NF-kappaB inhibitors in Crohn's colitis model. Am J Physiol Gastrointest Liver Physiol. 2003;285:G483–G493. doi: 10.1152/ajpgi.00038.2003. [DOI] [PubMed] [Google Scholar]
- 7.Doyle DA, Morais Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 8.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) British journal of pharmacology. (3rd edition) 2008;153 Suppl 2:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbarali HI. Signal-transduction pathways that regulate smooth muscle function. II. Receptor-ion channel coupling mechanisms in gastrointestinal smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2005;288:G598–G602. doi: 10.1152/ajpgi.00402.2004. [DOI] [PubMed] [Google Scholar]
- 10.Mikami A, Imoto K, Tanabe T, et al. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 11.Koch WJ, Ellinor PT, Schwartz A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. The Journal of biological chemistry. 1990;265:17786–17791. [PubMed] [Google Scholar]
- 12.Lyford GL, Strege PR, Shepard A, et al. alpha(1C) (Ca(V)1.2) L-type calcium channel mediates mechanosensitive calcium regulation. Am J Physiol Cell Physiol. 2002;283:C1001–C1008. doi: 10.1152/ajpcell.00140.2002. [DOI] [PubMed] [Google Scholar]
- 13.Soldatov NM. Molecular diversity of L-type Ca2+ channel transcripts in human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4628–4632. doi: 10.1073/pnas.89.10.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlinson WJ, Stea A, Bourinet E, Charnet P, Nargeot J, Snutch TP. Functional properties of a neuronal class C L-type calcium channel. Neuropharmacology. 1993;32:1117–1126. doi: 10.1016/0028-3908(93)90006-o. [DOI] [PubMed] [Google Scholar]
- 15.Saada N, Dai B, Echetebu C, Sarna SK, Palade P. Smooth muscle uses another promoter to express primarily a form of human Cav1.2 L-type calcium channel different from the principal heart form. Biochem Biophys Res Commun. 2003;302:23–28. doi: 10.1016/s0006-291x(03)00097-4. [DOI] [PubMed] [Google Scholar]
- 16.Wegener JW, Schulla V, Koller A, Klugbauer N, Feil R, Hofmann F. Control of intestinal motility by the Ca(v)1.2 L-type calcium channel in mice. Faseb J. 2006;20:1260–1262. doi: 10.1096/fj.05-5292fje. [DOI] [PubMed] [Google Scholar]
- 17.Kang M, Morsy N, Jin X, Lupu F, Akbarali HI. Protein and gene expression of Ca2+ channel isoforms in murine colon: effect of inflammation. Pflugers Arch. 2004;449:288–297. doi: 10.1007/s00424-004-1339-5. [DOI] [PubMed] [Google Scholar]
- 18.Shi XZ, Sarna SK. Homeostatic and Therapeutic Roles of Vip in Smooth Muscle Function: Myo-Neuro-Immune Interactions. Am J Physiol Gastrointest Liver Physiol. 2009 doi: 10.1152/ajpgi.00194.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dittmer A, Dittmer J. Beta-actin is not a reliable loading control in Western blot analysis. Electrophoresis. 2006;27:2844–2845. doi: 10.1002/elps.200500785. [DOI] [PubMed] [Google Scholar]
- 20.Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods. 2008;172:250–254. doi: 10.1016/j.jneumeth.2008.05.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costello CM, Mah N, Hasler R, et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005;2:e199. doi: 10.1371/journal.pmed.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman J, Yu JG, Suntres Z, et al. ADOA3R as a therapeutic target in experimental colitis: proof by validated high-density oligonucleotide microarray analysis. Inflamm Bowel Dis. 2006;12:766–789. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Augustin O, Merlos M, Zarzuelo A, Suarez MD, de Medina FS. Disturbances in metabolic, transport and structural genes in experimental colonic inflammation in the rat: a longitudinal genomic analysis. BMC Genomics. 2008;9:490. doi: 10.1186/1471-2164-9-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi XZ, Pazdrak K, Saada N, Dai B, Palade P, Sarna SK. Negative transcriptional regulation of human colonic smooth muscle Cav1.2 channels by p50 and p65 subunits of nuclear factor-kappaB. Gastroenterology. 2005;129:1518–1532. doi: 10.1053/j.gastro.2005.07.058. [DOI] [PubMed] [Google Scholar]
- 25.Miles AM, Grisham MB. Antioxidant properties of aminosalicylates. Methods Enzymol. 1994;234:555–572. doi: 10.1016/0076-6879(94)34128-1. [DOI] [PubMed] [Google Scholar]
- 26.Abriel H, Staub O. Ubiquitylation of ion channels. Physiology (Bethesda) 2005;20:398–407. doi: 10.1152/physiol.00033.2005. [DOI] [PubMed] [Google Scholar]
- 27.Galamb O, Gyorffy B, Sipos F, et al. Inflammation, adenoma and cancer: objective classification of colon biopsy specimens with gene expression signature. Dis Markers. 2008;25:1–16. doi: 10.1155/2008/586721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009;137:569–578. doi: 10.1053/j.gastro.2009.04.012. 578 e561. [DOI] [PubMed] [Google Scholar]
- 29.Mustafa AK, Gadalla MM, Sen N, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beyak MJ, Vanner S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurones: the role of voltage-gated ion channels. Neurogastroenterol Motil. 2005;17:175–186. doi: 10.1111/j.1365-2982.2004.00596.x. [DOI] [PubMed] [Google Scholar]
- 31.Bhave G, Gereau RWt. Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- 32.Yue DT, Herzig S, Marban E. Beta-adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 34.Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiological reviews. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greiser M, Halaszovich CR, Frechen D, et al. Pharmacological evidence for altered src kinase regulation of I (Ca,L) in patients with chronic atrial fibrillation. Naunyn-Schmiedeberg's archives of pharmacology. 2007;375:383–392. doi: 10.1007/s00210-007-0174-6. [DOI] [PubMed] [Google Scholar]
- 36.Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. The Journal of biological chemistry. 1998;273:5337–5342. doi: 10.1074/jbc.273.9.5337. [DOI] [PubMed] [Google Scholar]
- 37.Davis MJ, Wu X, Nurkiewicz TR, et al. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2001;281:H1835–H1862. doi: 10.1152/ajpheart.2001.281.5.H1835. [DOI] [PubMed] [Google Scholar]
- 38.Kang M, Ross GR, Akbarali HI. COOH-terminal association of human smooth muscle calcium channel Ca(v)1.2b with Src kinase protein binding domains: effect of nitrotyrosylation. Am J Physiol Cell Physiol. 2007;293:C1983–C1990. doi: 10.1152/ajpcell.00308.2007. [DOI] [PubMed] [Google Scholar]
- 39.Nakayama S, Ito Y, Sato S, Kamijo A, Liu HN, Kajioka S. Tyrosine kinase inhibitors and ATP modulate the conversion of smooth muscle L-type Ca2+ channels toward a second open state. Faseb J. 2006;20:1492–1494. doi: 10.1096/fj.05-5049fje. [DOI] [PubMed] [Google Scholar]
- 40.Aoyama M, Murakami M, Iwashita T, Ito Y, Yamaki K, Nakayama S. Slow deactivation and U-shaped inactivation properties in cloned Cav1.2b channels in Chinese hamster ovary cells. Biophys J. 2003;84:709–724. doi: 10.1016/S0006-3495(03)74890-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross GR, Kang M, Akbarali HI. Colonic inflammation alters Src kinase - dependent gating properties of single Ca2+ channels via tyrosine nitration. Am J Physiol Gastrointest Liver Physiol. doi: 10.1152/ajpgi.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Storr M. TRPV1 in colitis: is it a good or a bad receptor?--a viewpoint. Neurogastroenterol Motil. 2007;19:625–629. doi: 10.1111/j.1365-2982.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- 43.Nilius B, Voets T, Peters J. TRP channels in disease. Sci STKE 2005. 2005:re8. doi: 10.1126/stke.2952005re8. [DOI] [PubMed] [Google Scholar]
- 44.Jin X, Morsy N, Winston J, Pasricha PJ, Garrett K, Akbarali HI. Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am J Physiol Cell Physiol. 2004;287:C558–C563. doi: 10.1152/ajpcell.00113.2004. [DOI] [PubMed] [Google Scholar]
- 45.Bian K, Murad F. Nitric oxide (NO)--biogeneration, regulation, and relevance to human diseases. Front Biosci. 2003;8:d264–d278. doi: 10.2741/997. [DOI] [PubMed] [Google Scholar]
- 46.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Te Velde AA, Pronk I, de Kort F, Stokkers PC. Glutathione peroxidase 2 and aquaporin 8 as new markers for colonic inflammation in experimental colitis and inflammatory bowel diseases: an important role for H2O2? Eur J Gastroenterol Hepatol. 2008;20:555–560. doi: 10.1097/MEG.0b013e3282f45751. [DOI] [PubMed] [Google Scholar]
- 48.Prasad M, Goyal RK. Differential modulation of voltage-dependent K+ currents in colonic smooth muscle by oxidants. Am J Physiol Cell Physiol. 2004;286:C671–C682. doi: 10.1152/ajpcell.00137.2003. [DOI] [PubMed] [Google Scholar]
- 49.Prasad M, Matthews JB, He XD, Akbarali HI. Monochloramine directly modulates Ca(2+)-activated K(+) channels in rabbit colonic muscularis mucosae. Gastroenterology. 1999;117:906–917. doi: 10.1016/s0016-5085(99)70350-1. [DOI] [PubMed] [Google Scholar]
- 50.Prasad M, Goyal RK. Monochloramine selectively inhibits the transient outward potassium current in colonic smooth muscle. Surgery. 2003;134:319–328. doi: 10.1067/msy.2003.242. [DOI] [PubMed] [Google Scholar]
- 51.Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci. 2007;52:2015–2021. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- 52.Chuang HH, Lin S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20097–20102. doi: 10.1073/pnas.0902675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological reviews. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 55.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 56.Forrester MT, Foster MW, Benhar M, Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–195. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshida T, Inoue R, Morii T, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 59.Miampamba M, Sharkey KA. Temporal distribution of neuronal and inducible nitric oxide synthase and nitrotyrosine during colitis in rats. Neurogastroenterol Motil. 1999;11:193–206. doi: 10.1046/j.1365-2982.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- 60.Singer II, Kawka DW, Scott S, et al. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- 61.Zingarelli B, Szabo C, Salzman AL. Reduced oxidative and nitrosative damage in murine experimental colitis in the absence of inducible nitric oxide synthase. Gut. 1999;45:199–209. doi: 10.1136/gut.45.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross GR, Kang M, Shirwany N, Malykhina AP, Drozd M, Akbarali HI. Nitrotyrosylation of Ca2+ Channels Prevents c-Src Kinase Regulation of Colonic Smooth Muscle Contractility in Experimental Colitis. The Journal of pharmacology and experimental therapeutics. 2007;322:948–956. doi: 10.1124/jpet.107.123075. [DOI] [PubMed] [Google Scholar]
- 63.Kang M, Ross GR, Akbarali HI. The effect of tyrosine nitration of L-type Ca(2+) channels on excitation-transcription coupling in colonic inflammation. British journal of pharmacology. doi: 10.1111/j.1476-5381.2009.00599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bubolz AH, Wu Q, Larsen BT, Gutterman DD, Liu Y. Ebselen reduces nitration and restores voltage-gated potassium channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293:H2231–H2237. doi: 10.1152/ajpheart.00717.2007. [DOI] [PubMed] [Google Scholar]
- 65.Ohashi M, Faraci F, Heistad D. Peroxynitrite hyperpolarizes smooth muscle and relaxes internal carotid artery in rabbit via ATP-sensitive K+ channels. Am J Physiol Heart Circ Physiol. 2005;289:H2244–H2250. doi: 10.1152/ajpheart.00254.2005. [DOI] [PubMed] [Google Scholar]
- 66.Zanelli SA, Ashraf QM, Mishra OP. Nitration is a mechanism of regulation of the NMDA receptor function during hypoxia. Neuroscience. 2002;112:869–877. doi: 10.1016/s0306-4522(02)00141-0. [DOI] [PubMed] [Google Scholar]
- 67.Irie Y, Saeki M, Kamisaki Y, Martin E, Murad F. Histone H1.2 is a substrate for denitrase, an activity that reduces nitrotyrosine immunoreactivity in proteins. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5634–5639. doi: 10.1073/pnas.1131756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang M, Akbarali HI. Denitration of L-type calcium channel. FEBS Lett. 2008;582:3033–3036. doi: 10.1016/j.febslet.2008.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Linden DR, Levitt MD, Farrugia G, Szurszewski JH. Endogenous Production of H(2)S in the Gastrointestinal Tract: Still in Search of a Physiologic Function. Antioxid Redox Signal. doi: 10.1089/ars.2009.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gallego D, Clave P, Donovan J, et al. The gaseous mediator, hydrogen sulphide, inhibits in vitro motor patterns in the human, rat and mouse colon and jejunum. Neurogastroenterol Motil. 2008;20:1306–1316. doi: 10.1111/j.1365-2982.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- 71.Teague B, Asiedu S, Moore PK. The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. British journal of pharmacology. 2002;137:139–145. doi: 10.1038/sj.bjp.0704858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang G, Wu L, Wang R. Interaction of Hydrogen Sulfide with Different Ion Channels. Clin Exp Pharmacol Physiol. 2009 doi: 10.1111/j.1440-1681.2010.05351.x. [DOI] [PubMed] [Google Scholar]
- 73.Jiang B, Tang G, Cao K, Wu L, Wang R. Molecular Mechanism for H(2)S-Induced Activation of K(ATP) Channels. Antioxid Redox Signal. doi: 10.1089/ars.2009.2894. [DOI] [PubMed] [Google Scholar]
- 74.Koch TR, Carney JA, Go VL, Szurszewski JH. Spontaneous contractions and some electrophysiologic properties of circular muscle from normal sigmoid colon and ulcerative colitis. Gastroenterology. 1988;95:77–84. doi: 10.1016/0016-5085(88)90293-4. [DOI] [PubMed] [Google Scholar]
- 75.Koutroubakis IE, Malliaraki N, Dimoulios PD, Karmiris K, Castanas E, Kouroumalis EA. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig Dis Sci. 2004;49:1433–1437. doi: 10.1023/b:ddas.0000042242.22898.d9. [DOI] [PubMed] [Google Scholar]