

Abstract
Single nucleotide polymorphisms (SNPs) in the multiple drug resistance protein 1 (MRP1) and P-glycoprotein 1 (MDR1) genes modulate their ability to mediate drug resistance. We therefore sought to retrospectively evaluate their influence on outcomes in relapsed and/or refractory myeloma patients treated with bortezomib or bortezomib with pegylated liposomal doxorubicin (PLD). The MRP1/R723Q polymorphism was found in five subjects among the 279 patient study population, all of whom received PLD + bortezomib. Its presence was associated with a longer time to progression (TTP; median 330 vs. 129 days; p = 0.0008), progression-free survival (PFS; median 338 vs. 129 days; p = 0.0006), and overall survival (p = 0.0045). MDR1/3435(C > T), which was in Hardy–Weinberg equilibrium, showed a trend of association with PFS (p = 0.0578), response rate (p = 0.0782) and TTP (p = 0.0923) in PLD + bortezomib patients, though no correlation was found in the bortezomib arm. In a recessive genetic model, MDR1/3435 T was significantly associated with a better TTP (p = 0.0405) and PFS (p = 0.0186) in PLD + bortezomib patients. These findings suggest a potential role for MRP1 and MDR1 SNPs in modulating the long-term outcome of relapsed and/or refractory myeloma patients treated with PLD + bortezomib. Moreover, they support prospective studies to determine if such data could be used to tailor therapy to the genetic makeup of individual patients.
Keywords: Bortezomib, MDR1, MRP1, Multiple myeloma, Pegylated liposomal doxorubicin, SNP
Introduction
Prior to the development of novel biologically based therapies for multiple myeloma, relapses after successive treatment regimens resulted in progressively shorter response durations [1], which in part reflected the occurrence of drug resistance. Recently, the advent of novel therapies, including regimens incorporating either proteasome inhibitors [2] or immunomodulatory agents [3], has significantly improved the median survival for patients with advanced disease [4]. To further improve the outcome of patients in this setting, combination regimens with both conventional drugs and novel agents are applied. One example is that of the immunomodulatory agent lenalidomide which, when combined with dexamethasone, is superior to corticosteroids alone [5, 6]. A second such combination regimen incorporates the proteasome inhibitor bortezomib (VELCADE®) with pegylated liposomal doxorubicin (PLD; DOXIL®) [7, 8]. In an international randomized phase III trial, PLD with bortezomib was superior to bortezomib monotherapy for relapsed or refractory multiple myeloma, in that it significantly improved the median time to progression, duration of response, and 15-month survival rate [9].
Although several prognostic factors have been identified for newly diagnosed myeloma, and for patients with relapsed and/or refractory disease [10, 11], genetic factors that may influence responses to therapy, especially in the latter setting, have been less well defined. One possible factor is variant multiple drug-resistant (MDR) genotypes, which have been associated with differential expression of the MDR1 gene and its 170-kDa protein product, P-glycoprotein, also known as the ATP-binding cassette Transporter B1 (ABCB1). MDR1 is a transmembrane protein that acts as an energy-dependent drug efflux pump for chemotherapeutic drugs commonly used against hematologic malignancies [12]. Therefore, inborn genetic variations leading to a modified expression of these transporters could influence patient outcomes [13]. In a previous study of the effect of the MDR1 C3435T single nucleotide polymorphism (SNP) in myeloma patients treated with dexamethasone, doxorubicin and vincristine (DAV), followed by autologous stem cell transplantation (SCT), this SNP was predictive of the overall survival (OS), but did not modify the response to treatment [14]. Indeed, the three most common MDR1 SNPs, C3435T, C1236T, and G2677T/A, have been found to influence the outcomes of myeloma patients treated with DAV and then SCT [15]. Moreover, overexpression of MDR1 in myeloma cells might contribute to treatment failure in patients receiving proteasome inhibitors [16], since P-glycoprotein conferred bortezomib resistance in some preclinical models [17, 18]. Therapy resistance may also be mediated by another multidrug-resistance associated protein, also known as ATP-binding cassette, subfamily C, member 1 (MRP1, or ABCC1). Like MDR1, it confers resistance to anthracyclines [19], and because the MRP1 mutation Arg723Gln has an effect on MRP1 expression and trafficking, it significantly reduced MRP1-mediated resistance to a wide spectrum of drugs [20]. Previous studies investigated whether efflux pump inhibitors would modulate bortezomib activity, and the combination of MDR1-specific or MRP1-specific inhibitors did enhance the antitumor effects of bortezomib [21].
To further evaluate the possibility that SNPs in these multidrug resistance genes could influence long-term outcomes in PLD + bortezomib-treated patients, we analyzed DNA samples from patients who were treated in a phase III study comparing bortezomib alone with PLD + bortezomib. Specifically, we tested for the presence of the MDR1 polymorphisms 1236C > T, 2677G > W (W = T or A), and 3435C > T and the MRP1 gene R723Q polymorphism. These were then compared to study endpoints, including time to progression (TTP), progression-free survival (PFS), and OS, and comparisons were made between the two treatment arms of the study.
Methods
Patients
The current analysis involved patients from a previously reported randomized, parallel-group, open-label, multicenter, phase III study (DOXIL-MMY-3001) [9]. This trial compared PLD plus bortezomib with bortezomib alone in 646 bortezomib-naïve patients with multiple myeloma who had suffered disease progression after an initial response to at least one line of prior therapy or who were refractory to initial treatment. Patients who had received prior doxorubicin, PLD, or other anthracyclines were eligible providing that they had not previously progressed on that regimen. However, patients were excluded if they had received a cumulative anthracycline dose of more than 240 mg/m2 or had a left ventricular ejection fraction below normal institutional limits. Review boards at participating institutions approved the study, which was conducted according to the Declaration of Helsinki, the International Conference on Harmonization, and the Guidelines for Good Clinical Practice. All patients provided written, informed consent and were randomized to receive either PLD at a dose of 30 mg/m2 as a 1-h intravenous infusion on day 4 and bortezomib at a dose of 1.3 mg/m2 as a 5-s intravenous bolus dose on days 1, 4, 8, and 11 of every 21-day cycle or bortezomib alone at a dose of 1.3 mg/m2 on the same dose and schedule. Treatment continued for at least two cycles beyond a complete remission (CR), or until disease progression or unacceptable toxicity occurred, for a maximum of eight cycles. However, patients with levels of paraprotein that continued to decrease by >25% from course to course after eight cycles were allowed to continue treatment for as long as treatment was tolerated.
Analyses
The objectives of the pharmacogenomic analyses were to test whether the MDR1 gene polymorphisms—3435C > T (RefSNP ID rs1045642), 2677G > W (W = T or A; RefSNP ID rs2032582), and 1236C > T (RefSNP ID rs1128503), and the MRP1 gene R723Q polymorphism (RefSNP ID rs4148356), were associated with a different overall response rate (CR + partial response) and response durability (TTP, PFS, and OS). MDR1 and MRP1 variants were analyzed using both the TaqMan® real-time polymerase chain reaction and by direct sequencing. All subjects in the intention to treat set who consented to DNA testing, and who produced at least one useable genotype, were included in the pharmacogenomics analysis set. The genotyping was performed by EPIDAUROS Biotechnologie AG (Bernried, Germany) according to Good Laboratory Practice Standards. Two types of controls were introduced in defined, additional positions of the processing batch: (a) water (=negative control) as an indicator of contamination and (b) DNAs of defined genotype (=positive control) as indicators for the assay quality.
Polymorphisms which had a p value of 0.05 or less by logrank test were classified as “significant.” Summary statistics for age, sex, race, height, weight, body surface area, and baseline left ventricular ejection fraction were calculated; while for continuous variables, the mean, standard deviation, median, and range were determined. For categorical variables, the frequency and percent of each category were calculated, and summary statistics were calculated after stratifying by treatment group. Hardy–Weinberg equilibrium (HWE) was tested with an exact test using 10,000 permutations. A severe deviation from HWE (p < 0.001) could be caused by genotyping error. For each polymorphism, genotype frequencies were tabulated after stratifying by treatment arm, and the frequency and percentage of subjects with each endpoint were calculated after stratifying by genotype. The association of each endpoint and the genotype of each polymorphism were tested using the exact 2-sided Cochran–Armitage trend test after stratifying by treatment arm.
Results
Study population
Usable genotype data and clinical data were available for 301 subjects, of whom 279 were Caucasian. Because genetic transmission patterns differ among racial groups, it is a general practice to treat each racial group separately in a genetic analysis. Twenty-two non-Caucasian subjects for whom data were available in this study belonged to at least three racial groups (African American, Asian American, and other). These subject samples were excluded from the analyses, and from subsequent analyses, since the sample size was too small to reach statistical significance. As a result, this report focused on the Caucasian population only (n = 279).
Demographic and baseline data from the cohorts that were treated with either bortezomib or PLD + bortezomib, and for whom pharmacogenomic data were available, are shown in Table 1. The characteristics of the baseline parameters for the pharmacogenomic analysis datasets were well balanced and were similar to the intent-to-treat cohorts for the study population as a whole [9].
Table 1.
Baseline characteristics of the study populations
| Variable | Category | PLD + Bortezomib | Bortezomib |
|---|---|---|---|
| Age (Years) | n | 142 | 137 |
| Mean (SD) | 63.0 (9.61) | 62.2 (9.27) | |
| Median | 62.0 | 63.0 | |
| Range | (37.0, 85.0) | (40.0, 84.0) | |
| Height (cm) | n | 142 | 137 |
| Mean (SD) | 167.2 (12.31) | 167.3 (10.85) | |
| Median | 169 | 169 | |
| Range | (115, 194) | (142, 189) | |
| Weight (kg) | n | 142 | 137 |
| Mean (SD) | 79.0 (16.97) | 77.2 (15.31) | |
| Median | 76.4 | 76.0 | |
| Range | (44.5, 140) | (46.0, 129) | |
| Body surface area (m2) | n | 142 | 137 |
| Mean (SD) | 1.9 (0.24) | 1.9 (0.23) | |
| Median | 1.9 | 1.9 | |
| Range | (1.4, 2.5) | (1.4, 2.4) | |
| Baseline LVEF (%) | n | 142 | 134 |
| Mean (SD) | 62.6 (7.76) | 66.5 (51.33) | |
| Median | 62.0 | 62.0 | |
| Range | (43.0, 88.0) | (48.0, 651) | |
| Sex (n, %) | Female | 61 (43.0) | 49 (35.8) |
| Male | 81 (57.0) | 88 (64.2) |
cm centimeters, kg kilograms, LVEF left ventricular ejection fraction, m 2 meter squared in body surface area, PLD pegylated liposomal doxorubicin, SD standard deviation
A Hardy–Weinberg equilibrium test was performed on all four loci, and both allele frequencies and genotype frequencies were calculated (Table 2). Since the MDR1 locus has three alleles, the A and T alleles were combined as W so that this locus could be treated as two alleles. It was apparent that these four genetic markers were indeed in Hardy–Weinberg equilibrium. In particular, consistent with previous reports [15], the three MDR1 variants were fairly common in trial subjects. However, the MRP1 marker was close to monoallelic since the minor allele frequency was extremely low, and the study population contained 274 homozygous wild-type patients, but only five heterozygous patients (Table 2), all of whom were randomized to the PLD + bortezomib arm of this phase III trial (Table 3).
Table 2.
Summary statistics of the studied genetic markers
| Marker | Frequency minor allele | Hardy–Weinberg equilibrium p value | n/Genotype frequencies of homozygous wild type | n/Genotype frequencies of heterozygous | n/Genotype frequencies of homozygous variant | Missing data |
|---|---|---|---|---|---|---|
| MDR1 1236C > T | 0.4355 | 0.6110 | 91 (0.3262) | 133 (0.4767) | 55 (0.1971) | 0 |
| MDR1 2677G > W | 0.4373 | 0.7430 | 87 (0.3118) | 140 (0.5018) | 52 (0.1864) | 0 |
| MDR1 3435C > T | 0.4910 | 0.8145 | 73 (0.2626) | 137 (0.4928) | 68 (0.2446) | 1 |
| MRP1 R723Q | 0.0090 | 0.8800 | 274 (0.9821) | 5 (0.0179) | 0 (0) | 0 |
Table 3.
Clinical characteristics of patients with the MRP1 R723Q polymorphism
| Patient | Prior therapies | Paraprotein type and level | Cytogenetics |
|---|---|---|---|
| 1 | 1. Idarubicin and dexamethasone induction | IgA κ, 3.7 g/dL | Normal |
| 2. Autologous stem cell transplantation with high dose melphalan followed by prednisolone and interferon maintenance | |||
| 3. Thalidomide | |||
| 2 | 1. Vincristine, doxorubicin, methyl prednisolone induction | IgG λ, 1.6 g/dL | Normal |
| 2. Autologous stem cell transplantation with high dose melphalan followed by prednisone maintenance | |||
| 3 | 1. Vincristine, doxorubicin, and dexamethasone induction | IgA κ, 3.9 g/dL | Not done |
| 2. Autologous stem cell transplantation with high dose melphalan | |||
| 4 | 1. Melphalan and prednisone | IgA λ, 3.2 g/dL | Normal |
| 2. Melphalan and prednisone | |||
| 3. Thalidomide and dexamethasone | |||
| 5 | 1. Vinblastine and prednisolone | IgG κ, 5.3 g/dL | Not done |
| 2. Vincristine, BCNU, melphalan, cyclophosphamide, prednisone | |||
| 3. Doxorubicin, melphalan, cyclophosphamide |
Influence of MDR1 and MRP1 SNPs
The possible presence of associations between 1236C > T, 2677G > W (W = T or A), and 3435C > T in MDR1 and R723Q in MRP1 and the response rate of the bortezomib or PLD + bortezomib groups, as well as response durability measures, were then evaluated. Genotype p value was based on Cochran–Armitage trend test, and allele p value was based on Fisher’s exact test. Both tests were for responders (complete + partial responses) versus nonresponders (minor response + stable disease + progressive disease), and these response categories were defined according to the European Bone Marrow Transplantation criteria [22]. In the present study, the MRP1 gene polymorphism R723Q was significantly associated with time to progression (Fig. 1a; p = 0.0008), progression-free survival (Fig. 1b; p = 0.0006), and overall survival (Fig. 1c; p = 0.0045) in subjects who received PLD + bortezomib. With regard to the MDR1 gene polymorphism at 3435(C > T), this was associated with progression-free survival (Fig. 2; p = 0.0578). Also, this SNP may have been linked to response rate (p = 0.0782) and time to progression (p = 0.0601) in subjects who received PLD + bortezomib treatment, though neither of these associations reached the level of statistical significance.
Fig. 1.
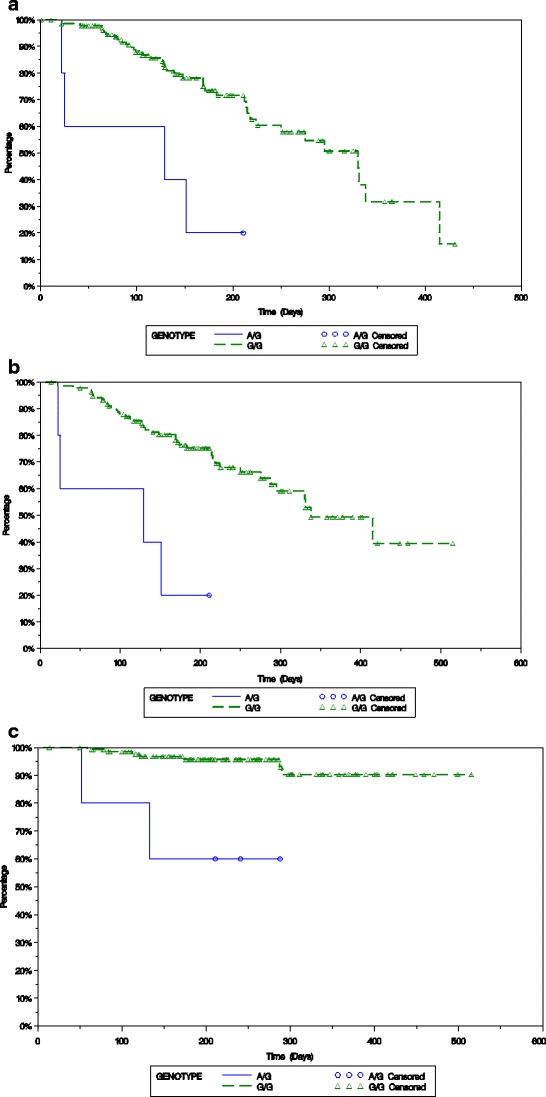
Time to progression (panel a), progression-free survival (panel b), and overall survival (panel c) of patients treated for their relapsed and/or refractory multiple myeloma with PLD + bortezomib is plotted based on the presence of either the A/G or G/G SNP at R723Q in MRP 1 using the Kaplan–Meier method
Fig. 2.
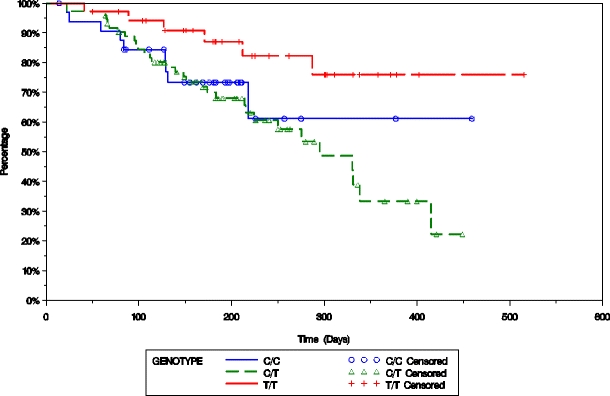
Progression-free survival of patients treated for their relapsed and/or refractory multiple myeloma with PLD + bortezomib is plotted based on the presence of either the C/C, C/T, or T/T SNP at C3435T in MDR1 using the Kaplan–Meier method
To further investigate the genetic influence of the above polymorphisms on response, a haplotype analysis was conducted that combined the three individual MDR1 SNPs based on their genetic transmission patterns, also known as linkage disequilibrium. Haplotype analysis indicated that the three most frequent haplotypes showed a trend of association with response rate in subjects with relapsed multiple myeloma who received PLD + bortezomib treatment (p = 0.0775), though not at the level of statistical significance. The most frequent diplotype that contained 3435 T (i.e. T–W–T/C–G–T or H2/H3) may have been associated with time to progression (p = 0.0819) and progression-free survival (p = 0.0891) in subjects treated with PLD + bortezomib when compared to the most frequent diplotype (C–G–C/T–W–T or H1/H2), though not at the level of statistical significance.
Discussion
The introduction of novel agents, and their use in rationally designed combination regimens, has revolutionized the therapy of multiple myeloma and contributed to an increasing overall survival [23, 24]. However, myeloma remains incurable, is still characterized by a decreasing response duration with each successive salvage therapy [1], and in the relapsed and especially the refractory settings, is associated with a median survival of 6 months or less [25]. This behavior is associated in part with the acquisition of a drug-resistant phenotype [4, 26, 27]. One major mechanism for this phenotype has been attributed to efflux pumps such as MDR1 and MRP1, which can reduce the intracellular drug concentrations of agents used as part of our chemotherapeutic armamentarium. Doxorubicin, vincristine, and etoposide are examples of drugs known to be MDR1 and/or MRP1 substrates that have activity against myeloma [28, 29]. This prompted us to examine the possibility that such SNPs could influence outcomes of myeloma patients treated with either bortezomib or the PLD + bortezomib combination.
In the present retrospective study, the MRP1 gene polymorphism R723Q was significantly associated with TTP (Fig. 1a), PFS (Fig. 1b), and OS (Fig. 1c) in subjects who received PLD + bortezomib. Studies of the MRP1 mutation Arg723Gln have not shown that it impacts on model substrate transport [30], but it does reduce resistance to agents such as daunorubicin and doxorubicin [19], and thus it may exert its effect instead through MRP1 expression and trafficking. MRP1 also plays a cytoprotective role by contributing to the cellular defense system against oxidative stress [31], possibly through its transport of glutathione and its conjugates [32]. Since anthracyclines exert their antitumor effects in part through the induction of just such stress [33], it may be differences in the ability of the MRP1 R723Q isoform to play such a role that increased the sensitivity to PLD + bortezomib, though this hypothesis will require laboratory testing. Moreover, further studies of the impact of the MRP1 R723Q allele will be needed to confirm our findings, given the few subjects with this genotype that were present in our population.
With regard to MDR1 gene polymorphisms, the SNP at 3435(C > T) was associated with PFS (Fig. 2), and may have been associated with response rate and TTP in subjects who received PLD + bortezomib, though not at the level of statistical significance. Also, haplotype analysis indicated that the three most frequent haplotypes may have been associated with the response rate. The most frequent diplotype that contained 3435 T (i.e., T–W–T/C–G–T or H2/H3) may have been associated with TTP (p = 0.0819) and PFS (p = 0.0891; Fig. 3) in subjects who received PLD + bortezomib when compared to the most frequent diplotype (C–G–C/T–W–T or H1/H2), though again not at the level of statistical significance.
Fig. 3.
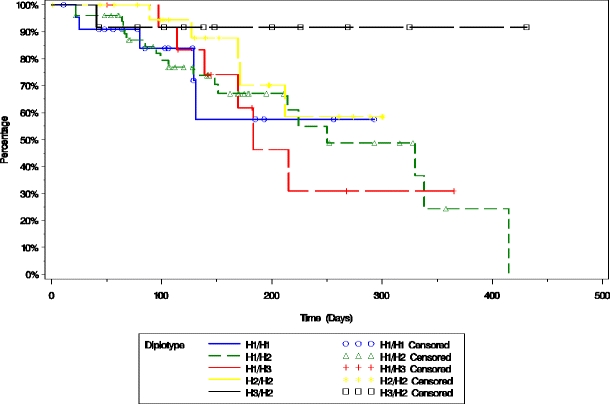
Time to progression of patients treated for their relapsed and/or refractory multiple myeloma with PLD + bortezomib is plotted based on the presence of the five most common MDR1 diplotypes using the Kaplan–Meier method
Multidrug resistance genes have previously been identified as independent prognostic factors for overall survival in patients with acute myeloid leukemia and multiple myeloma at diagnosis [14] and relapse [16]. However, this report suggests that there may be a predictive value of MDR1 genotypes in modifying the outcome of advanced myeloma. In particular, in the MDR1 C3435T SNP, the T/T allele has been found to be associated with weaker drug efflux activity [34]. It is therefore possible that myeloma cells carrying the T/T allele would experience a greater level of cytotoxicity from PLD because of less efficient doxorubicin efflux, resulting in a longer drug exposure than tumor cells with the C/C or C/T genotype. As such, T/T allele carriers might be expected to show a better response to chemotherapy, but additional preclinical and clinical data would be needed to confirm this possibility [14, 35].
While MDR1 is known to play a relevant role in the efflux of anthracyclines, the possibility that it plays a similar role with respect to bortezomib efflux still needs to be clarified. Some preclinical studies have suggested that bortezomib sensitivity may be influenced by modulation of the function of these gene products [18, 21]. Others, however, have not shown any impact on bortezomib of these ATP-cassette binding proteins [36, 37], and bortezomib has not been felt to be a substrate for these transporters [38]. Interestingly, our current studies did not reveal an association between outcomes and MDR1 or MRP1 polymorphisms in the cohort of patients treated with single-agent bortezomib, further supporting the likelihood that this proteasome inhibitor is not an MDR substrate.
In conclusion, our study does indicate that MDR1 and MRP1 polymorphisms may influence outcomes of patients with relapsed and/or refractory myeloma receiving PLD + bortezomib. If confirmed by prospective studies, these data suggest the possibility that such SNP analysis could be used to identify patients with an increased likelihood of benefiting from this rationally designed regimen. Using such approaches, it may be possible in the future to tailor therapy in the relapsed and/or refractory setting to each individual based not only on the molecular characteristics of their disease, but also on the patient’s own genetic makeup.
Acknowledgments
The authors would like to thank Dr. John Sun for his independent review of the statistical analyses presented herein, which were performed by Dr. C. Chris Huang.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
These data were presented in part at the 51st Annual Meeting and Exposition of the American Society of Hematology in New Orleans, LA, USA, December 6, 2009.
References
- 1.Kumar SK, Therneau TM, Gertz MA, et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc. 2004;79:867–874. doi: 10.4065/79.7.867. [DOI] [PubMed] [Google Scholar]
- 2.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–1979. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimopoulos MA, Kastritis E, Rajkumar SV. Treatment of plasma cell dyscrasias with lenalidomide. Leukemia. 2008;22:1343–1353. doi: 10.1038/leu.2008.123. [DOI] [PubMed] [Google Scholar]
- 4.Laubach JP, Mahindra A, Mitsiades CS, et al. The use of novel agents in the treatment of relapsed and refractory multiple myeloma. Leukemia. 2009;23:2222–2232. doi: 10.1038/leu.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- 6.Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133–2142. doi: 10.1056/NEJMoa070596. [DOI] [PubMed] [Google Scholar]
- 7.Orlowski RZ, Voorhees PM, Garcia RA, et al. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105:3058–3065. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- 8.Biehn SE, Moore DT, Voorhees PM, et al. Extended follow-up of outcome measures in multiple myeloma patients treated on a phase I study with bortezomib and pegylated liposomal doxorubicin. Ann Hematol. 2007;86:211–216. doi: 10.1007/s00277-006-0220-3. [DOI] [PubMed] [Google Scholar]
- 9.Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol. 2007;25:3892–3901. doi: 10.1200/JCO.2006.10.5460. [DOI] [PubMed] [Google Scholar]
- 10.Richardson P, Mitsiades C, Schlossman R et al. (2007) The treatment of relapsed and refractory multiple myeloma. Hematology Am Soc Hematol Educ Program 317–323. [DOI] [PubMed]
- 11.Reece DE, Leitch HA, Atkins H, et al. Treatment of relapsed and refractory myeloma. Leuk Lymphoma. 2008;49:1470–1485. doi: 10.1080/10428190802064941. [DOI] [PubMed] [Google Scholar]
- 12.Zhou SF. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica. 2008;38:802–832. doi: 10.1080/00498250701867889. [DOI] [PubMed] [Google Scholar]
- 13.Obata H, Yahata T, Quan J, et al. Association between single nucleotide polymorphisms of drug resistance-associated genes and response to chemotherapy in advanced ovarian cancer. Anticancer Res. 2006;26:2227–2232. [PubMed] [Google Scholar]
- 14.Buda G, Maggini V, Galimberti S, et al. MDR1 polymorphism influences the outcome of multiple myeloma patients. Br J Haematol. 2007;137:454–456. doi: 10.1111/j.1365-2141.2007.06605.x. [DOI] [PubMed] [Google Scholar]
- 15.Maggini V, Buda G, Martino A, et al. MDR1 diplotypes as prognostic markers in multiple myeloma. Pharmacogenet Genomics. 2008;18:383–389. doi: 10.1097/FPC.0b013e3282f82297. [DOI] [PubMed] [Google Scholar]
- 16.Buda G, Martino A, Maggini V, et al. MDR1 C3435T polymorphism indicates a different outcome in advanced multiple myeloma. Acta Haematol. 2009;122:42–45. doi: 10.1159/000235820. [DOI] [PubMed] [Google Scholar]
- 17.Minderman H, Zhou Y, O'Loughlin KL, Baer MR. Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother Pharmacol. 2007;60:245–255. doi: 10.1007/s00280-006-0367-6. [DOI] [PubMed] [Google Scholar]
- 18.Rumpold H, Salvador C, Wolf AM, et al. Knockdown of PgP resensitizes leukemic cells to proteasome inhibitors. Biochem Biophys Res Commun. 2007;361:549–554. doi: 10.1016/j.bbrc.2007.07.049. [DOI] [PubMed] [Google Scholar]
- 19.Lepper ER, Nooter K, Verweij J, et al. Mechanisms of resistance to anticancer drugs: the role of the polymorphic ABC transporters ABCB1 and ABCG2. Pharmacogenomics. 2005;6:115–138. doi: 10.1517/14622416.6.2.115. [DOI] [PubMed] [Google Scholar]
- 20.Yin JY, Huang Q, Yang Y, et al. Characterization and analyses of multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphisms in Chinese population. Pharmacogenet Genomics. 2009;19:206–216. doi: 10.1097/FPC.0b013e328323f680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura T, Tanaka K, Matsunobu T, et al. The mechanism of cross-resistance to proteasome inhibitor bortezomib and overcoming resistance in Ewing's family tumor cells. Int J Oncol. 2007;31:803–811. [PubMed] [Google Scholar]
- 22.Bladé J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 23.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kastritis E, Zervas K, Symeonidis A, et al. Improved survival of patients with multiple myeloma after the introduction of novel agents and the applicability of the International Staging System (ISS): an analysis of the Greek Myeloma Study Group (GMSG) Leukemia. 2009;23:1152–1157. doi: 10.1038/leu.2008.402. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Bladé J, Crowley J et al. (2009) Natural history of multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter International Myeloma Working Group study. Blood 114: Abstract 2878. [DOI] [PMC free article] [PubMed]
- 26.Raab MS, Podar K, Breitkreutz I, et al. Multiple myeloma. Lancet. 2009;374:324–339. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Barlogie B, Shaughnessy JD., Jr The molecular characterization and clinical management of multiple myeloma in the post-genome era. Leukemia. 2009;23:1941–1956. doi: 10.1038/leu.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucci M, Quatraro C, Dammacco F, Silvestris F. Role of active drug transporters in refractory multiple myeloma. Curr Top Med Chem. 2009;9:218–224. doi: 10.2174/156802609787521625. [DOI] [PubMed] [Google Scholar]
- 29.Turk D, Szakacs G. Relevance of multidrug resistance in the age of targeted therapy. Curr Opin Drug Discov Devel. 2009;12:246–252. [PubMed] [Google Scholar]
- 30.Letourneau IJ, Deeley RG, Cole SP. Functional characterization of non-synonymous single nucleotide polymorphisms in the gene encoding human multidrug resistance protein 1 (MRP1/ABCC1) Pharmacogenet Genomics. 2005;15:647–657. doi: 10.1097/01.fpc.0000173484.51807.48. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K, Shibata T, Oba T, et al. Multidrug-resistance-associated protein plays a protective role in menadione-induced oxidative stress in endothelial cells. Life Sci. 2009;84:211–217. doi: 10.1016/j.lfs.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 32.Keppler D, Leier I, Jedlitschky G, Konig J. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance protein MRP1 and its apical isoform MRP2. Chem Biol Interact. 1998;111–112:153–161. doi: 10.1016/S0009-2797(97)00158-0. [DOI] [PubMed] [Google Scholar]
- 33.Gianni L, Herman EH, Lipshultz SE, et al. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. 2008;26:3777–3784. doi: 10.1200/JCO.2007.14.9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hitzl M, Drescher S, van der Kuip H, et al. The C3435T mutation in the human MDR1 gene is associated with altered efflux of the P-glycoprotein substrate rhodamine 123 from CD56+ natural killer cells. Pharmacogenetics. 2001;11:293–298. doi: 10.1097/00008571-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Mizutani T, Masuda M, Nakai E, et al. Genuine functions of P-glycoprotein (ABCB1) Curr Drug Metab. 2008;9:167–174. doi: 10.2174/138920008783571756. [DOI] [PubMed] [Google Scholar]
- 36.Gil L, Styczynski J, Dytfeld D, et al. Activity of bortezomib in adult de novo and relapsed acute myeloid leukemia. Anticancer Res. 2007;27:4021–4025. [PubMed] [Google Scholar]
- 37.Wiberg K, Carlson K, Aleskog A, et al. In vitro activity of bortezomib in cultures of patient tumour cells—potential utility in haematological malignancies. Med Oncol. 2009;26:193–201. doi: 10.1007/s12032-008-9107-6. [DOI] [PubMed] [Google Scholar]
- 38.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/CNV-120030218. [DOI] [PubMed] [Google Scholar]