

Abstract
Genomes are subject to constant threat by damaging agents that generate DNA double-strand breaks (DSBs). The ends of linear chromosomes need to be protected from DNA damage recognition and end-joining, and this is achieved through protein-DNA complexes known as telomeres. The Mre11-Rad50-Nbs1 (MRN) complex plays important roles in detection and signaling of DSBs, as well as the repair pathways of homologous recombination (HR) and non-homologous end joining (NHEJ). In addition, MRN associates with telomeres and contributes to their maintenance. Here we provide an overview of MRN functions at DSBs, and examine its roles in telomere maintenance and dysfunction.
Keywords: MRN complex, DNA damage, DNA repair, DSBs, telomeres
1. Introduction
DNA double strand breaks (DSBs) can be generated by chemical and physical damage inflicted by ionizing radiation, select chemotherapy drugs, and metabolic byproduct reactive oxygen species. DSBs can also result from errors during replication, and are produced by programmed enzymatic activities during meiosis and V(D)J recombination. Regardless of how they are generated, DSBs differ from all other types of DNA lesions in that the sequence information requisite for guiding repair is no longer contained within a contiguous duplex molecule. If left unrepaired, DSBs are among the most deleterious DNA lesions, with the potential to generate chromosomal translocations, aneuploidy, and increased incidence of malignancy. The importance and centrality of DSB repair pathways during the course of evolution is demonstrated by conservation of the core components from yeast to mammals. There are two major competing pathways for DSB repair: homologous recombination (HR) and non-homologous end-joining (NHEJ). Although we do not fully understand the regulation of pathway choice, the relative extent to which these two pathways are employed depends on the cell type, the phase of the cell cycle in which the DNA damage is encountered, and also varies between species [1,2].
At least three distinct functionalities are required for repair of DSBs: detection of the damage, an ability to control the cell cycle and transcriptional programs in response to the damage, and mechanisms for catalyzing repair of the lesion. The Mre11-Rad50-Nbs1 (MRN) complex sits at the hub of the eukaryotic DSB response mechanism, and has emerged as a crucial player in each of these three facets of DSB repair. This complex of proteins acts as DSB sensor, co-activator of DSB-induced cell cycle checkpoint signaling, and as a DSB repair effector in both the HR and NHEJ pathways [3–7]. The MRN complex has also been found to associate with telomeres at the ends of linear chromosomes, where it contributes to their maintenance. Since MRN promotes sensing and repair of DNA ends, its presence at chromosome termini appears paradoxical. In this review, we first provide an overview of MRN’s constituents, structure, catalytic activities, protein binding partners, and signal transduction roles in the context of DSB repair within non-telomeric regions of the chromosome. We then discuss the emerging roles of the MRN complex in telomere maintenance and dysfunction.
2. The MRN Complex
Mre11, Rad50, and Xrs2 (the Saccharomyces cerevisiae homolog of vertebrate-specific Nbs1) were first identified via screens for yeast genes involved in meiotic recombination, and resistance to DNA damage induced by UV light and X-rays. Consistent with the nearly identical phenotypes resulting from defects in these three genes, Ogawa and coworkers demonstrated that Mre11, Rad50, and Xrs2 belong to the same epistasis group [8], and subsequently these proteins were shown to associate physically with each other in both yeast and mammals [9–11]. Here we describe specific features of each of the main components of the complex.
Mre11
Mre11 is a highly conserved 70–90 kDa protein composed of an N-terminal Mn2+/Mg2+-dependent [12] phosphoesterase domain, and two distinct C-terminal DNA-binding domains [13,14] (Fig. 1A). Isolated Mre11 forms stable dimers [15] that possess a number biochemical activities including: (i) intrinsic DNA binding activity [9,16,17] with the specific ability to synapse DSB termini [15], and (ii) endo- and exonuclease activities against a variety of single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) substrates [17,18]. While these nuclease activities contribute to both NHEJ [19] and HR [20], it should be noted that Mre11 conspicuously lacks the 5′ → 3′ exonuclease activity requisite for generating the long 3′ ssDNA overhangs necessary for HR. Although it has possible that a protein-binding partner might switch the polarity of the exonuclease activity of Mre11, it is more likely that Mre11 facilitates the activity of additional DSB processing factors. In both yeast and mammals a number of 5′ → 3′ exonucleases have been identified [12,21] that are capable of contributing to the generation of 3′ overhangs during HR, giving weight to the idea that other enzymes act in concert with MRN [22].
Fig. 1. Characteristics of the MRN complex.

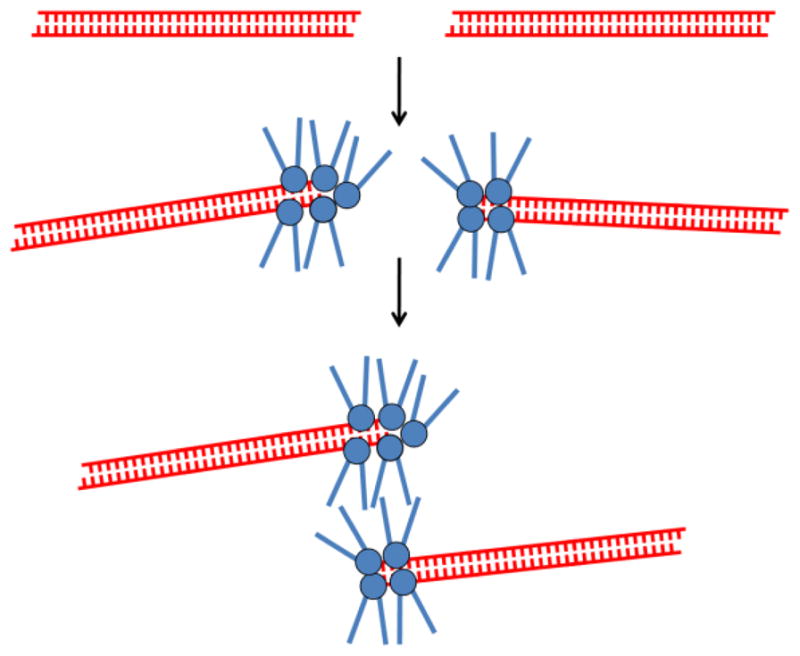
(A) Domain organization of Mre11, Rad50, and Nbs1. (B) Model of intermolecular interactions within the MRN·DNA ternary complex. While there is evidence that multiple MRN complexes can cluster at DNA termini, for simplicity only a single complex is depicted. See text for details. (C) Clustering of MRN complexes at DNA termini, and the subsequent tethering of multiple DNA molecules via the coiled-coil arms of Rad50. The DNA-binding globular heads of the MRN complexes are depicted by spheres.
In vivo, Mre11 exists in Mre112Rad502 “core” complexes where each Mre11 molecule binds a single Rad50 (Fig. 1B). The Nbs1 or Xrs2 proteins bind this core complex via interactions with Mre11 to give an overall stoichiometry of Mre112Rad502Nbs12, although there is some discrepancy over the number of Nbs1 proteins bound to the MR complex [11,23]. Supporting the importance of these interactions to MRN complex stability and function, Mre11 mutations that destabilize the MRN complex result in significantly decreased levels of Rad50 and Nbs1 in vivo [17,24], and knockdown of individual components of MRN can produce decreases in the other two members [25]. In reconstitution studies the addition of Rad50 enhances the affinity of Mre11 for DNA and stimulates its nuclease activity, and this is further enhanced by the addition of Nbs1 [17].
Rad50
Rad50 is a ~150 kDa protein displaying both sequence and structural homology to structural maintenance of chromosome (SMC) family members that control the higher-order structure and dynamics of chromatin. The N-terminal Walker A and C-terminal Walker B nucleotide binding motifs stably associate with one another to form a bipartite ATP-binding cassette (ABC)-type ATPase domain [26,27] that preferentially binds and partially unwinds dsDNA termini [28]. The intervening ~575 amino acids form an anti-parallel coiled-coil that spans ~500 angstroms and terminates with a zinc-hook (CxxC) motif [29] (Figs. 1A–B). Formation of the stable Mre112Rad502 core complex is achieved by each unit of the Mre11 dimer binding a Rad50 molecule at the intersection of its globular and coiled-coil domains [27,28]. This results in a spatial juxtaposition of the DNA-binding/termini-unwinding capacities of Rad50 with the DNA-binding, DSB tethering, and nuclease activities of Mre11 (Fig. 1B).
Independent of the Mre11-mediated dimerization of Rad50, biochemical analyses indicate that isolated Rad50 is able to form robust dimers under certain conditions [30]. Consistent with this, a crystal structure demonstrated that the globular ATPase/DNA-binding domains from two Rad50 molecules can associate [26]. Importantly, this interaction is ATP-dependent, with two ATP molecules getting sandwiched within the Rad50-Rad50 interface (Fig. 1B). Hopfner and coworkers deemed ATP hydrolysis by this complex too inefficient to support motor or helicase functions, and therefore proposed a “switch” function instead [26]. This “switch hypothesis” predicts that within the context of the Mre112Rad502Nbs12 complex, the two Rad50 molecules toggle between states in which their ATPase/DNA-binding domains are associated or disassociated (Fig. 1B). Since ATP binding-induced structural changes dramatically enhance the affinity of Rad50 for linear double stranded DNA [26], an ATP binding/hydrolysis cycle may be a mechanism for modulating the length of time the MRN complex remains bound to a substrate or product.
Scanning force microscopy has demonstrated that while the globular head of the Mre112Rad502 complex associates with the termini of linear dsDNA, the two coiled-coil regions of Rad50 are flexible “arms”, and project outward away from the DNA [28] (Fig. 1B). With increasing concentration, Mre112Rad502 complexes oligomerize at dsDNA termini, where the coiled-coil arms of Mre112Rad502 complexes mediate interactions between DNA termini [28] (Fig. 1C). Since this tethering of DNA termini was not observed at lower protein concentrations where only a single Mre112Rad502 complex was associated with a given DNA terminus, it was suggested that this may be a mechanism for achieving a robust and specific DSB tethering effect through the use of multiple weak interactions [28]. Crystallographic studies subsequently demonstrated that it is the zinc hook of Rad50 that mediates interaction between the coiled-coil arms of Rad50 molecules [29] (Fig. 1B). Mutation of the zinc-coordinating cysteine residues of Rad50 resulted in significant DSB repair defects in yeast, and complete replacement of the zinc hook with an FKBP dimerization cassette mitigated this repair defect [31]. This suggests that the ability of Rad50 molecules to bind one another via the distal terminus of their coiled-coil arms is important for MRN function.
The available structural and functional data indicate that a major role of the MRN complex is to mediate spatial juxtaposition of DNA molecules, and that this involves two distinct modes of binding. In the current model, the Mre11 dimer facilitates short-range synapsis of the two ends of a DSB, while Rad50 enables long-range tethering of two DNA molecules (such as a broken chromosome and its sister chromatid) by dimerizing via its zinc hook (Fig. 1B) [15,29]. In its fully extended conformation, the zinc-mediated Rad50 dimer places the two DNA molecules being bound by each Rad50/Mre11 globular head ~1200 angstroms from each other [29]. Importantly, however, the coiled-coils of Rad50 are extremely flexible, suggesting that even while they remain tethered by Rad50 the sister chromatids would be capable of achieving a tighter spatial proximity more conducive to homology-mediated repair.
Nbs1
The third member of the MRN complex is Nbs1, a 65–85 kDa protein. Nbs1 consists of an FHA domain and two adjacent BRCT domains at its N-terminus, in addition to an Mre11-interaction domain at its C-terminus [32,33] (Figs. 1A–B). The FHA domain binds phosphorylated threonine residues in Ser-X-Thr motifs present in DNA damage proteins, including Mdc1 and Ctp1. The BRCT domains in human Nbs1 bind Ser-X-Thr motifs when the serine residue is phosphorylated. These phospho-dependent interactions are important for recruiting repair and checkpoint proteins to DNA breaks [32,33]. By virtue of its nuclear localization signal and its interaction with Mre11, Nbs1 is responsible for translocation of the MRN complex into the nucleus. This can be observed with Nbs1 mutants lacking the Mre11-binding domain, which are themselves nuclear, while the Mre112Rad502 core complex remains cytoplasmic [34].
Limited proteolysis of Nbs1 yields a stable N-terminal fragment and C-terminal degradation products, suggesting that the FHA/BRCT core of Nbs1 is linked to Mre11 via a flexible tether [33] (Fig. 1B). While Nbs1 stimulates the DNA binding and nuclease activities of the MR complex [17], it does not itself possess a known enzymatic activity. Rather, Nbs1 contributes to DSB repair primarily by mediating protein-protein interactions at DNA breakage sites. The central region of Nbs1 possesses several SQ motifs that are phosphorylated by the ATM kinase as part of the DNA damage response. The C-terminus also contains a domain that interacts with ATM and recruits it to DSBs [35,36], and is required for the induction of apoptosis in response to damage [37].
3. MRN Mutations: Human Diseases and Mouse Models
In humans, hypomorphic mutations in the NBS1 gene result in Nijmegen breakage syndrome (NBS) [38,39], a rare autosomal recessive disorder characterized by microcephaly, immunodeficiency, and predisposition to cancer [40]. The most common mutation in NBS patients is 657del5, which results in truncated proteins that partially maintain some functions of the full length protein: a short N-terminal fragment that includes the FHA/BRCT domains can be detected, and in some cell types there is also a 70 kD C-terminal fragment which is capable of interacting with Mre11. Hypomorphic mutations in human MRE11 lead to ataxia-telangiectasia-like disorder (A-TLD) [24], in which patients display ataxia and neurodegeneration, resembling the phenotypes of ATM deficiency [41]. A single patient has been reported with a hypomorphic mutation in RAD50, who exhibited phenotypes similar to NBS [42]. Patients with NBS are prone to developing malignancies such as lymphoma and leukemia [40], and somatic alterations in MRN may contribute more widely to carcinogeneisis [43]. Cell lines derived from NBS and A-TLD patients have been valuable in dissecting the functions of MRN and the consequences of compromised function. Consistent with the involvement of MRN in cell cycle checkpoint signaling and DNA repair, cells from patients with NBS and A-TLD display increased radio-sensitivity and are defective for checkpoint activation [24,38].
Animal models of MRN defects have been useful in understanding the pathology of human disease. Null mutations in members of the MRN complex lead to embryonic lethality in mice, and therefore mouse models have been generated to mimic the hypomorphic mutations identified in human. Models have been made for A-TLD (Mre11ATLD1/ATLD1) [44] and NBS (by generating truncated proteins NBSΔb/Δb and NBSm/m, as well as a humanized NBS syndrome mouse model hNBS657Δ5) [45,46]. In addition, insights have been gleaned from mice engineered to express either Nbs1 mutants lacking specific domains [37,47] or the Rad50S hypermorphic mutation initially described in S. cerevisae (Rad50S/S) [48]. While the disease phenotypes of these mouse models are less severe than those in the corresponding human disorders, together with cell and tissue specific knockouts, they can individually recapitulate various aspects of the human disease including immunodeficiency, cancer predisposition, and germline and neuronal defects [46,49,50].
Immunological deficiency, including defects in both humoral and cellular immunity, is a hallmark of human NBS. Specifically, human B-cells display decreased variability in IgG, and IgA versus IgM subtypes, and an increased susceptibility to lymphogenesis. Conditional mouse models with targeted deletion of NBS in B lymphocytes revealed that the decreased immunoglobulin variability is due to a defect in class switch recombination (CSR) [51]. Since CSR requires repair of the DSBs induced as intermediates in the process, the basis for CSR defects in MRN deficient cells is likely due to the functions of MRN in NHEJ, although signaling defects could also contribute.
Cancer predisposition and chromosomal instability in NBS patients appears in the form of lymphomas, particularly B-cell lymphomas. NBS, ATLD, and Rad50S/S mice display increased tumorigenesis in a p53 null background, while Nbs1 heterozygotes are susceptible to various types of cancers independent of p53 [46,52]. Cells derived from the model animals are sensitive to ionizing radiation, defective for checkpoint activation and display increased amounts of chromosomal aberrations [45,48,52,53].
Germ line defects in NBS patients include infertility and compromised sexual maturation. The hNBS657Δ5 mouse recapitulates these effects when compared to littermate controls [46]. The male mice have smaller testes with histological degeneration, increased apoptosis, and delayed appearance of germ cells. The adult female mice fail to breed and have small ovaries devoid of oocytes. Analysis of meiotic chromosome spreads in oocytes at birth reveals depletion in the diplotene stage, suggesting that Nbs1 is required for meiotic progression. Analysis of meiotic events in mice harboring the hypomorphic Mre11 and Nbs1 mutations also revealed defects in synapsis of homologous chromosomes and crossovers, and suggested that MRN contributes to normal sex-specific differences in meiosis [54].
Neurological defects are a hallmark of MRN mutation, with microcephaly and ataxia observed in NBS patients, and neurodegeneration detected in A-TLD patients [55]. These different neuropathies are probably reflective of the fact that the respective disease-causing mutations differentially impact DNA damage signaling in the brain. Conditional disruption of the murine ortholog of the human NBS1 gene in the mouse central nervous system (CNS) causes ataxia, microcephally, cerebellar disorganization and disruption of the visual system [50,56]. Although the hypomorphic NBS and A-TLD mice did not exhibit neurological defects, when damage was introduced during development these mice displayed distinct neurological phenotypes which were attributed to differential activation of an apoptotic response [44,52,57]. The MRN complex is required for activation of the ATM-dependent p53 apoptotic response either during neural development, or under conditions of DNA damage [56,57]. NBS mice expressing the C-terminal domain of Nbs1 retain the ability of Nbs1 to interact with Mre11 and ATM, and this is sufficient to activate an ATM-dependent DNA damage response [35–37], which can lead to apoptosis during neuronal development and result in microcephaly [57]. In contrast, A-TLD mice subjected to ionizing radiation do not exhibit normal ATM signaling, p53 activation, and apoptosis. Therefore, accumulation of mutations and genomic damage in these cells may be responsible for the neurodegeneration seen in A-TLD patients [57].
4. The Multiple Roles of MRN in DSB Repair
The MRN complex plays critical roles in pathways involved in DNA damage repair, checkpoint activation, telomere maintenance, meiosis and DNA replication. As a prelude to our discussion of MRN functions in telomere maintenance, here we focus on the activities of the complex most relevant to detection and repair of DSBs. We refer the reader to excellent recent articles that review additional aspects of MRN function in meiosis, replication and checkpoint signaling [13,58–60].
MRN-mediated DSB Detection and Activation of Signal Transduction
Coordination of the DNA damage surveillance and repair systems functions to prevent transmission of genetic mutations [61]. Sensors detect damaged DNA and activate protein kinases that launch a network of signal transduction cascades that form the DNA damage response (DDR) [62]. This network prevents an array of human diseases, and when compromised it can lead to genomic instability and cancer [61]. The primary signaling kinases are the ataxia telangiectasia mutated protein (ATM), the ATM and Rad3-related kinase (ATR), and the DNA-dependent protein kinase (DNA-PK). When these DNA damage kinases are activated they phosphorylate a specific serine residue of histone H2AX at the breakage site and flanking chromatin [63,64]. Proteins involved in repair and checkpoint activation subsequently accumulate at the DSB to form foci that are visible by fluorescent microscopy. Post-translational modifications to histones and repair proteins determines the temporal order of accumulation at the sites of damage [65,66]. Recruited proteins are involved in signal amplification, chromatin modification, and repair of the DSB. The kinase pathways induced by DSBs in human cells result in phosphorylation of more than 700 different proteins, including the mediators Mdc1 and 53BP1, downstream checkpoint kinases Chk1 and Chk2, and proteins with a diverse array of functions [67]. Signaling in response to DNA damage activates three distinct arrests to cell cycle progression: the G1/S checkpoint prevents cells from entering S phase, the intra-S checkpoint inhibits replication during S phase, and the G2/M checkpoint prevents damaged cells from entering mitosis. Checkpoint activation can be accompanied by changes in cellular transcription profiles and apoptotic death pathways.
The MRN complex plays a role very early in the DDR, acting as a sensor of DSBs [68]. It has been demonstrated that mutation, knockdown, degradation, or mislocalization of MRN components leads to defective ATM signaling [24,69–71]. In addition to being present diffusely throughout the nucleus [72], a fraction of the MRN complex is also sequestered within sub-nuclear compartments known as promyelocytic leukemia (PML) bodies [73]. Consistent with the role of MRN as a DSB sensor, the concentration of MRN components remains constant throughout the cell cycle [72]. Immunofluorescence (IF) analyses indicate that upon DSB formation, the MRN complex rapidly relocalizes from both the diffuse pool and PML bodies to breakage sites [73,74]. MRN can be detected within these repair foci using standard IF because hundreds, if not thousands, of copies of the protein complex accumulate within the vicinity of the DSB [75]. Rapid recruitment of fluorescently labeled Nbs1 has also been demonstrated in live human cells with DSBs generated by laser microirradiation, which is restricted to small sub-nuclear areas [76]. Potential benefits of such rapid detection are that it minimizes (i) the time that free DNA termini remain vulnerable to non-specific degradation, and (ii) the amount of time that DSB termini are able to diffuse away from one another. In the presence of multiple DSBs, this serves to maximize the probability of MRN synapsing the two termini that were originally contiguous.
In undamaged cells, ATM exists as a homodimer that is incapable of phosphorylating its substrates [77], presumably because dimerization occludes substrate binding. Upon induction of DSBs by ionizing radiation ATM is recruited to DSBs, at least in part, via a direct interaction with a C-terminal motif in Nbs1 [35,36] (Fig 1A). This evolutionarily conserved C-terminal region of Nbs1 may not be absolutely required for recruitment and activation of ATM [37,47], since its loss appears to be compensated for by mediators such as 53BP1 [78]. By monitoring laser micro-irradiation-induced DSBs in real time, it was demonstrated that an MRN-dependent accumulation of ATM occurs within seconds [79]. This MRN-ATM interaction, which is optimal in the presence of dsDNA termini [35,80] and is stimulated by Rad50-mediated melting of the duplex terminus [80], increases the effective concentration of ATM in the vicinity of the DSB and also promotes the autophosphorylation of ATM dimers [80]. Autophosphorylation of ATM at serine 1981 triggers dissociation of the inactive dimer into kinase active monomers [77]. Autophosphorylation of ATM is essential to the DSB response not only because of the subsequent dissociation to active monomers, but also because it enables ATM to be retained at the breakage site [81]. The S1981A mutant of human ATM, which forms dimers that cannot be activated via autophosphorylation, is initially recruited to DSBs at a rate similar to that of wild-type ATM but is released from the break [79], and is consequently incapable of catalyzing essential downstream phosphorylation events [82].
At low doses of radiation the presence of functional MRN complex enhances activation of ATM, although there are also MRN-independent mechanisms of ATM activation. Treatment of cells with mild hypotonic solution or with the topoisomerase II inhibitor chloroquine, both of which induce chromatin structural changes without inducing DSBs, triggers ATM activation to a degree similar to that achieved by ionizing radiation [77]. Moreover, it was recently shown that modification of chromatin structure by inhibition of histone deacetylases can also activate ATM [83,84]. Activation of ATM via these types of chromatin modification (in the absence of DSBs) results in ATM-mediated phosphorylation of p53 [77] but not of proteins such as Nbs1 or SMC1 that are integral to the DSB coping mechanism [82]. This raises the possibility that upon DSB formation, ATM is actually responding to a change in the higher order structure of chromatin. In addition to MRN, other proteins recruited through ATM interactions, such as Tip60 [85], may contribute to ATM activation and modification of chromatin at DSBs. Stimuli that alter chromatin and induce ATM activation in the absence of DSBs may also involve additional ATM-binding proteins, such as ATMIN, that could compete with Nbs1 [86].
In addition to recruitment of ATM and activation of it’s signaling, MRN also participates in the early steps of end resection at DSBs (see below) and this leads to subsequent activation of the ATR kinase [87]. Processing of DSBs is required to generate the substrates that lead to ATM-dependent activation of the ATR kinase [88]. It is proposed that ATM activation leads to resection of the blunt DNA ends at breaks [87], and that production of single-stranded tails transforms the ends from ATM substrates into ATR substrates [88]. In support of this model, the nuclease activity of Mre11 has been shown to contribute to ATR activation in a mouse model [89]. In response to other types of DNA damage, functional MRN complex also promotes ATM-independent activation of the ATR kinase, although the mechanism by which this occurs is not clear [25,90–93]. It has also been shown in Xenopus extracts that MRN-dependent processing of DSBs leads to the accumulation of short single-stranded DNA oligonucleotides that stimulate ATM activity [94], and these could have relevance for ATR signaling.
Activation of ATM and ATR at DSBs initiates a signaling network that (i) provides regulation of the cell cycle (via the protein kinases Chk1 and Chk2) [95], (ii) promotes chromatin remodeling that is necessary for allowing the repair machinery access to the lesion [81,96], and (iii) contributes to the recruitment and retention of additional proteins responsible for repairing the break. The DSB-induced signaling cascade that mediates checkpoint activation and lesion repair involves a large number of proteins and post-translational modifications (phosphorylation, acetylation, methylation, ubiquitinylation, sumoylation) that have been recently reported and reviewed elsewhere [66,85,95,97–100].
Repair Pathway Selection: HR versus NHEJ
The relative extent to which DSBs are repaired via HR versus NHEJ varies among different species and cell types. In yeast and the simpler eukaryotes, which possess compact genomes and a relative paucity of repetitive sequences, HR makes a greater contribution to DSB repair. However, in mammals, where intergenic regions are larger and repetitive regions more abundant, it has been suggested that NHEJ is faster and more efficient [101]. This predisposition towards NHEJ in mammals may reflect the fact that gross chromosomal rearrangements can arise if the wrong region is utilized during HR, in a templating molecule containing multiple regions of repetitive sequence [102]. Although a given organism may display a general preference for HR or NHEJ, the extent to which each pathway is employed temporally fluctuates depending on the phase of the cell cycle [103].
There are multiple HR sub-pathways [104], but the defining feature of HR in mitotic cells is that it utilizes the sister chromatid to guide repair of a DSB during the S and G2 phases of the cell cycle [2]. In meiotic cells, HR utilizes either the homologous chromosome or the sister chromatid to guide repair during the first and second meiotic divisions, respectively [105]. During HR, exonucleolytic processing generates long 3′ ssDNA overhangs on each side of the DSB which invade and base pair with the homologous regions of the intact sister chromatid (or homologous chromosome), and prime DNA synthesis [22]. Resolution of this crossed over complex yields two intact chromosomes of identical sequence, ensuring high fidelity during the HR repair process.
In contrast to HR, NHEJ is suppressed during meiosis and is mainly employed during the G0, G1, and early S-phases of the mitotic cell cycle, when sister chromatids are not present to guide repair [1]. During NHEJ the ends of the break are joined irrespective of their sequence, and this pathway is therefore inherently error-prone. In NHEJ repair the two termini of the DSB are either directly ligated in classical NHEJ (C-NHEJ), or distal regions of microhomology (consisting of 1–4 nucleotides) on each side of the DSB are utilized to align the fragments prior to ligation during the alternative NHEJ (A-NHEJ) pathway (Figs. 2A and 2B). In both types of NHEJ, processing of the termini via nuclease-mediated removal of nucleotides or polymerase-mediated gap filling may be employed. This ability to repair DSBs whose termini display little or no homology is both the strength and the weakness of NHEJ, in that a lethal DSB is traded for a small deletion or insertion. In the presence of multiple DSBs, NHEJ repair can give rise to gross chromosome rearrangements, such as inversions and translocations [106], because there is no mechanism for determining which of the multiple DNA termini were originally contiguous.
Fig. 2. Models of NHEJ.

(A) C-NHEJ, also known as direct end joining, is carried out with minimal processing of the DSB termini prior to ligation. Both blunt and protruding termini are amenable to C-NHEJ. A single DSB can be processed via multiple routes, with the product sequence being determined by the chronology in which ligase, nuclease, and polymerase activities are employed. Blue regions denote gaps filled in by a DNA polymerase. (B) A-NHEJ involves modest resection of DSB ends (<100 nucleotides) until regions of microhomology are encountered which can guide reattachment of the DNA ends. Regions of microhomology are depicted in green.
Resection at DSBs plays a central role in determining the outcome of the competition between HR and NHEJ. During the S and G2 phases of the cell cycle, DSBs are resected to give extensive 3′ ssDNA overhangs on each side of the break [87,107,108]. This serves to generate a substrate for the HR-specific ssDNA-binding factors RPA and Rad51, and thus ensures that the DSB will be repaired via HR [106]. It also reduces the efficiency of NHEJ because the NHEJ-specific DNA termini binding factor Ku70/80 has poor affinity for ssDNA [109]. In contrast, during the G0 and G1 phases, when sister chromatids are not present to facilitate HR, the cellular DSB resection activity is downregulated [110], which gives the NHEJ machinery opportunity to bind and process the break.
The Contribution of MRN to Repair Pathway Selection
The MRN complex, via its DNA end-processing activities, plays a pivotal function in initiating the processes that direct a DSB down the most appropriate repair pathway [22,106]. Although the MRN complex plays a key role, it is not sufficient and requires collaboration with other factors. The exonuclease activity of Mre11 in vitro operates in the opposite polarity to that required for HR resection in vivo. This has prompted several groups to search for additional factors that work with MRN to facilitate DSB processing. In budding yeast S. cerevisiae the Sae2 protein is required in conjunction with MRX for processing meiotic DSBs and promoting resection [111]. Sae2 has been suggested to be an endonuclease that cooperates with Mre11 to cleave certain DNA structures [112] and process covalent protein-linked DSBs [113]. Ctp1 in the fission yeast S. pombe [114] and CtIP in human cells [115,116] have been proposed to be functional counterparts of Sae2 for resection and repair pathway choice. Human CtIP undergoes DSB-induced phosphorylation, localizes to DSB repair foci, directly binds the MRN complex, and catalyzes or confers upon MRN the ability to catalyze 5′ → 3′ resection at DSBs [117]. By binding to DSBs after ATM activation, CtIP appears to facilitate the transition from DSB sensing to end-processing [118]. CtIP depletion results in attenuated recruitment of RPA and ATR to damage induced by laser micro-irradiation [116]. Since RPA binds the extensive 3′ single-stranded overhangs generated by DSB resection, and ATR is the protein kinase that signals the presence of ssDNA, these data were used to suggest that CtIP is integral to the DSB resection process [116]. This is consistent with the reduction in HR frequency observed with CtIP depletion. Simultaneous depletion of both CtIP and Mre11 reduces HR frequency to the same degree as CtIP depletion alone [116], suggesting that these proteins function within the same pathway. In further support of this, deletion of the C-terminal region of CtIP that mediates its interaction with MRN abrogates the ability of CtIP to promote ssDNA formation [116]. Important questions remain relating to the specific role played by MRN/CtIP during the DSB resection step of HR in mammalian cells. Human CtIP shares only a very small stretch of homology with Sae2 and it has not yet been demonstrated whether CtIP has nuclease activity.
After initial processing by a complex containing MRN and CtIP, further resection by the combined action of other helicases and nucleases generates the large regions of ssDNA required to complete the HR pathway [22,106]. In S. cerevisiae, MRX/Sae2 catalyzes the initial removal of a few hundred nucleotides from a DSB [119,120] and subsequently the processive 5′ → 3′ ExoI exonuclease or the Dna2 exonuclease (in conjunction with the Sgs1 helicase) continue resection to give kilobase-sized 3′ ssDNA overhangs [119,120]. DSB resection in vertebrates likely proceeds via a similar mechanism where resection is initiated by MRN/CtIP and then completed by ExoI in conjunction with the BLM helicase [121]. Upon DSB induction, specific damage signals propagate outwards from the break along chromatin, raising the possibility that it is these modifications (and the resultant alteration of chromatin structure) that restrict the MRX/Sae2- and ExoI-mediated phases of resection to ~100 nucleotides and ~2–3 kb, respectively.
The mechanisms responsible for the cell cycle dependence of the DSB resection activity are starting to come into focus. The ability to direct DSBs towards HR during S/G2 but not during G1 appears to be rooted in both the cellular concentration of CtIP and its phosphorylation state [117]. Consistent with CtIP’s role in the DSB resection step of HR, and the minimally detectable DSB resection activity seen in G1 extracts, CtIP is barely detectable during G0/G1 but displays maximal concentration during S/G2 [122]. A similar situation is seen for the S. pombe Ctp1 protein [114]. In chicken cells, phosphorylation of CtIP specifically in S/G2 is essential for it’s function in the DSB resection step of HR [123]. It has recently been shown in S. pombe that recruitment of Ctp1 by the N-terminus of Nbs1 involves phosphorylation [33] and CK2 has been suggested to mediate this phospho-dependent interaction [124]. Phosphorylation may also play a role in recruitment of CtIP in humans [115] and Sae2 in budding yeast [125], and in both cases phosphorylation mediated by CDK provides an explanation for the cell-cycle control of DSB resection.
The extensive repertoire of structural and catalytic functions attributed to MRN is utilized differently within the HR and NHEJ pathways. The initial contribution of MRN to HR is that of DSB sensing. In contrast, a unique set of NHEJ proteins are employed for detecting, synapsing, and processing DSBs. Similar to MRN, these NHEJ repair factors are present and active throughout the cell cycle [126]. It is presently unclear how MRN out-competes the NHEJ machinery for its place at DSB termini during the S and G2 phases of the cell cycle. A significant body of data indicates that Ku suppresses HR [127–129] and reduces DSB resection during HR [130], while loss or mutation of Ku increases the frequency at which DSBs are processed via the HR pathway [131]. These data support a model in which Ku and MRN, simultaneously present throughout the cell cycle, compete for DSB termini. This raises the issue of whether the initial DSB binding event is completely random or alternatively whether there are mechanisms that promote Ku binding during G1 and MRN binding during S/G2. If MRN binds a DSB during G1, 5′ → 3′ resection is prevented by both the phosphorylation state and low concentration of MRN-associated CtIP. Whether an analogous mechanism for suppressing NHEJ if Ku binds a DSB during S/G2 remains to be determined. If Ku or MRN bind a DSB during an “inappropriate” phase of the cell cycle, having a mechanism in place for actively removing them would appear to be important. Indeed, it was recently shown that removal of Ku from an unrepaired DSB is dependent on functional MRN, and requires the ATP-binding function of Rad50 [132].
Repair Functions of MRN in HR and NHEJ
The crystal structure of the Mre11 dimer bound to two DNA termini suggests that a single MRN complex spatially juxtaposes the ends of a broken chromosome [15] (depicted in Fig. 1B). While this short-range tethering (i.e. synapsis) function could conceivably be useful in NHEJ, where termini undergo minimal processing prior to ligation, it is not obvious that this would be beneficial to HR where DNA ends are resected to give expansive tracks of ssDNA. It is unclear whether DSB termini must be synapsed in order to initiate HR 5′ → 3′ resection or whether the termini can be bound and resected independently of one another.
Although Mre11 endows the MRN complex with both ssDNA endonuclease and dsDNA 3′ → 5′ exonuclease capabilities, these nuclease activities do not have equal importance during HR. Williams and coworkers identified point mutations that abrogate either the exonuclease activity or both the exo- and endonuclease activities of Mre11 [15]. Studying these mutants in S. pombe indicates that while 3′ → 5′ exonuclease activity is dispensable for HR, loss of endonuclease activity results in a severe HR defect approaching that observed for Mre11 knockout [15]. IR is capable of producing DSBs in which the 3′ moiety does not contain the 3′-hydroxyl necessary for polymerase-mediated extension after strand invasion/base pairing. Therefore, the 3′ → 5′ exo activity of Mre11 would appear to be ideal for removing these 3′ “blocking groups”. The dispensability of the 3′ → 5′ exonuclease activity of Mre11 in this experiment suggests that either (i) the assay is not sensitive enough to detect defects in processing these 3′ blocking groups, which may be present in only a fraction of IR induced DSBs, (ii) that the endonuclease activity of Mre11 can be utilized for cleaning up 3′ termini, or (iii) that a different nuclease is employed in these situations. Evidence from a nuclease deficient Mre11 mouse model (Mre11H129N/Δ) suggests a role for the nuclease activity in early events at DSBs during HR in higher eukaryotes [89].
Initial analyses of the importance of MRN to NHEJ produced conflicting results [133,134], but emerging data have now firmly established roles for MRN in both C- and A-NHEJ. In C-NHEJ, broken termini are initially bound by the ring-shaped Ku70/Ku86 heterodimer. These Ku-bound termini recruit and activate the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which phosphorylates repair factors and checkpoint proteins in a manner analogous to, but distinct from, that of ATM during HR [135,136]. While Ku and DNA-PKcs mediate synapsis of the two DNA termini, the Artemis nuclease cleans up the termini via both endo- and exonuclease activities [137,138]. Finally, the XRCC4-DNA ligase 4 complex associates with the terminal complexes and catalyzes sealing of both strands. C-NHEJ, also known as direct end-joining, is carried out with minimal processing of the DSB termini prior to ligation (Fig. 2A). In the absence of C-NHEJ factors such as XRCC4, Ku, and DNA-PKcs, the recently described alternative A-NHEJ pathway becomes more prevalent. A-NHEJ involves modest resection of DSB ends (<100 nucleotides) until regions of microhomology are encountered which can guide reattachment of the DNA ends (Fig. 2B). Multiple groups have demonstrated that deletion of the XRCC4 gene both decreases the efficiency of NHEJ and causes a shift from usage of C-NHEJ to utilization of A-NHEJ instead [5,7]. Sequencing the joints of repaired molecules can determine which pathway has been employed. Recent studies show that siRNA-mediated knockdown of Mre11 results in reduced end-joining efficiency in both XRCC4+/+ and XRCC4−/− backgrounds [4,5]. This indicates that MRN functions in both the C-NHEJ and A-NHEJ pathways. The function of MRN in NHEJ appears to be independent of ATM signaling since these results can be reproduced in the presence of a chemical inhibitor of ATM [5].
Although C-NHEJ is used predominantly in an XRCC4+/+ background, imprecise (i.e. deletion-prone) microhomology-mediated A-NHEJ products are also detected. Depletion of Mre11 in a mouse ES XRCC4+/+ background reduces the frequency at which DSBs are repaired using microhomology, suggesting that Mre11 facilitates resection in the search for microhomologies distal to the break [5]. Nuclease-dead mutants can be used to determine whether the role of Mre11 in this C-NHEJ-competent background is due to tethering versus resection activities. In C-NHEJ-competent S. cerevisiae, end-joining defects associated with Mre11 deletion can be rescued by nuclease-defective Mre11, indicating a structural rather than catalytic function for yeast Mre11 in this context [139]. When the C-NHEJ system is compromised, as in the mouse ES XRCC4−/− background, knockdown of Mre11 results in a decrease in the length of resection tracks prior to end-joining [5]. Whether Mre11 is itself catalyzing resection in the search for microhomology or is simply facilitating this process has not been determined. Depletion of CtIP in asynchronous SV40 transformed human fibroblasts causes a significant decrease in end-joining efficiency [4], consistent with a facilitative role for MRN. These recent studies indicate that in contrast to the situation with HR, knowledge of the roles of MRN in NHEJ is just beginning to take shape. Among the important questions that will need to be addressed are how MRN facilitates repair, why it is not sterically occluded by the presence of the NHEJ machinery, and what the specific roles of Mre11 (and other nucleases) are in resection prior to end-joining.
5. MRN in Telomere Maintenance and Dysfunction
The evolutionary transition from circular to linear chromosomes brought with it two new challenges to genome stability. The first of these, known as the “end replication problem”, relates to the loss of nucleotides from the 5′ terminus of the lagging strand after every round of DNA replication. The second challenge to genome stability associated with linear chromosomes is that of preventing the chromosome termini from being recognized and processed as DSBs. The specialized repetitive sequences and protein-DNA complexes that comprise telomeres function both to maintain the chromosome ends and to provide protection from the DNA repair machinery.
Removal of the RNA primers from lagging strand Okazaki fragments results in a gap of missing nucleotides at the 5′ terminus, which cannot be filled by DNA polymerases due to the strict 5′ → 3′ polarity of their synthesis activity. Consequently, in the absence of a prophylactic mechanism, continuous cycles of replication would cause the genomes of individual organisms to grow progressively shorter and genes would be lost [140]. The addition of telomeres, non-coding repetitive DNA sequences, to the termini of chromosomes overcomes this problem. Telomerase, a specialized reverse transcriptase that carries its own RNA template, synthesizes telomeres of sufficient size to ensure that genetic information is not lost due to the end replication problem. After telomerase has synthesized a guanine-rich 3′ single stranded extension (composed of TTAGGG repeats in humans) at the chromosome terminus, the cytosine-rich complementary strand is synthesized by traditional semi-conservative replication. In yeast, telomerase is active at each round of DNA replication, thereby ensuring that telomere length maintains a steady state. In contrast, during vertebrate development and within stem cell populations, telomerase synthesizes telomeres with a length that is sufficient to sustain numerous future replication cycles. Since telomerase is inactive in somatic cells, telomeres grow progressively shorter over time until they reach a critical length, at which point cell senescence or apoptosis is triggered. Vertebrate telomeres therefore provide a solution to the end replication problem in a temporally finite manner. This brings the added benefit of tumor suppression, in that unchecked cellular replication associated with cancer is thwarted when these cells reach their critical telomere length.
Throughout Eukarya, telomeres are bound by specific proteins that sequester the free chromosome termini within a nucleoprotein cap [141]. In higher eukaryotes these same telomere-binding proteins additionally promote the formation of a unique lariat-like structure called the “t-loop”, which provides a second layer of protection from the DSB repair machinery [140,142]. Critical to t-loop formation is the generation of a 3′ single-stranded overhang (termed the “G-overhang” because it is present on the G-rich strand of the telomere) at each chromosome terminus. Since the newly synthesized lagging strand is missing a portion of its 5′ terminus, a short 3′ G-overhang (~10 nucleotides) is inherently present at this end of a newly synthesized chromosome. In contrast, leading strand synthesis generates a blunt chromosome end. In higher eukaryotes both this blunt end and the short G-overhang at the opposite chromosome end are processed by an unknown nuclease to generate mature G-overhangs 50–300 nucleotides in length [140]. Folding each chromosome terminus back upon itself enables the G-overhang to invade and base pair with the complementary strand (analogous to what occurs during HR), giving rise to the t-loop lariat (Fig. 3). Although G-overhangs are also present at telomeres in S. cerevisiae, they are only 12–14 nucleotides in length [143] and t-loops have not been observed. Regardless of whether or not t-loops are employed, the importance of sequestering telomeres into nucleoprotein caps is made clear by the fact that cap disruption can result in shortening or lengthening of telomeres, telomere fusion, telomere loss, elevated levels of recombination, and checkpoint activation [144].
Fig. 3. Human telomere structure.

Specific interactions between shelterin components, and between these proteins and specific regions of telomeric DNA are highlighted. Looping back the 3′ G-overhang enables it to invade distal, duplex regions of the telomere and base pair with the complementary C-rich strand, giving rise to the t-loop structure. See text for details.
In vertebrates stabilization of telomeres and formation of the t-loop are facilitated by the shelterin complex, which consists of the following six proteins: Telomeric Repeat-binding Factors 1 and 2 (TRF1 and TRF2), repressor and activator protein 1 (RAP1), TRF1-interacting nuclear protein 2 (TIN2), protection of telomeres 1 (POT1), and TIN2- and POT1-interacting protein (TPP1) [141]. Whereas TRF1 and TRF2 bind the double-stranded region of the telomere, POT1 has affinity specifically for the single-stranded G-overhang (Fig. 3). TIN2 bridges TRF1 and TRF2, while TPP1 bridges TIN2 and POT1. RAP1 is recruited to telomeres via its interaction with TRF2 [140]. Details regarding the vertebrate shelterin complex, its homologs in other organisms, and other facets of telomere structure and function are examined elsewhere in this issue.
The importance of the MRX complex for normal telomere maintenance was first recognized many years ago when deletion or disruption of S. cerevisiae Rad50 [145], Mre11, or Xrs2 [146] were reported to result in shortened telomeres and cell senescence [147]. Lundblad and coworkers subsequently demonstrated MRX to be in the same S. cerevisiae epistasis group as telomerase [148]. Consistent with this notion, S. cerevisiae MRX associates with telomeres in late S-phase [149] when yeast telomeres are synthesized, and in the absence of functional MRX the single-stranded telomeric DNA binding protein Cdc13 is unable to bind to telomeres [149,150]. Multiple groups have independently demonstrated that in both S. cerevisiae and S. pombe telomere length [151] and the specific formation of G-overhangs [149,152] are unaffected by the nuclease-inactivating D56N or H125N mutations of Mre11. Moreover, telomerase-mediated replication of telomeres is defective in Mre11 null cells but not in the Mre11-D56N or Mre11-H125N backgrounds [153]. Targeting S. cerevisiae telomerase to telomeres via fusion with Cdc13 overcomes telomere maintenance defects resulting from a non-functional MRX complex [153]. Collectively, these observations suggest that MRX facilitates the recruitment of telomerase to telomeres. Consistent with this, in S. cerevisiae the specific recruitment of telomerase to telomeres during the S phase is abolished by the absence of Mre11 [149]. While the MRX-mediated recruitment of yeast telomerase to telomeres could involve a direct interaction between these proteins, it could also simply be a consequence of MRX promoting the processing of telomeres into a form suitable for telomerase sequestration. Considering the well established role of MRX as a promoter of 5′ → 3′ resection at DSBs, the specificity of Cdc13 for ssDNA, and the affinity of Cdc13 for telomerase, it is tempting to speculate that MRX works with Sae2 to generate short 3′ overhangs that are bound by Cdc13 and subsequently recruit telomerase.
The first indication that MRN may also contribute to telomere maintenance in higher eukaryotes was the identification of Nbs1 and Mre11 sequestered at telomeres in meiotic human fibroblasts [154]. It was subsequently shown that MRN specifically associates with the TRF2 component of shelterin, and that Nbs1 accumulates at telomeres in S phase but not during G1 or G2 [155]. Since TRF2 does not associate with ionizing radiation induced DSBs, this interaction appears to occur exclusively within the telomere micro-environment.
Cultured human NBS fibroblasts display shortened telomeres. In these cells, the coexpression of Nbs1 along with telomerase results in longer telomeres than the expression of telomerase alone [156]. This suggests that, analogous to the situation in yeast, human MRN facilitates telomerase activity at telomeres. Further supporting this notion, knockdown of Mre11 or Nbs1 in cultured human cancer cells specifically reduces the length of 3′ G-overhangs, but has no effect in cells that do not express telomerase [157]. Collectively, these data demonstrate that MRN facilitates the action of telomerase at chromosome termini in mammalian cells. Whether MRN facilitates telomerase activity by modifying telomere ends, opening up the t-loop, altering chromatin structure, or by directly associating with telomerase remains to be determined. In telomerase-negative primary human cells, chromatin immuno-precipitation revealed that Mre11, phosphorylated Nbs1, and ATM were bound to telomeres in the G2 phase of the cell cycle [158]. This study suggested that telomeres become accessible in G2 and are recognized as DNA damage. A localized DNA damage response at telomeres may therefore be required for recruiting the processing machinery that is responsible for formation of the end protection complex.
Telomeres can be rendered dysfunctional by removal of the telomere DNA binding protein TRF2 from the shelterin complex. These “uncapped” telomeres are recognized as DSBs, and result in ATM activation, phosphorylation of Chk2 and H2AX, the formation of 53BP1-associated telomere-induced DNA damage foci (TIF), and NHEJ-mediated chromosome fusion [159]. The role of MRN at dysfunctional telomeres has recently been studied in embryonic fibroblasts derived from mice harboring MRN mutations and deletions, combined with TRF2 removal by conditional deletion or shRNA knockdown [160–162]. These studies demonstrate that MRN is required for ATM signaling in response to telomere dysfunction. When TRF2 levels are depleted by shRNA, ATM activation and TIF formation are reduced in the Mre11Δ/Δ null background but remain robust in the nuclease deficient Mre11H129N/Δ background [162]. This suggests that the MRN complex functions to detect and signal the presence of dysfunctional uncapped telomeres, and that this capability does not depend on the Mre11 nuclease activity.
Studies of dysfunctional telomeres performed in TRF2 deficient cells, have revealed a complex role for MRN in telomere fusion by NHEJ. Depending on the stage of the cell cycle and the specific structure of the telomere terminus, MRN can either promote or suppress the NHEJ-mediated fusion of dysfunctional telomeres. Conditional double-knockout of Nbs1 and TRF2 results in abrogated fusions in G1, due to defects in ATM-dependent signaling [161]. Although in G1 the MRN complex may promote NHEJ, the role of MRN after replication in G2 appears to be very different [161], and here the ability of MRN to promote end-to-end chromosome fusions at uncapped telomeres depends on Mre11’s nuclease activity [162]. In another study employing TRF2 knockdown cells, the number of chromosome fusions in the Mre11Δ/Δ and Mre11H129N/Δ backgrounds was 15-fold lower than that observed in an Mre11 active background [162]. To test whether Mre11 promotes NHEJ of TRF2-uncapped telomeres by removing the 3′ G-overhang, an in-gel hybridization assay was employed. In contrast to cells with functional Mre11 where the 3′ overhang is rapidly degraded, in Mre11Δ/Δ and Mre11H129N/Δ cells the overhang persists [162]. This suggests that the Mre11 nuclease activity is required for processing of 3′ overhangs to allow efficient NHEJ of telomere ends when rendered dysfunctional by TRF2 removal. An especially illuminating finding of these TRF2-uncapped telomere studies was that even though they occurred much less frequently, telomere-telomere fusions still occurred in the absence of functional MRN [160–162]. Importantly, the majority (~90%) of these residual telomere fusions involved the leading strands of sister chromatids. In the Mre11H129N/Δ background only ~60% of telomere-telomere fusions involved the leading strands of sister chromatids [162]. The authors suggested that this can be explained by the structural differences between telomere termini generated by leading versus lagging-strand replication. Leading-strand replication generates a blunt-ended telomere terminus that can readily be fused via NHEJ without prior nuclease processing. In contrast, lagging-strand replication generates a 3′ telomeric overhang, which is incompatible with DNA ligation [159] and would therefore require nuclease processing prior to fusion. Thus, within a TRF2 deficient background the MRN complex appears to prevent the fusion of newly replicated leading strand telomeres by promoting 5′ end resection to give NHEJ-incompatible 3′ overhangs. It will be interesting to determine the extent to which CtIP and other end-processing factors are involved here.
The cellular response to dysfunctional telomeres is in many ways similar to the response induced by non-telomeric DSBs. In both of these contexts MRN is required for ATM activation, and many of the factors that accumulate at ionizing radiation-induced foci (IRIF) also accumulate at TIF. Moreover, both damage-induced DSBs and dysfunctional telomeres can lead to the same signaling pathways, cell cycle arrest, and apoptosis [163,164]. Differences between damage-induced DSBs and uncapped telomeres include (i) the fact that TRF2 suppresses ATM activation only within the telomere microenvironment since it is abundant at chromosome ends but not elsewhere in the nucleus [165], and (ii) DNA processing is not required for the ATM-mediated damage response at telomeres [159].
6. MRN and Exogenous DNA
In addition to the situation at the termini of linear eukaryotic chromosomes, the issues of end recognition and protection also emerge when exogenous extra-chromosomal DNA is encountered in the nucleus. For example, when the genome of a linear DNA virus is delivered to the nucleus of an infected cell it may be perceived as a DSB, and therefore has the potential to trigger the endogenous DNA damage response [166]. Some viruses have therefore evolved elaborate schemes to ensure that detrimental processing of their genomic termini does not take place during viral replication. In addition to its role as a sensor of cellular DNA ends, the MRN complex has emerged as a detector of viral genomes, with a central role in the cellular response to viral DNA [166]. This ability of the MRN complex to detect DNA ends can be either beneficial or detrimental to the virus lifecycle.
One of the first examples of a virus that interacts with the MRN complex came from the dsDNA virus Adenovirus. It has been suggested that one way the linear Adenovirus genome is protected is through virally-encoded proteins that induce the degradation and mislocalization of components of the cellular DNA damage machinery, including the MRN complex [167]. Mutants of Adenovirus unable to attenuate functions of the MRN complex are defective in viral replication and progeny production [168–171]. One phenotype of these mutant viruses is that the viral genome is ligated into concatemers through end-to-end joining in a process that requires both MRN and NHEJ factors [167,172]. During infection with Adenovirus mutants unable to inactivate the MRN complex, the cellular DNA damage signaling responses are also activated [69]. Attenuation of signaling and checkpoint activation by viral proteins that degrade or mislocalize MRN demonstrated upstream functions of this complex in both ATM and ATR signaling pathways [69,90]. The termini of the Adenovirus genome are also protected by a covalently-attached, virally-encoded terminal protein, which is important for initiation of viral DNA replication. We have suggested that removal of this protein from the end of the viral genome is analogous to removal of Spo11 from DSBs during meiotic recombination [167], and we propose that the combined action of the MRN complex together with CtIP is required for processing of the protein-blocked ends.
Beyond Adenovirus, the MRN complex has been implicated in the response to many different viruses [166]. It is found at viral replication centers during the early stages of SV40 infection, and components of the complex may be specifically downregulated late during infection in an ATM-dependent manner [173]. Nbs1 was shown to interact with the SV40 encoded T-antigen, leading to the discovery that Nbs1 can suppress rereplication of genomes during S phase [174]. The MRN complex is also found at virally-induced replication centers in the nuclei of cells infected by many different viruses. For example, MRN is present within the globular compartments formed during HSV-1 infection, and in this example Mre11 is beneficial to the early stages of viral replication [175]. There is also evidence that MRN may be downregulated late during HSV-1 infection, similar to the scenario with SV40 [176]. In addition to activation of signaling in response to viral genetic material, the MRN complex also plays a role in circularization, episomal maintenance and integration of viral genomes. These studies of the MRN complex in the context of virus infection have revealed its role as part of an anti-viral defense and have highlighted ways in which cellular repair pathways can be exploited by invading genomes [166].
7. Conclusion
The MRN complex sits at a central position in a complex network that senses, signals, and ultimately facilitates repair of DNA damage. It has crucial roles as a sensor of DSBs, and in activating the signal transduction cascades that lead to cell cycle checkpoints. It also plays pivotal roles in regulating repair pathway selection, as well as the actual DNA repair processes of both NHEJ and HR. A better understanding of the activities of this multifaceted complex will explain the requirements for maintaining genomic integrity, and the malignancies that arise in patients with genome instability disorders. This will also open up new opportunities to consider chemical ways to disrupt specific branches of the pathways controlled by MRN, and in this way sensitize tumor cells to DNA damaging cancer therapeutics. A forward chemical genetic screen has already identified an inhibitor of MRN that prevents MRN-dependent activation of ATM, the G2/M checkpoint, and homology-directed repair in mammalian cells [177].
The equilibrium between telomere function and dysfunction depends on a large number of factors, and the roles of MRN in this balance are only just beginning to be elucidated. While MRN can promote telomere function by recruiting telomerase to properly capped telomeres and by preventing the fusion of newly replicated leading strand telomeres, it can also exacerbate telomere dysfunction by degrading the G-overhang and thereby promoting telomere fusion. The dual functions of MRN in DNA processing and activation of damage signaling, are evident in its response to both genomic DSBs and telomeres. The importance of MRN to telomere metabolism has been demonstrated in a wide range of organisms, including S. pombe, K. lactis, A. thaliana, D. melanogaster, and H. sapiens. Differences between these systems may be exploited to generate a detailed model of MRN function at telomeres, and will help us to navigate the complicated relationship between telomeres and the DNA damage machinery.
Acknowledgments
We apologize to the many groups whose primary research papers could not be cited due to space constraints. We thank our colleagues in the field of DNA repair for helpful discussions and members of the Weitzman lab for comments on the manuscript. Work on the MRN complex in the Weitzman lab has been supported by grants from the National Institutes of Health (AI067952, CA097093 and AI051686) and a Pioneer Developmental Chair from the Salk Institute. BJL is supported by a postdoctoral Ruth L. Kirschstein National Research Service Award (NIH/NCI T32 CA009523) and NIO is supported in part by a gift from the H.A. & Mary K. Chapman Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein KA, Rothstein R. At loose ends: resecting a double-strand break. Cell. 2009;137:807–10. doi: 10.1016/j.cell.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor EM, Cecillon SM, Bonis A, Chapman JR, Povirk LF, Lindsay HD. The Mre11/Rad50/Nbs1 complex functions in resection-based DNA end joining in Xenopus laevis. Nucleic Acids Res. 2010;38:441–54. doi: 10.1093/nar/gkp905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 5.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–13. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zha S, Boboila C, Alt FW. Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol. 2009;16:798–800. doi: 10.1038/nsmb0809-798. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa H, Johzuka K, Nakagawa T, Leem SH, Hagihara AH. Functions of the yeast meiotic recombination genes, MRE11 and MRE2. Adv Biophys. 1995;31:67–76. doi: 10.1016/0065-227x(95)99383-z. [DOI] [PubMed] [Google Scholar]
- 9.Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell. 1998;95:705–16. doi: 10.1016/s0092-8674(00)81640-2. [DOI] [PubMed] [Google Scholar]
- 10.Dolganov GM, Maser RS, Novikov A, Tosto L, Chong S, Bressan DA, Petrini JH. Human Rad50 is physically associated with human Mre11: identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol Cell Biol. 1996;16:4832–41. doi: 10.1128/mcb.16.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem. 1998;273:21447–50. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 12.Hopkins BB, Paull TT. The P. furiosus mre11/rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell. 2008;135:250–60. doi: 10.1016/j.cell.2008.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85:509–20. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 14.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–27. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 15.Williams RS, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jager M, Dronkert ML, Modesti M, Beerens CE, Kanaar R, van Gent DC. DNA-binding and strand-annealing activities of human Mre11: implications for its roles in DNA double-strand break repair pathways. Nucleic Acids Res. 2001;29:1317–25. doi: 10.1093/nar/29.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–88. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–79. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 19.Zhuang J, Jiang G, Willers H, Xia F. Exonuclease function of human Mre11 promotes deletional nonhomologous end joining. J Biol Chem. 2009;284:30565–73. doi: 10.1074/jbc.M109.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milman N, Higuchi E, Smith GR. Meiotic DNA double-strand break repair requires two nucleases, MRN and Ctp1, to produce a single size class of Rec12 (Spo11)-oligonucleotide complexes. Mol Cell Biol. 2009;29:5998–6005. doi: 10.1128/MCB.01127-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farah JA, Cromie GA, Smith GR. Ctp1 and Exonuclease 1, alternative nucleases regulated by the MRN complex, are required for efficient meiotic recombination. Proc Natl Acad Sci U S A. 2009;106:9356–61. doi: 10.1073/pnas.0902793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–95. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Linden E, Sanchez H, Kinoshita E, Kanaar R, Wyman C. RAD50 and NBS1 form a stable complex functional in DNA binding and tethering. Nucleic Acids Res. 2009;37:1580–8. doi: 10.1093/nar/gkn1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart GS, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 25.Zhong H, Bryson A, Eckersdorff M, Ferguson DO. Rad50 depletion impacts upon ATR-dependent DNA damage responses. Hum Mol Genet. 2005;14:2685–93. doi: 10.1093/hmg/ddi302. [DOI] [PubMed] [Google Scholar]
- 26.Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- 27.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–85. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 28.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–35. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 29.Hopfner KP, et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–6. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- 30.Cahill D, Carney JP. Dimerization of the Rad50 protein is independent of the conserved hook domain. Mutagenesis. 2007;22:269–74. doi: 10.1093/mutage/gem011. [DOI] [PubMed] [Google Scholar]
- 31.Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol. 2005;12:403–7. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd J, et al. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139:100–11. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams RS, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desai-Mehta A, Cerosaletti KM, Concannon P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol. 2001;21:2184–91. doi: 10.1128/MCB.21.6.2184-2191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 36.You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–79. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stracker TH, Morales M, Couto SS, Hussein H, Petrini JHJ. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature. 2007;447:218–21. doi: 10.1038/nature05740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carney JP, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 39.Varon R, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–76. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 40.Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K. Nijmegen breakage syndrome and functions of the responsible protein, NBS1. Genome Dyn. 2006;1:191–205. doi: 10.1159/000092508. [DOI] [PubMed] [Google Scholar]
- 41.Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst) 2004;3:1219–25. doi: 10.1016/j.dnarep.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Waltes R, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84:605–16. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dzikiewicz-Krawczyk A. The importance of making ends meet: mutations in genes and altered expression of proteins of the MRN complex and cancer. Mutat Res. 2008;659:262–73. doi: 10.1016/j.mrrev.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Theunissen JWF, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JHJ. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Molecular Cell. 2003;12:1511–23. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
- 45.Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. The EMBO Journal. 2002;21:1447–55. doi: 10.1093/emboj/21.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Difilippantonio S, et al. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005;7:675–85. doi: 10.1038/ncb1270. [DOI] [PubMed] [Google Scholar]
- 47.Difilippantonio S, et al. Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J Exp Med. 2007;204:1003–11. doi: 10.1084/jem.20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bender CF, et al. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002;16:2237–51. doi: 10.1101/gad.1007902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kracker S, et al. Nibrin functions in Ig class-switch recombination. Proc Natl Acad Sci USA. 2005;102:1584–9. doi: 10.1073/pnas.0409191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med. 2005;11:538–44. doi: 10.1038/nm1228. [DOI] [PubMed] [Google Scholar]
- 51.Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci USA. 2005;102:1590–5. doi: 10.1073/pnas.0406289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JHJ. A murine model of Nijmegen breakage syndrome. Curr Biol. 2002;12:648–53. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- 53.Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JH. The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr Biol. 2007;17:373–8. doi: 10.1016/j.cub.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frappart PO, McKinnon PJ. Ataxia-telangiectasia and related diseases. Neuromolecular Med. 2006;8:495–511. doi: 10.1385/NMM:8:4:495. [DOI] [PubMed] [Google Scholar]
- 56.Baranes K, et al. Conditional inactivation of the NBS1 gene in the mouse central nervous system leads to neurodegeneration and disorganization of the visual system. Exp Neurol. 2009;218:24–32. doi: 10.1016/j.expneurol.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 57.Shull ERP, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JHJ, McKinnon PJ. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev. 2009;23:171–80. doi: 10.1101/gad.1746609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–58. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- 59.Borde V, Cobb J. Double functions for the Mre11 complex during DNA double-strand break repair and replication. Int J Biochem Cell Biol. 2009;41:1249–53. doi: 10.1016/j.biocel.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 60.Borde V. The multiple roles of the Mre11 complex for meiotic recombination. Chromosome Res. 2007;15:551–63. doi: 10.1007/s10577-007-1147-9. [DOI] [PubMed] [Google Scholar]
- 61.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 64.Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. H2AX: functional roles and potential applications. Chromosoma. 2009;118:683–92. doi: 10.1007/s00412-009-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schleker T, Nagai S, Gasser SM. Posttranslational modifications of repair factors and histones in the cellular response to stalled replication forks. DNA Repair (Amst) 2009;8:1089–100. doi: 10.1016/j.dnarep.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 66.Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18:8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 67.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 68.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–62. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 69.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–20. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Girard PM, Riballo E, Begg AC, Waugh A, Jeggo PA. Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene. 2002;21:4191–9. doi: 10.1038/sj.onc.1205596. [DOI] [PubMed] [Google Scholar]
- 71.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–21. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maser RS, Monsen KJ, Nelms BE, Petrini JH. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087–96. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–8. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–2. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 75.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–95. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 76.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5:255–60. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 77.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 78.Lee JH, Goodarzi AA, Jeggo PA, Paull TT. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 2010;29:574–85. doi: 10.1038/emboj.2009.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–90. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 81.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 82.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–38. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee JS. Activation of ATM-dependent DNA damage signal pathway by a histone deacetylase inhibitor, trichostatin A. Cancer Res Treat. 2007;39:125–30. doi: 10.4143/crt.2007.39.3.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jang ER, Choi JD, Park MA, Jeong G, Cho H, Lee JS. ATM modulates transcription in response to histone deacetylase inhibition as part of its DNA damage response. Exp Mol Med. 2010;42:195–204. doi: 10.3858/emm.2010.42.3.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930–6. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanu N, Behrens A. ATMINistrating ATM signalling: regulation of ATM by ATMIN. Cell Cycle. 2008;7:3483–6. doi: 10.4161/cc.7.22.7044. [DOI] [PubMed] [Google Scholar]
- 87.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 88.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–58. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carson CT, et al. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009;28:652–62. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manthey KC, Opiyo S, Glanzer JG, Dimitrova D, Elliott J, Oakley GG. NBS1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. J Cell Sci. 2007;120:4221–9. doi: 10.1242/jcs.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olson E, Nievera CJ, Lee AYL, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J Biol Chem. 2007;282:22939–52. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- 93.Stiff T, Reis C, Alderton GK, Woodbine L, O’Driscoll M, Jeggo PA. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 2005;24:199–208. doi: 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 2008;27:1953–62. doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–83. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–82. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conde F, Refolio E, Cordon-Preciado V, Cortes-Ledesma F, Aragon L, Aguilera A, San-Segundo PA. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics. 2009;182:437–46. doi: 10.1534/genetics.109.101899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–9. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol. 2009;187:319–26. doi: 10.1083/jcb.200908074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008;7:1765–71. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 103.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 104.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–68. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ding DQ, Haraguchi T, Hiraoka Y. From meiosis to postmeiotic events: alignment and recognition of homologous chromosomes in meiosis. FEBS J. 2010;277:565–70. doi: 10.1111/j.1742-4658.2009.07501.x. [DOI] [PubMed] [Google Scholar]
- 106.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–6. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–75. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dynan WS, Yoo S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998;26:1551–9. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Y, Shim EY, Davis M, Lee SE. Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair (Amst) 2009;8:1235–41. doi: 10.1016/j.dnarep.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–8. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- 112.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–51. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–7. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–46. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–65. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402–9. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.You Z, et al. CtIP links DNA double-strand break sensing to resection. Mol Cell. 2009;36:954–69. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem. 2000;275:18541–9. doi: 10.1074/jbc.M909494199. [DOI] [PubMed] [Google Scholar]
- 123.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dodson GE, Limbo O, Nieto D, Russell P. Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle. 2010;9:1516–1522. doi: 10.4161/cc.9.8.11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–92. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–7. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wasko BM, Holland CL, Resnick MA, Lewis LK. Inhibition of DNA double-strand break repair by the Ku heterodimer in mrx mutants of Saccharomyces cerevisiae. DNA Repair (Amst) 2009;8:162–9. doi: 10.1016/j.dnarep.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fukushima T, et al. Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S-G2 phase DNA double-strand break repair. J Biol Chem. 2001;276:44413–8. doi: 10.1074/jbc.M106295200. [DOI] [PubMed] [Google Scholar]
- 129.Clikeman JA, Khalsa GJ, Barton SL, Nickoloff JA. Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKU-dependent and -independent mechanisms. Genetics. 2001;157:579–89. doi: 10.1093/genetics/157.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y, et al. Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol. 2007;14:639–46. doi: 10.1038/nsmb1261. [DOI] [PubMed] [Google Scholar]
- 131.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu D, Topper LM, Wilson TE. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–49. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang J, Dynan WS. Reconstitution of the mammalian DNA double-strand break end-joining reaction reveals a requirement for an Mre11/Rad50/NBS1-containing fraction. Nucleic Acids Res. 2002;30:667–74. doi: 10.1093/nar/30.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Di Virgilio M, Gautier J. Repair of double-strand breaks by nonhomologous end joining in the absence of Mre11. J Cell Biol. 2005;171:765–71. doi: 10.1083/jcb.200506029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 136.Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol Rev. 2004;200:132–41. doi: 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- 137.Gu J, et al. DNA-PKcs regulates a single-stranded DNA endonuclease activity of Artemis. DNA Repair (Amst) 9:429–37. doi: 10.1016/j.dnarep.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yannone SM, Khan IS, Zhou RZ, Zhou T, Valerie K, Povirk LF. Coordinate 5′ and 3′ endonucleolytic trimming of terminally blocked blunt DNA double-strand break ends by Artemis nuclease and DNA-dependent protein kinase. Nucleic Acids Res. 2008;36:3354–65. doi: 10.1093/nar/gkn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang X, Paull TT. The Mre11/Rad50/Xrs2 complex and non-homologous end-joining of incompatible ends in S. cerevisiae. DNA Repair (Amst) 2005;4:1281–94. doi: 10.1016/j.dnarep.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 140.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 11:171–81. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 142.Denchi EL. Give me a break: how telomeres suppress the DNA damage response. DNA Repair (Amst) 2009;8:1118–26. doi: 10.1016/j.dnarep.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 143.Larrivee M, LeBel C, Wellinger RJ. The generation of proper constitutive G-tails on yeast telomeres is dependent on the MRX complex. Genes Dev. 2004;18:1391–6. doi: 10.1101/gad.1199404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lydall D. Hiding at the ends of yeast chromosomes: telomeres, nucleases and checkpoint pathways. J Cell Sci. 2003;116:4057–65. doi: 10.1242/jcs.00765. [DOI] [PubMed] [Google Scholar]
- 145.Kironmai KM, Muniyappa K. Alteration of telomeric sequences and senescence caused by mutations in RAD50 of Saccharomyces cerevisiae. Genes Cells. 1997;2:443–55. doi: 10.1046/j.1365-2443.1997.1330331.x. [DOI] [PubMed] [Google Scholar]
- 146.Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–28. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chamankhah M, Xiao W. Formation of the yeast Mre11-Rad50-Xrs2 complex is correlated with DNA repair and telomere maintenance. Nucleic Acids Res. 1999;27:2072–9. doi: 10.1093/nar/27.10.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, Haber JE, Lundblad V. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol. 1998;8:657–60. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- 149.Takata H, Tanaka Y, Matsuura A. Late S phase-specific recruitment of Mre11 complex triggers hierarchical assembly of telomere replication proteins in Saccharomyces cerevisiae. Mol Cell. 2005;17:573–83. doi: 10.1016/j.molcel.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 150.Diede SJ, Gottschling DE. Exonuclease activity is required for sequence addition and Cdc13p loading at a de novo telomere. Curr Biol. 2001;11:1336–40. doi: 10.1016/s0960-9822(01)00400-6. [DOI] [PubMed] [Google Scholar]
- 151.Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–66. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tomita K, et al. Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol. 2003;23:5186–97. doi: 10.1128/MCB.23.15.5186-5197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tsukamoto Y, Taggart AK, Zakian VA. The role of the Mre11-Rad50-Xrs2 complex in telomerase-mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol. 2001;11:1328–35. doi: 10.1016/s0960-9822(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 154.Lombard DB, Guarente L. Nijmegen breakage syndrome disease protein and MRE11 at PML nuclear bodies and meiotic telomeres. Cancer Res. 2000;60:2331–4. [PubMed] [Google Scholar]
- 155.Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347–52. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 156.Ranganathan V, et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires NBS and telomerase catalytic subunit. Curr Biol. 2001;11:962–6. doi: 10.1016/s0960-9822(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 157.Chai W, Sfeir AJ, Hoshiyama H, Shay JW, Wright WE. The involvement of the Mre11/Rad50/Nbs1 complex in the generation of G-overhangs at human telomeres. EMBO Rep. 2006;7:225–30. doi: 10.1038/sj.embor.7400600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 2005;20:551–61. doi: 10.1016/j.molcel.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 159.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–8. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 160.Attwooll CL, Akpinar M, Petrini JH. The mre11 complex and the response to dysfunctional telomeres. Mol Cell Biol. 2009;29:5540–51. doi: 10.1128/MCB.00479-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dimitrova N, de Lange T. Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol. 2009;29:5552–63. doi: 10.1128/MCB.00476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deng Y, Guo X, Ferguson DO, Chang S. Multiple roles for MRE11 at uncapped telomeres. Nature. 2009;460:914–8. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 164.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–5. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 165.Karlseder J, Hoke K, Mirzoeva OK, Bakkenist C, Kastan MB, Petrini JH, de Lange T. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004;2:E240. doi: 10.1371/journal.pbio.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15:119–26. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 167.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–52. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 168.Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol. 2005;79:6207–15. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mathew SS, Bridge E. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology. 2007;365:346–55. doi: 10.1016/j.virol.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 170.Mathew SS, Bridge E. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology. 2008;374:11–22. doi: 10.1016/j.virol.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lakdawala SS, Schwartz RA, Ferenchak K, Carson CT, McSharry BP, Wilkinson GW, Weitzman MD. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J Virol. 2008;82:8362–72. doi: 10.1128/JVI.00900-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263:307–12. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- 173.Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–28. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18:1305–16. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102:5844–9. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gregory DA, Bachenheimer SL. Characterization of mre11 loss following HSV-1 infection. Virology. 2008;373:124–36. doi: 10.1016/j.virol.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dupre A, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–25. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]