

Abstract
Tissue plasminogen activator (t-PA) is one of the fibrin-specific serine proteases that play a crucial role in the fibrinolytic system. The rapid clearance of the drug from the circulation, caused by its active uptake in the liver, has lead to complicated clinical applications. Different forms of plasminogen activators have been developed to treat thrombotic disease. Deletion of the first three domains of t-PA by gene manipulation techniques has shown a significant increase in its plasma half life. In order to compensate the disadvantage of higher bleeding risk, a novel chimeric truncated form of t-PA with 394 amino acids and more fibrin affinity compared to the truncated form was designed to be expressed in Chinese Hamster Ovarian (CHO) cells. The recombinant chimeric plasminogen activator consists of kringle 2 and serine protease (K2S) domains of t-PA, namely GHRP-SYQ-K2S. The level of expression was found to be 752 IU/ml with 566,917 IU/mg specific activity, based on amidolytic activity. The fibrin binding of this novel chimeric truncated t-PA was 86% of the full length t-PA at a fibrinogen concentration of 0.2 mg/ml. This could be a promising approach with more desirable pharmacodynamic properties compared to existing commercial forms.
1. Introduction
Myocardial infarction (MI) is an irreversible necrosis or death of heart muscles due to prolonged ischemia [1]. According to the third monitoring report of the World Health Organization, cardiovascular diseases cause 12 million deaths throughout the world each year [2, 3]. Thrombolytic drugs play a crucial role in the management of patients with acute myocardial infarction, pulmonary embolism, deep vein thrombosis, arterial thrombosis, acute thrombosis of retinal vessel, extensive coronary emboli, and peripheral vascular thromboembolism [4]. Recognition of the importance of fibrinolytic system in thrombus resolution has resulted in the development of various fibrinolytic agents and plasminogen activators (PAs) with different pharmacokinetic and pharmacodynamic properties [4]. Three different generations of PAs have been introduced to the market. The first generation agents are Streptokinase and Urokinase. The second generation agents are Alteplase and Acylated plasminogen streptokinase activator complex (APSAC). The third generation agents are Vampire bat plasminogen activator (BatPA), Reteplase, Tenecteplase, Lanatoplase, and Staphylokinase [4].
Tissue-type plasminogen activator (t-PA) is the dominant PA involved in fibrinolysis [5]. T-PA is a glycoprotein with 67 kDa, 527 amino acids, which promotes conversion of plasminogen to plasmin in the presence of fibrin. The protein molecule is divided into five structural domains: finger domain (F) followed by a growth factor domain (EGF) near the N-terminal region and the two kringle 1 (K1) and kringle 2 (K2) domains. Next to kringle 2 domain is the serine protease domain with the catalytic site at the C terminus. Both finger and kringle 2 bind to the fibrin and accelerate t-PA activation on plasminogen [6]. However, full length t-PA has some major disadvantages among which the rapid clearance from plasma due to the recognition of structural elements on first three N-terminal domains by certain hepatic receptors is the most important [7–9]. Furthermore, prokaryotic production and refolding process of full length form is challenging [10]. These two substantial reasons, have lead to synthesis of the smallest active molecule such as Reteplase and Lanatoplase that are commercially available [4].
Reteplase is a deletion mutant of t-PA with prolonged half-life, in which the F, EGF, and K1 region of wild type t-PA molecular have been deleted. Finger domain is the responsible domain for fibrin affinity. Therefore, Reteplase has weaker affinity for fibrin and causes more fibrinogen depletion than full length forms [4, 11, 12]. The chimeric tetrapeptide selection was based on following evidences. Human fibrinogen can be converted to fibrin through thrombin-catalyzed release of small peptides from the amino-terminal segments of the K and L chains that are named fibrinopeptides A and B, respectively [13, 14]. The tetrapeptide GHRP can interact with a complementary site on the L lobe of fibrin monomers and prevent polymerization [13, 14]. Furthermore, it has been reported that histidine-16 of the BL chain plays an important role in the association of fibrin [14–16].
In this study, the first three domains of t-PA were deleted in order to increase half life. The other crucial change made to the protein was that a chimeric tertapeptide Gly-His-Arg-Pro (GHRP) with high fibrin affinity, was added upstream of K2S to compensate for diminished fibrin affinity due to F domain deletion. Therefore, a novel truncated form t-PA was designed to be expressed in Chinese hamster ovarian (CHO). This experiment was primarily designed and analyzed based on in silico analysis of the molecule.
2. Materials and Methods
DG44 transfection kit and Zeocin selection marker were obtained from Gibco-Invitrogen cooperation (CA, USA). Plasminogen was received from Sigma (MO, USA); Chromolize t-PA Assay Kit was purchased from Biopool (Trinity Biotech plc. Ireland). Rabbit polyclonal antibody to t-PA was supplied by Abcam (MA, USA) and goat anti rabbit IgG-HRP conjugated was purchased from Santa Cruz biotechnology (CA, USA). All reagents for SDS/polyacrylamide gel electrophoresis were from Bio-Rad, (CA, USA).
2.1. In Silico Predictions
All of the molecular modeling and structure visualizations were done on a dual processor windows-based platform, workstation using the Deepview/Swiss PDB viewer program version 3.7, 2001 (http://www.expasy.org/spdbv) and VMD 1.8.7 (http://www.ks.uiuc.edu), DPM (double prediction Method) algorithm [17]. Secondary structure prediction was used to predict the secondary structure of our novel protein. Atomic coordinates of kringle 2 and serine protease domain of t-PA were retrieved from the Protein Data Bank (http://www.rcsb.org/pdb/cgi) [entries 1tpk and 1bda, resp.] and 1fzv and 2flh both were used as templates for the 7 amino acid chimeric part: GHRP-SYQ. The 1fzv and 2flh entries were chosen since they had GDRPSY and PSYQ in their sequence, respectively, and their pdb files did not cause any problem. To produce theoretical chimeric truncated t-PA using above templates a model was designed by Modeller 9v1, via automodel package. Energy minimization was done by GROMOS96 implementation of Swiss-Pdbviewer http://www.igc.ethz.ch/GROMOS/index). The Ramachandran plots analysis was performed by RAMPAGE online Ramachandran plot analysis software (http://mordred.bioc.cam.ac.uk/~rapper/rampage).
2.2. Construction of Desired Gene Cassette Using Splicing by Overlap Extension PCR (SOEing-PCR)
Full-length human t-PA (GenBank accession number 101047) was amplified using the CHO 1–15 cell line (ATCC-CRL 9606) genomic DNA and cloned into pTZ57R during our previous work [18]. The general mechanism of SOE is illustrated in (Figure 1). The segments to be joined; fragment AB: signal sequence from upstream of t-PA full length gene and fragment CD: K2S from downstream of the gene) are amplified in separate PCRs (“1” and “2”). The primers used to be “SOEn” (B: CGGCCTATGACCTCTAGCTCCTCTTCTGAATCGGG and C: GGTCATAGGCCGTCATATCAAGGAAACAGTG) are made partially complementary to one another and also include nucleotides to be inserted (GHRP-SYQ) between signal sequence and K2S. Forward and reverse primers are as follows: A, GATCTGCCACCATGGATG, and D, TGGTCTAGATCACGGTCGCATGTTG. The PCR products of these first reactions overlap via their homologous sequences at the ends which contain the desired insertion (GHRP-SYQ) as well. The three domains were deleted and a 21 bp fragment was inserted simultaneously. The products of the first step are mixed in a SOE reaction (reaction 3), and subjected to repeated rounds of denaturation, reannealing, and primer extension by DNA polymerase. By presence of the appropriate primers (“A” and “D”) in the reaction, the recombinant product is formed. The first step PCR reactions was done in the following conditions for AB and CD fragments: first reaction: 1 μL t-PA DNA (22.7 ng/μL), 0.2 mM DNTP, 1 μL primer A (0.4 μM), 2 μL primer B (0.4 μM), 21 μL Buffer, and 2 mM MgSO4; Second reaction: 1 μL t-PA DNA (26 ng/μL), 0.2 mM DNTP, 1 μL primer D (0.4 μM), 2 μL primer C (0.4 μM), 21 μL Buffer, and 2 mM MgSO4. The SOEing PCR reaction was as follows: Fragment AB as the first template 1 μL (54 ng/μL) and fragment CD as second template 1 μL (58 ng/μL) were combined and the PCR reaction was run without the presence of primers for 3 cycles (94°C, 4 min, 94°C, 1 min, and 72°C 1 min), then the procedure completed in 94°C 1 min, 68°C 1 min, 72°C 1 min for 30 cycles, and a final 5 min 72°C extension time in the presence of A and D primers.
Figure 1.

Schematic overview of primer designed for SOEing PCR process.
2.3. Construction of Expression Vector pTracer-SV40-GHRP-SYQ-K2S
After performing SOEing PCRs, the SOEn 1200 bp gene was cleaned up by a QIAquick PCR Purification kit (Qiagen Germany), cloned in an intermediate vector pJET1.2/blunt Cloning Vector via CloneJET PCR Cloning Kit (Fermentas, Vilnius, Lithuania) based on the manufacturer's procedures. After confirming the proper sequence arrangement by bidirectional sequencing, two upstream and downstream BglII restriction sites of pJET were used due to accomplish cloning into CHO expression vector, EcoRV site in pTracer-SV40's. BglII sticky ends were converted to blunt ends using CloneJET PCR Cloning Kit DNA blunting enzyme. The recombinant pTracer-SV40-chimeric-truncated-t-PA plasmid was purified by EndoFree Plasmid Mega kit (Qiagen Germany) and the presence and right orientation of the gene was confirmed by SmaI restriction enzyme digestion (results not shown).
2.4. CHO Transfection and Transient Expression
DG44 Transfection Kit (GIBCO Invitrogen USA) was utilized to transfect DG44 CHO cells by LIPOFECTAMINE 2000 CD, based on manufacturer's 12 well plate's protocol. Transfected cells were examined under fluorescent microscope GFP filters with excitation wavelength of 400 nm and emissions wavelength of 510 nm for transient expression of pTracer_SV40 GFP protein which is partially correlated with t-PA gene expression.
2.5. Stable Expression
Zeocin sensitivity of CHO DG44 cells was determined based on pTracer-SV40 manual guide. The pTracer-SV40-chimeric-truncated t-PA was linearized from BglII restriction site. Linierized plasmid was then transected to the cells by DG44 transfection kit, and stable integrants were selected above the predetermined Zeocin sensitivity concentration (500 μg/mL).
2.6. SDS-PAGE and Western Blot Analysis
Supernatants from stable transfected CHO culture medium were harvested and concentrated using Amicon filtering system (Millipore, USA). SDS-PAGE was carried out in a 12% resolving polyacrylamide gel according to the Laemmli method. Western blot analysis of culture media was performed according to Sambrook et al. Electroblotting was performed in a semidry blotting system. Proteins were transferred to a nitrocellulose membrane, and antigen-antibody complexes were visualized by DAB-HRP system. Primary polyclonal rabbit anti-t-PA antibody (Abcam. USA) were diluted 1/1000, the secondary antibody, peroxidase conjugated goat antirabbit antibody (Santa Cruz. USA) was used in a 1/1500 dilution.
2.7. Copolymerized Plasminogen Polyacrylamide Gel Electrophoresis
An 11% resolving polyacrylamide gel was copolymerized with plasminogen and gelatin as previously described by Heussen and Dowdle [19]. The stacking gel was prepared as 4% concentration without plasminogen and gelatin. Electrophoresis was performed at 40°C at a constant current of 8 mA. The residual SDS in gel slab was removed after a gentle shaking at room temperature for 1 h in 2.5% Triton X-100. Then, the gel slab was incubated in 0.1 M glycine-NaOH, pH 8.3, for 5 h at 37°C. Finally, the gel slab was stained and destained by standard Coomassie brilliant blue (R-250) dying system. The location of the peptide harboring enzymatic activity was not stained by dye in contrast to blue-paint background.
2.8. Purification
The one-step purification procedure was performed using HiTrap Benzamidine FF (high sub) column. The column is specified for the purification of serine proteases. The buffers referred to are as follows: 0.05 mMTris-HCL, 0.5 M NaCl, PH 7.4 was used as binding and wash buffer. Elution buffers were performed using a step gradient of 0.5 M NaCl, 10 mM HCl, and PH 2.0, and 1 M NaCl, 10 mM HCl, and PH 2.0. Affinity bound substances were eluted by increasing the salt concentration. Step 1: after washing the column with 5 columns of distilled water to remove storage buffer, the column was equilibrated with 5-column volumes of binding buffer. Step 2: cell culture conditioned media was filtered through a 0.45 μm filter and loaded to the column at the same flow rate (1 mL/min). Step 3: the column was again washed with 10 column volumes of binding buffer until no material appeared in eluted fraction. Step 4: the t-PA was eluted from the column with 5 mL of each salt concentration (0.5 and 1 M NaCl ) of elution buffers. The pH and conductivity of the buffers were such that the t-PA bound selectively to the column electrostatically while the bulk of the other nonserine protease proteins material did not bind to the resin and were removed in the column flow through.
2.9. Quantification (Amidolytic Activity Test)
Biopool Chromolize t-PA Assay Kit is a biofunctional immunosorbent assay based on capturing t-PA by sp-322 monoclonal antibodies coated on the microtest wells. After fulfilling, the steps from the kit's manual samples were read at 405 nm and 492 nm. Absorbance at 492 nm is measured and subtracted from 405 nm. Various dilutions of each sample were assayed. The amount of developed colour is proportional to the amount of t-PA activity in the sample.
2.10. Fibrin Binding Assay
The binding of t-PAs was assessed by previously reported methods [20–22]. Briefly, various concentrations of fibrinogen (0–0.3 mg/mL) were mixed with bovine thrombin (0.5 U/mL) in buffer (0.05 M Tris-HCl, pH 7.4, 0.12 M NaCl, 0.01% Tween 80, and 1 mg/mL bovine serum albumin) to form fibrin clots. After incubating the mixture for 30 min at 37°C equal units (3000 unit) of truncated or full length t-PA were added. The mixture was incubated for 30 min at 37°C, and the clot was removed by centrifugation (15 min, 13 000 rpm, 4°C; Sigma 202 MD). The amount of enzyme bound to fibrin was calculated from the difference of the total amount of enzyme and the free enzyme in the supernatant, as determined by ELISA. In the ELISA assay, 100 microliters of full length and truncated t-PA are pipetted into each well. The covered plate is incubated for 15 minutes at 37°C, and the reaction is started by the addition of 100 microliters of a plasminogen/S-2251 mixture which contains two parts 10 mM S-2251, one part plasminogen solution (1 mcg/mL), and seven parts assay buffer. The mixture is prepared immediately prior to use. (Assay buffer is Tris saline prepared by dissolving 605 mg Tris in 800 mL distilled water and adjusting the pH to 8.8 with 6 normal HC1, then dissolving 5.84 grams NaCl and 0.1 grams Triton X-100 into the buffer and diluting to 1000 mL.) The absorbance (405 nm) is then measured at 20, 40, and 60 minutes [17, 23, 24].
3. Results
3.1. In Silico Predictions
3.1.1. Secondary Structure Prediction
Double prediction method (DPM) algorithm [17] was used to predict secondary structure of the chimeric truncated t-PA and full length protein. Percentage of alpha helix, extended strand, beta turn and random coil were 27.66, 26.9, 13.45, and 31.9 in chimeirc-truncated t-PA and 26.87, 26.69, 15.84, and 30.6 in full-length t-PA, respectively. As could be expected, no significant differences in secondary structure were detected between chimeric truncated t-PA and full-length protein.
3.1.2. Tertiary Structure Prediction
Chimeric-truncated-t-PA (GHRP-SYQ-K2S) was successfully modeled via MODELLER 9V1 automodel package using templates mentioned in Section 2 (Figures 2(a) and 2(b)). We superimposed chimeric-truncated model both on K2 (1TPK) and S (1BDA). The predicted model for chimeric-truncated t-PA in this study showed desirable calculated RMSDs after superimposing to the 1tpk (2.79, 2.64, and 2.94 for A, B, and C chains, resp.) and also 0.53 after superimposing to the 1bda structure. As RMSD values below 2 were acceptable in our criteria, the results of 1bda superimposing which represents functional domain of t-PA showed that the mentioned genetic manipulations do not change the main structure drastically. Therefore, it is predicted that designed novel molecule would not loose thrombolytic activity. The results of Ramachandran plot analysis showed desirable results of almost 1% of residues in outliner region for both K2S and chimeric form which is acceptable in this software's criteria.
Figure 2.

Cartoon Ribbon display in VMD 1.8.7 program for two different views of GHRP-SYQ-K2S modeled with MODELLER 9V1. (a) Left back view for a ribbon model of GHRP-SYQ-K2S modeled with MODELLER 9V1 automodel package (chimeric part shown in orange). (b) Left bottom view for a ribbon model of GHRP-SYQ-K2S modeled with MODELLER 9V1 automodel package; purple: alpha helix, blue: 3_10 helix, yellow: extended beta, tan: bridge beta, cyan: turn, and white: coil.
3.2. Preparing Desired Cassette Gene
3.2.1. SOEing PCR
We succeeded in deleting the first three domains of t-PA (Finger, EGF, and K1), and juxtaposing signal peptide upstream of K2S. A 21 base pare DNA sequence, related to GHRP-SYQ amino acids, was inserted between signal sequence and K2S domains by using 2 sets of primers in one SOEing reaction, simultaneously. The PCR products for the forward and reverse reactions which were used to generate the chimeric-truncated gene are shown in Figure 3. The PCR products with 134 bp (lane1), 1089 bp (lane3), and 1223 bp (lane4) represent signal sequence-GHRP, GHRP-SYQ-K2S and full-length mutated sequence (signal sequence + GHRP + SYQ + K2S), respectively. These findings are in accordance with the theoretical calculated lengths.
Figure 3.

Gel-electrophoresis analysis of PCR-products of chimeric truncated t-PA on 1% agarose gel. Lane 1: 134 bp forward PCR product; signal sequence + GHRP. Lane 3: 1089 bp Reverse PCR product; GHRP-SYQ-K2S. Lane 4: 1223 bp full-length SOEn PCR product signal sequence + GHRP-SYQ-K2S. Lane 2&5: 1 kb ladder.
3.2.2. Construction of Truncated-t-PA-pTracer-SV40 Fusion Plasmid
The full-length fused fragment was successfully amplified and ligated into the pTracer-SV40 vector (Invitrogen-USA) using EcoR-V and BglII restriction enzyme sites on vector and inserted gene, respectively. Furthermore, ligation was confirmed by digestion with restriction endonucleases (data not shown). To confirm absence of any kind of mutation during gene manipulation and also to determine desired chimeric conjunction, the cloned gene was sequenced. The sequence showed the desired chimeric conjunction at the 36th to the 39th residue, and first 3 amino acids of native t-PA, exactly before K2S fragment and just after signal sequence (data not shown).
3.3. Transfections
3.3.1. Transient Expression
After transfection of CHO cells with nonlinear plasmid, the cells were allowed to recover for 24 to 48 hours and then assayed by fluorescence microscopy. The results showed 10%–20% transfection efficiency (data not shown).
3.3.2. Stable Transfection
As mentioned in Section 2, chimeric truncated t-PA was cloned in pTracer-SV40 expression vector for transformation. The positive clones were selected based on Zeocin resistance and PCR amplification of the integrated t-PA gene was performed. The proper band related to chimeric truncated t-PA gene is shown in Figure 4.
Figure 4.
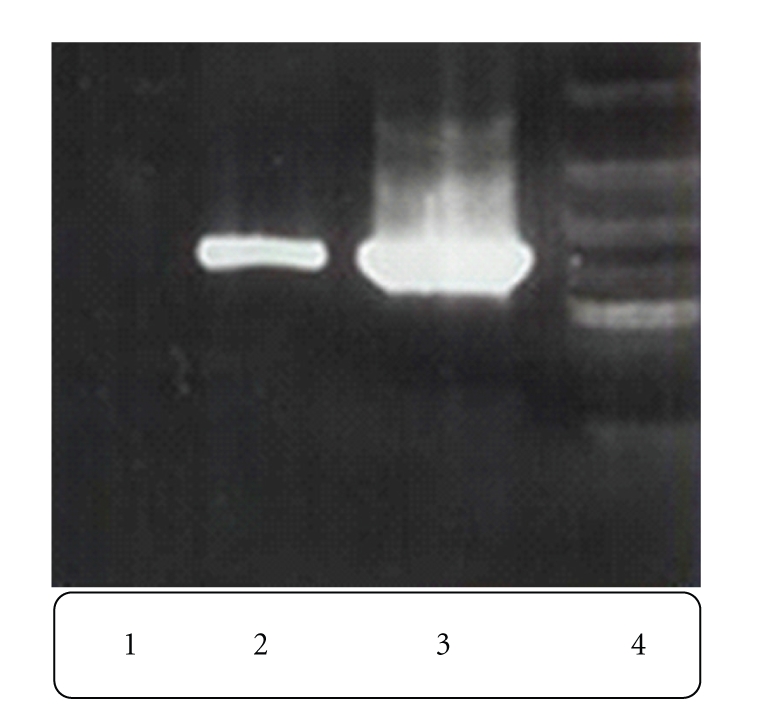
Gel-electrophoresis analysis of PCR products performed on stable transfected CHO cells and control cell line. Lane 1: PCR product from nontransfected CHO genomic DNA. Lane 2: PCR product from transfected CHO genomic DNA. Lane 3: PCR product from another transfected CHO genomic DNA. Lane 4: 1 kb ladder
3.4. Expression Analysis
Expression of chimeric truncated t-PA gene is shown in Figure 5(a); a 43 kd protein bond is shown in lane 1 Figure 5(a). This band is related to chimeric truncated t-PA based on theoretical calculations, 359 amino acid sequences. The full-length t-PA protein is shown in lane 2, 65 kDa. This finding was confirmed using antibody against t-PA protein in Western blot analysis, Figure 5(b). Lane 1 and 2 in Figure 5(b) belong to different clones of the chimeric truncated t-PA, and lane 3 is commercially available t-PA, Actylase.
Figure 5.

(a) SDS-PAGE analysis from supernatant of transfected CHO cells, 12% gel-coumasei blue stained. Right line: truncated t-PA CHO cell culture medium. Middle line: full length t-PA. Left line: protein marker. (b) Western Blot analysis from supernatant of transfected CHO cells. Lane 1&2: chimeric truncated t-PA CHO cell culture medium. Lane 3: full length t-PA.
3.5. Electrophoretic Activity Analysis
Using the standard electrophoretic procedure as described in Section 2, the supernatants of cultured media of transfected CHO cells showed enzymatic activity as clear zones of proteolysis against a blue background of undegraded stained gelatin. As shown in Figure 6, clear zone for truncated form is similar to what is demonstrated in the positive control well, commercial t-PA medicine.
Figure 6.

Identification of plasminogen activators and proteases in 11% polyacrylamide gel containing gelatin. The gel contains 11% polyacrylamide, 0.1% SDS, 0.1% copolymerized gelatin and 13 μg/ml plasminogen. Samples were electrophoresed at a constant current of 8 mA in 4°C temperature as follows: Lane3 Reference protein mixture lane 1: Actylase (full length t-PA), lane 2: Actylase +polyclonal anti-t-PA rabbit antibody, lane 4: negative control, lane 6: transfected CHO cells supernatant, lane 5: transfected CHO cells supernatant+anti-t-PA antibody. After electrophoresis the gels were washed in Triton X-100, incubated in glycine-NaOH buffer at 37°C for 3 h and fixed, stained, and destained as described under Method.
3.6. Quantitative Analysis
Our utilized affinity purification procedure resulted in more than 80% purified samples (data not shown). For quantitative determination of human tissue plasminogen activator activity in purified supernatant samples of transfected CHO cell, a biofunctional immunosorbent assay was performed. Based on Biopool's Chromolize t-PA assay kit, amidolytic unit on days 7 and 9 of culture was found to be 540 unit/mL and 752 unit/mL, respectively. This expression is much higher than what has been shown previously in E. coli, 7 unit/mL [25]. The estimated specific activity was 566,917 unit/mg. This activity is comparable to what is reported for full length t-PA: 580,000 unit/mg and truncated form: 575,000 unit/mg.
3.7. Fibrin Affinity Assay
As it is illustrated in Figure 7, in 0.2 mg/mL fibrinogen concentration the chimeric truncated protein (GHRP-SYQ-t-PA) showed a fibrin affinity of 38%, compared to full length form with 44%. In 0.3 mg/mL fibrinogen, chimeric-truncated form had 46% affinity to fibrin compared to full-length form with the value of 53%. Therefore, assuming that the control full-length form has 100% of fibrin affinity, the chimeric-truncated form represented 86% of fibrin affinity of full length protein.
Figure 7.

Binding to fibrin clots. Different concentrations of fibrin were incubated with full length (♦) or chimeric-truncated t-PA (∗) Data represent means obtained from three experiments.
4. Discussion
Plasminogen activators (PAs) are used in treating cardiovascular and cerebrovascular obstructions [12, 26–28]. Apart from laborious production and refolding process, short plasma half life has complicated full-length t-PA's clinical application [8, 29, 30]. Deletion-variants of t-PA such as Lanotaplase and Reteplase are nonglycosylated truncated forms constructed to increase plasma half life [4, 31, 32]. It seems that deletion of these regions could be an advantage. In addition, presence of GHRP amino acids sounds beneficial for improving fibrin affinity [16]. Taking advantage of these two factors a chimeric truncated form of t-PA was designed based on in silico analysis. The analysis showed that the molecule preserves the crucial regions of full length t-PA. As previously reported the first 3 amino acids of t-PA (SYQ) are important for the protein activity, and we decided to conjugate this part to the K2S (functional domain) the same as what is done in Reteplase (truncated form of t-PA). This truncated form has proven to be active while SYQ is juxtaposed to K2S domain without any other interfering amino acids. So, we proposed that SYQ-K2S arrangement is responsible for the serine protease activity. On the other hand, the aim of this study was to improve some pharmacodynamic properties of an existing truncated t-PA (Reteplase). So, in order to compare our proposed form with the previous one we should keep all other parts intact; that is, SYQ-K2S should have been persevered to observe the sole effect of GHRP addition in fibrin-binding affinity. The chimeric GHRP was designed to increase fibrin affinity compared to Reteplase. We proposed that in order to have better access to fibrin cloths in purpose of improved interaction of the chimeric protein with fibrin cloths it is better to have the chimeric GHRP situated in the N-terminus and not between other residues the same as what is done in Jiao et al.'s article.
Apart from cell line development [33] and synergism strategies [33–35] improving efficacy by protein engineering is greatly promising. Principal focus of these efforts is eliminating regions to reduce the size of molecule and increase half life. Deleting F, E, and k1 domains reduces both fibrin binding and fibrin enhancement of plasminogen activator [36, 37]. In order to compensate this disadvantage, inserting additional fibrin-binding sites into the molecule enhances specifity.
Our chimeric-truncated form showed desirable protein stability based on Ramachandran plot predictions and RSMD values. In Ramachandran plot analysis, nearly, 1% of the residues, including residues 4 and 5 were found to be in outliner region. Given that the residues 4 and 5 are representing an unnatural, chimeric junction, less than 1% of residues are not modeled well which is favorable compared to similar strategies [38]. Additionally, the best RMSD value was the result of superimposing our model on 1BDA template which consists of 300 amino acids; 76% of total protein residues and the active site as well.
The experimental analysis confirmed our in silico analysis findings. Chimeric-truncated t-PA was expressed in CHO DG44 cells. The level of expression was found to be 752 IU/mL in optimized condition. This level of expression is promising compared to enzymatic activity of produced t-PA in E. coli (3–7 IU/mL) [10], nonmodified rCHO (50 IU/mL) [37], Aspergillus nidulans (0.1 μg/mL) [39], or Leishmania tarentolae (70 IU/mL) [18].
Enhancing productivity of cell lines is crucial for reducing costs. Among nonrecombinant mammalian cell producers Bowes melanoma cell line is perhaps the best choice with the productivity more than 30 IU per 106 cells per day (0.1 mg/L conditioned medium) [37, 40]. Epithelial cells like guinea pig ear keratocytes (GPK) have yields of production ranged over 5–10 mg/L from perfusion cultures with (7–10)×106 anchored cells per milliliter [37, 40]. Between recombinant mammalian cells with inserted copies of human t-PA gene, mouse C127 fibroblasts (with 55 mg/L production rate ), rat myeloma (with 52 mg/L production rate), and CHO cells (with 65 mg/L production rate) in perfused or fed-batch systems are worth mentioning [37, 41].
Considering other recombinant cultures, Saccharomyces cervisiae has a low yield of production with extensive hyper glycosylated and heterogenic forms [42]. Bacterium Escherchia coli produces t-PA as inclusion bodies consisting 5%–10% of cell proteins [43]. Because of low recovery of active t-PA from inclusion bodies, bacterial production process does not seem feasible. In one study, coexpressing of native or heterologous cysteine oxidoreductases increased expression levels to 180 μg/L [44]. Despite the complexity of renaturation from inclusion bodies [45], E. coli fermentation is far less demanding than animal cell culture. Furthermore, glycosylation is not necessary for biological activity; also nonglycosylated forms show increased plasma half lives, hence, t-PA.
Production in bacteria is still desirable and perhaps become feasible either with advances in renaturation methods or with reduction in number of disulfide bonds via protein engineering. The suggested chimeric-truncated form is promising due to potential of production in prokaryotic systems similar to its nonglycosylated commercial relative, Reteplase.
We designed our cassette gene by having t-PA signal sequence upstream of chimeric-truncated molecule via SOEing PCR in order to take advantage of proper folding and glycosylation in CHO. Different sizes of templates is a problematic matter in applying SOEing PCR procedure and closer template sizes are preferred in this strategy [46]. Here, owing to appropriate primer design, two remarkably different sizes of templates, 134 bp and 1089 bp, were successfully sued by using sensible molar ratios of both templates and primers. In addition, deletion and insertion were performed concurrently.
The protein migrated with an apparent molecular mass of 43 kDa on SDS-PAGE; also, an 86 kDa bond was obvious while western blotting was performed. All the dimer form was converted to monomer form by dissolving the protein in 8 M urea, and just one bond at 43 KD was observed on Western Blot (data not shown). These data confirmed that the doubled molecular mass size could be due to aggregation of truncated t-PA.
Biological activity was tested with electrophoretic zymography analysis. Previous studies have shown that the main extracellular protease band secreted by the CHO was approximately 92 or 95 KDa [47, 48]. This indicates that the extra higher size bonds are probably from CHO itself. However, presence of an obvious band of almost 43 KDa and its drastic diminish after inhibition by antibody confirms that the proteolytic activity was exclusively due to t-PA and not other probably existing serine proteases. As it is shown in Section 3, (lane 5 of Figure 6), significant decline of proteolytic zone in the presence of special t-PA antibody is apparent. The degree of inhibition by antibody is dependent upon the t-PA concentration, but is nevertheless dramatically reduced when compared to the equivalent concentration in samples without antibody [19, 48].
Both fibrin specifity and fibrin affinity are touted as desirable properties in PAs [21, 23]. The fibrin-specific agents are much more likely to bind to fibrin bound plasminogen as opposed to free plasminogen. Therefore, the lack of systemic plasminemia limits systemic fibrinogenolysis, which is associated with bleeding complications [32]. Reteplase was initially thought to have desired fibrin-specifity [32], but in practice it was less fibrin specific than full-length form [4]. Subsequently, higher frequency of bleeding was reported by Reteplase [4]. Previous studies showed that Reteplase has the in vitro fibrin binding equal to 30% of that of full-length form [12]. The fibrin-binding experiment demonstrated that chimeric-truncated t-PA had 86% of fibrin-binding capacity of full-length form, Actylase, suggesting that our novel protein could be targeted to fibrin clot with GHRP. However, considering controversies now besetting advantages of fibrin affinity, to what extent this increase of fibrin affinity is beneficial for drug is now under investigation in our group for further steps of research. Nevertheless, as the chimeric protein has yet less fibrin affinity than full-length one, it can be said that this degree of increase is not at least pernicious.
Acknowledgment
This paper was supported by a grant from the Pasteur Institute of Iran.
References
- 1.Boersma E, Mercado N, Poldermans D, Gardien M, Vos J, Simoons ML. Acute myocardial infarction. The Lancet. 2003;361(9360):847–858. doi: 10.1016/S0140-6736(03)12712-2. [DOI] [PubMed] [Google Scholar]
- 2.Beaglehole R, Saracci R, Panico S. Cardiovascular diseases: causes, surveillance and prevention. International Journal of Epidemiology. 2001;30(1):S1–S4. doi: 10.1093/ije/30.suppl_1.s1. [DOI] [PubMed] [Google Scholar]
- 3.The World Health Report. Geneva, Switzerland: World Health Organiztion; 2000. Health system:Improving performance. [Google Scholar]
- 4.Baruah DB, Dash RN, Chaudhari MR, Kadam SS. Plasminogen activators: a comparison. Vascular Pharmacology. 2006;44(1):1–9. doi: 10.1016/j.vph.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Camiolo SM, Thorsen S, Astrup T. Fibrinogenolysis and fibrinolysis with tissue plasminogen activator, urokinase, streptokinase-activated human globulin, and plasmin. Proceedings of the Society for Experimental Biology and Medicine. 1971;138(1):277–280. doi: 10.3181/00379727-138-35878. [DOI] [PubMed] [Google Scholar]
- 6.Rijken DC. Relationships between structure and function of tissue-type plasminogen activator. Klinische Wochenschrift. 1988;66(supplement 12):33–39. [PubMed] [Google Scholar]
- 7.Kuiper J, Van’t Hof A, Otter M, Biessen EAL, Rijken DC, van Berkel TJC. Interaction of mutants of tissue-type plasminogen activator with liver cells: effect of domain deletions. Biochemical Journal. 1996;313(3):775–780. doi: 10.1042/bj3130775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emeis JJ, van den Hoogen CM, Jense D. Hepatic clearance of tissue-type plasminogen activator in rats. Thrombosis and Haemostasis. 1985;54(3):661–664. [PubMed] [Google Scholar]
- 9.Hotchkiss A, Refino CJ, Leonard CK, et al. The influence of carbohydrate structure on the clearance of recombinant tissue-type plasminogen activator. Thrombosis and Haemostasis. 1988;60(2):255–261. [PubMed] [Google Scholar]
- 10.Pennica D, Holmes WE, Kohr WJ. Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature. 1983;301(5897):214–221. doi: 10.1038/301214a0. [DOI] [PubMed] [Google Scholar]
- 11.Philippe C. Biotechnology for cardiovascular diseases. 2007, www.bioimpact.org.
- 12.Weaver WD. The role of thrombolytic drugs in the management of myocardial infarction. Comparative clinical trials. European Heart Journal. 1996;17:9–15. doi: 10.1093/eurheartj/17.suppl_f.9. [DOI] [PubMed] [Google Scholar]
- 13.Bailey K, Bettelheim FR, Lorand L, Middlebrook WR. Action of thrombin in the clotting of fibrinogen. Nature. 1951;167(4241):233–234. doi: 10.1038/167233a0. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa N, Sasaki S. Location of the binding site “b” for lateral polymerization of fibrin. Thrombosis Research. 1990;57(2):183–195. doi: 10.1016/0049-3848(90)90318-7. [DOI] [PubMed] [Google Scholar]
- 15.Pandya BV, Gabriel JL, O’Brien J, Budzynski AZ. Polymerization site in the βchain of fibrin: mapping of the B β 1–55 sequence. Biochemistry. 1991;30(1):162–168. doi: 10.1021/bi00215a024. [DOI] [PubMed] [Google Scholar]
- 16.Jiao J, Yu M, Ru B. Characterization of a recombinant chimeric plasminogen activator with enhanced fibrin binding. Biochimica et Biophysica Acta. 2001;1546(2):399–405. doi: 10.1016/s0167-4838(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 17.Deleage G, Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Engineering. 1987;1(4):289–294. doi: 10.1093/protein/1.4.289. [DOI] [PubMed] [Google Scholar]
- 18.Soleimani M, Mahboudi F, Davoudi N, et al. Expression of human tissue plasminogen activator in the trypanosomatid protozoan Leishmania tarentolae. Biotechnology and Applied Biochemistry. 2007;48(1):55–61. doi: 10.1042/BA20060217. [DOI] [PubMed] [Google Scholar]
- 19.Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Analytical Biochemistry. 1980;102(1):196–202. doi: 10.1016/0003-2697(80)90338-3. [DOI] [PubMed] [Google Scholar]
- 20.van Zonneveld A-J, Veerman H, Pannekoek H. On the interaction of the finger and the kringle-2 domain of tissue-type plasminogen activator with fibrin. Inhibition of kringle-2 binding to fibrin by ε-amino caproic acid. The Journal of Biological Chemistry. 1986;261(30):14214–14218. [PubMed] [Google Scholar]
- 21.Nesheim M, Fredenburgh JC, Larsen GR. The dissociation constants and stoichiometries of the interactions of Lys-plasminogen and chloromethyl ketone derivatives of tissue plasminogen activator and the variant Δ FEIX with intact fibrin. The Journal of Biological Chemistry. 1990;265(35):21541–21548. [PubMed] [Google Scholar]
- 22.Higgins DL, Vehar GA. Interaction of one-chain and two-chain tissue plasminogen activator with intact and plasmin-degraded fibrin. Biochemistry. 1987;26(24):7786–7791. doi: 10.1021/bi00398a038. [DOI] [PubMed] [Google Scholar]
- 23.Green George DJ. purified type I and type II t-PA. US Patent WO 89/09820, 1989.
- 24.Kohnert U, Rudolph R, Verheijen JH, et al. Biochemical properties of the kringle 2 and protease domains are maintained in the refolded t-PA deletion variant BM 06.022. Protein Engineering. 1992;5(1):93–100. doi: 10.1093/protein/5.1.93. [DOI] [PubMed] [Google Scholar]
- 25.Manosroi JMA. Methods for large scale protein production in prokaryotes. WO/2002/040696, 2002.
- 26.Levine GN, Ali MN, Schafer AI. Antithrombotic therapy in patients with acute coronary syndromes. Archives of Internal Medicine. 2001;161(7):937–948. doi: 10.1001/archinte.161.7.937. [DOI] [PubMed] [Google Scholar]
- 27.Ohman EM, Harrington RA, Cannon CP, Agnelli G, Cairns JA, Kennedy JW. Intravenous thrombolysis in acute myocardial infarction. Chest. 2001;119(supplement 1):253S–277S. doi: 10.1378/chest.119.1_suppl.253s. [DOI] [PubMed] [Google Scholar]
- 28.Russell D, Madden KP, Clark WM, Zivin JA. Tissue plasminogen activator cerebrovascular thrombolysis in rabbits is dependent on the rate and route of administration. Stroke. 1992;23(3):388–393. doi: 10.1161/01.str.23.3.388. [DOI] [PubMed] [Google Scholar]
- 29.Topol E, Califf R, Van de Werf F, et al. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. The New England Journal of Medicine. 1993;329(10):673–682. doi: 10.1056/NEJM199309023291001. [DOI] [PubMed] [Google Scholar]
- 30.Collen D. Towards improved thrombolytic therapy. The Lancet. 1993;342(8862):34–36. doi: 10.1016/0140-6736(93)91888-s. [DOI] [PubMed] [Google Scholar]
- 31.Bhana N, Spencer CM. Lanoteplase. BioDrugs. 2000;13(3):217–224. doi: 10.2165/00063030-200013030-00006. [DOI] [PubMed] [Google Scholar]
- 32.Ouriel K, Katzen B, Mewissen M, et al. Reteplase in the treatment of peripheral arterial and venous occlusions: a pilot study. Journal of Vascular and Interventional Radiology. 2000;11(7):849–854. doi: 10.1016/s1051-0443(07)61799-0. [DOI] [PubMed] [Google Scholar]
- 33.Klausner A. Researchers probe second generation t-PA. Biotechnology. 1986;4:706–711. [Google Scholar]
- 34.Noll G, Lammle B, Duckert F. Treatment with stanozolol before thrombolysis in patients with arterial occlusions. Thrombosis Research. 1985;37(4):529–532. doi: 10.1016/0049-3848(85)90098-2. [DOI] [PubMed] [Google Scholar]
- 35.Pierard LQL. Secretion of recombinant hybrid plasminogen activator by mouse myeloma cells. In: Spier RE, Griffiths JB, Stephenne J, Crooy PJ, editors. Advances in Animal Cell Biology and Technology for Bioprocesses. London, UK: Butterworths; 1989. pp. 475–480. [Google Scholar]
- 36.Cartwright T. Production of t-PA from animal cell culture. In: Spier RE, Griffiths JB, Stephenne J, Crooy PJ, editors. Animal Cell Biotechnology. New York, NY, USA: Academic Press; 1992. pp. 217–245. [Google Scholar]
- 37.Rouf SA, Moo-Young M, Chisti Y. Tissue-type plasminogen activator: characteristics, applications and production technology. Biotechnology Advances. 1996;14(3):239–266. doi: 10.1016/0734-9750(96)00019-5. [DOI] [PubMed] [Google Scholar]
- 38.Bagchi P, Mahesh M, Somashekhar R. Pharmaco-informatics: homology modelling of the target protein (GP1, 2) for Ebola hemorrhagic fever and predicting an Ayurvedic remediation of the disease. Journal of Proteomics and Bioinformatics. 2009;2(7):287–294. [Google Scholar]
- 39.Upshall A, Kumar AA, Bailey MC, et al. Secretion of active human tissue plasminogen activator from the filamentous fungus Aspergillus nidulans. Bio/Technology. 1987;5(12):1301–1304. [Google Scholar]
- 40.Datar RV, Cartwright T, Rosen C-G. Process economics of animal cell and bacterial fermentations: a case study analysis of tissue plasminogen activator. Nature Biotechnology. 1993;11(3):349–357. doi: 10.1038/nbt0393-349. [DOI] [PubMed] [Google Scholar]
- 41.Lubiniecki A, Arathoon R, Polastri G, et al. Selected strategies for manufacture and control of recombinant tissue plasminogen activator prepared from cell culture. In: Spier RE, Griffiths JB, Stephenne J, Crooy PJ, editors. Advances in Animal Cell Biology and Technology for Bioprocesses. London, UK: Butterworths; 1990. pp. 442–451. [Google Scholar]
- 42.Lemontt JF, Wei CM, Dackowski WR. Expression of active human uterine tissue plasminogen activator in yeast. DNA. 1985;4(6):419–428. doi: 10.1089/dna.1985.4.419. [DOI] [PubMed] [Google Scholar]
- 43.Griffiths JB, Electricwala A. Production of tissue plasminogen activators from animal cells. Advances in Biochemical Engineering. 1987;34:147–166. doi: 10.1007/BFb0000678. [DOI] [PubMed] [Google Scholar]
- 44.Manosroi J, Tayapiwatana C, Götz F, Werner RG, Manosroi A. Secretion of active recombinant human tissue plasminogen activator derivatives in Escherichia coli. Applied and Environmental Microbiology. 2001;67(6):2657–2664. doi: 10.1128/AEM.67.6.2657-2664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chisti Y, Moo-Young M. Separation techniques in industrial bioprocessing. Journal Institution of Chemical Engineers Symposium Series. 1994;137:135–146. [Google Scholar]
- 46.Li X, Qiu Y, Shen Y, et al. Splicing together different regions of a gene by modified polymerase chain reaction-based site-directed mutagenesis. Analytical Biochemistry. 2008;373(2):398–400. doi: 10.1016/j.ab.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Elliott P, Hohmann A, Spanos J. Protease expression in the supernatant of Chinese Hamster Ovary cells grown in serum-free culture. Biotechnology Letters. 2003;25(22):1949–1952. doi: 10.1023/b:bile.0000003992.09492.4b. [DOI] [PubMed] [Google Scholar]
- 48.Opdenakker GMM. Protease inhibitors, a DNA construct for the expression of a protease and a process for measuring proteases and/or protease inhibitors, EP0736302, 1996.