
Table 1. Phylogenetic signal in the 4 datasets for the three different traits.
| Dataset |

|
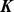 for log(spVL) for log(spVL) |
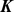 for dsCD4 for dsCD4 |
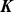 for prAZT for prAZT |
 for log(spVL) for log(spVL) |
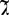 for dsCD4 for dsCD4 |
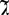 for prAZT for prAZT |
| MSM strict | 134 | 0.59
|
n.s. | 0.91
|
0.51 (0.27) | 0 | 1.07 (0.12) |
| all strict | 230 | n.s. | n.s. | n.s. | 0.17 | 0 | 0.88 (0.06) |
| MSM liberal | 404 | 0.09
|
n.s. | 0.82
|
0.13 (0.05) | 0 | 1.07 (0.015) |
| all liberal | 661 | n.s. | n.s. | 0.71
|
— | — | — |
We use two estimators (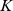 and
and 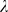 ) that lead to similar results. Significance code for the p-value of the randomisation test for
) that lead to similar results. Significance code for the p-value of the randomisation test for 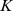 is ‘
is ‘ ’
’ 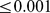 , ‘
, ‘ ’
’ 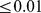 , ‘
, ‘ ’
’ 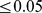 , and ‘n.s.’ indicates that the signal does not differ from that found on a random tree. The
, and ‘n.s.’ indicates that the signal does not differ from that found on a random tree. The 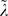 are obtained by taking the median value of
are obtained by taking the median value of 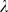 over 161 trees (see the Methods). We also show the standard deviation in brackets. ‘—’ indicates that the largest tree could not be computed with the Bayesian method because of the large number of patients.
over 161 trees (see the Methods). We also show the standard deviation in brackets. ‘—’ indicates that the largest tree could not be computed with the Bayesian method because of the large number of patients.  is the sample size of each dataset.
is the sample size of each dataset.