

Abstract
The proposal that molecules can perform electronic functions in devices such as diodes, rectifiers, wires, capacitors, or serve as functional materials for electronic or magnetic memory, has stimulated intense research across physics, chemistry, and engineering for over 35 years. Because biology uses porphyrins and metalloporphyrins as catalysts, small molecule transporters, electrical conduits, and energy transducers in photosynthesis, porphyrins are an obvious class of molecules to investigate for molecular electronic functions. Of the numerous kinds of molecules under investigation for molecular electronics applications, porphyrins and their related macrocycles are of particular interest because they are robust and their electronic properties can be tuned by chelation of a metal ion and substitution on the macrocycle. The other porphyrinoids have equally variable and adjustable photophysical properties, thus photonic applications are potentiated. At least in the near term, realistic architectures for molecular electronics will require self-organization or nanoprinting on surfaces. This review concentrates on self-organized porphyrinoids as components of working electronic devices on electronically active substrates with particular emphasis on the effect of surface, molecular design, molecular orientation and matrix on the detailed electronic properties of single molecules.
1. Introduction
Many current electronic technologies are rapidly approaching the limit of performance and miniaturization in a growing number of applications across science and technology because traditional inorganic materials and component architectures are optimized to the edges of the theoretical limits of performance. Most currently employed techniques for mass production of silicon based electronic devices involve multiple chemically and energetically wasteful procedures such as purification and crystallization of wafer materials, high energy radiation for patterning, toxic etchants, and vapor deposition of nanolayers. As a consequence of these methods, manufacturing large arrays of chips, each with millions of devices, fabrication facilities are stunningly expensive to build and operate. Additionally, most of the energy used to power these devices is lost as heat rather than used to perform intended functions, the dissipation of which requires additional production and energy costs. A great motivation towards the development of molecular electronics is part of efforts to increase performance while at the same time diminishing component size, reduce production costs, and minimizing the environmental impacts of production and operation [1]. Flexible display and electronics technologies, and ink-jet printing of circuitry will also benefit from molecule based electronics.
Molecules or collections of molecules functioning as electronic components have ample precedent in nature. Voltage, ligand, antibiotic, and other ion conducting channels are digital electronics self-assembled into biological membranes in that they have only ‘on’ or ‘off’ positions with unit conductance that are unique to a given channel [2]. Photosynthetic reaction centers transport electrons over about eight nm with remarkable efficiency. Ion pumps can also be gated. The photo-driven purple membrane pumps containing bacteriorhodopsin have been studied for many decades in terms of their potential as molecular electronic and photonic materials because they are very robust, can cycle many thousands of times, and the distinctive color changes upon oxidation and/or reduction impart a second functionality to these materials [3, 4]. However, the rate of conducting ions in channels and bacteriorhodopsin proteins, and the stability of the former, limit the usefulness of these constructs as components of complex electronic devices. The various photosynthetic systems can provide much inspiration, but are too fragile for real-world applications.
Research on molecular electronics focuses on the molecule, but melds concepts from diverse fields such as physics, chemistry, biophysics, and electrical engineering. See several recent reviews that focus on different aspects of molecular electronics ranging from surface chemistry to molecular design to theory, including a beautiful discussion of mechanical bonds with molecular electronics applications by Stoddart [5–9]. Electronics fabricated from organic materials are potentially much less toxic, easier to recycle, and scalable. In addition, molecular electronics have the potential to contribute to the continuation of Moore’s law in the miniaturization of electronic components, pending the further development of bottom up nanofabrication techniques suitable for mass production.
In general, classic Coulombic charging, the relative spacing of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), the spin, and the vibrational modes will determine single electron currents through molecules connected to electrodes with tunneling barriers [10]. Thus, the reversibility of accessible redox states of molecules is important, e.g. in single molecule transistors [10]. In the case of conducting polymers, the conductance is dependent on the structure, conjugation of the molecular system, and the length. A recent study shows that the mechanical characteristics and topology of a polyfluorene are also important. Pulling this polymer from a gold surface with an STM tip allows about a 20 nm change in length whereupon the conductance curves show an exponential decay with increasing length and oscillations that correspond to a each monomer unit detaching from the surface [11]. The molecular electronic properties are also dependent on the matrix surrounding the molecule and the domain size (number of copies of a molecule in a discrete domain). Many of the physical properties of ceramic semiconductor devices have analogies in hybrid molecular electronics. For example, image charges generated in the source and the drain can result in the localization of charges in molecules such as conjugated phenyls [10], or in ensembled domains, until a critical charge density is reached whereupon the transistor switches (see below).
There are several reviews of the potential applications of porphyrins and phthalocyanines in molecular electronics [12–16], and as sensitizers for solar cells [17]. This review will focus specifically on applications of porphyrin and porphyrinoid molecules as components of working molecular electronic devices on electronically active substrates from ca. the past decade. Particular emphasis will be placed on how the detailed molecular architecture dictates the electronic properties and how the performance of these molecules is affected by surface chemistry, attachment, orientation, and matrix around the electroactive species. Though there is excellent work on materials composed of porphyrin and phthalocyanine films and aggregates, for example as components of solar energy harvesting, electroluminescent and electronic devices [18], these are not included in this review per se, except as a comparison to single molecule measurements or in terms of attachment chemistry.
1.1. Porphyrinoids
Single, small conjugated molecules have shown Kondo resonance [19]. Large, stable aromatic systems are particularly attractive as molecular electronics because the HOMO-LUMO energy gaps are in a realistic operational range, they tend to be stable, and the oxidation and or reduction chemistry is reversible under appropriate conditions. Porphyrins and the related macrocycles such as phthalocyanines are large aromatic dyes that are about 1 and 1.5 nm across, respectively. The porphyrin cores have 11 pi bonds (Figure 1) and the phthalocyanines have an additional eight in the fused isoindole structure (Figure 2). This class of dyes has rich electrochemical and photophysical properties amenable to molecular electronics applications. These divalent ligands can bind almost every metal in the periodic table, which further modulates the electronic and photonic properties. There is an extensive literature on the chemistry, properties, and applications of porphyrin and phthalocyanines [20–22]. There are two notable early examples of supramolecular devices based on porphyrins. The self-assembly of photo generated porphyrin cations and lipophilic ions into ion chains inside membranes results in the formation of photo-gated ion conductors [23–26], and the metal ion mediated self-assembly of porphyrin tetramers insides membranes results in photo-gated transistors [27]. The work of Bard, Fox and coworkers is another early example of the fabrication of a porphyrin-based electronic device. In this case, thin films of mesogenic porphyrins on ITO electrodes served as read-write devices that could be cycled about 1.5 billion times and have an equivalent storage density of 8×1010 bits/cm2 [28, 29]. Some simple, symmetric porphyrins with potential for microelectronics are commercially viable [30], and some phthalocyanines are already used in many applications ranging from compact disks to display technologies [22]. Currently, substantial strides have been made in the design and fabrication of porphyrinoid dye-based photovoltaics [17, 31–34], and relevant to this review: molecular wires [35–39], transistors [40], logic gates, junctions, memory storage, and other molecule-base systems [13].
Figure 1.
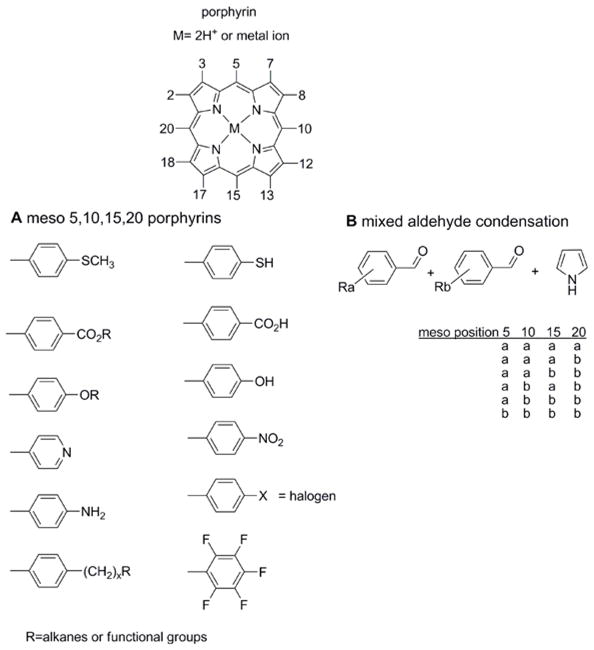
(A) Structure of the porphyrin macrocycles with a cadre of common meso (5,10,15,20) aryl derivatives. Note the meso alkane compounds are also readily accessible synthetically. (B) Much of the supramolecular chemistry of porphyrins uses less symmetric compounds, e.g. those used in the formation of SAMs on surfaces, rely on a mixed aldehyde synthesis wherein two aryl aldehydes are mixed with pyrrole to form a ‘combinatorial’ library of six compounds. The chromatographic separation of the compounds and isomers yields compounds that can be used to study molecular topologies, surface binding geometries, self-assembly into discrete arrays, or self-organization into films. Many of these compounds can also be made by more direct routes.
Figure 2.
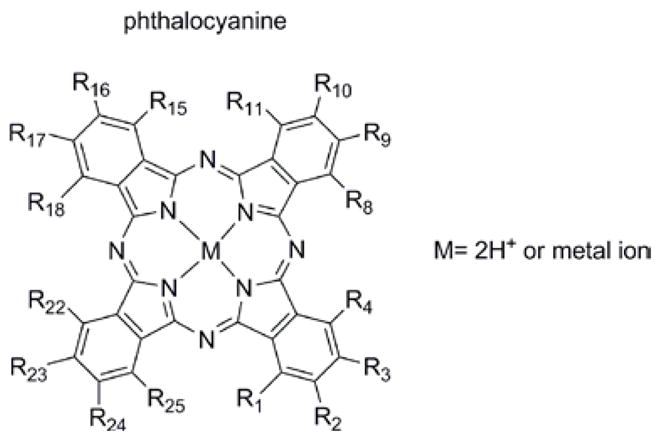
The phthalocyanine macrocycle and numbering scheme. The isoindole positions near the ring are often referred to as α (i.e. 1,4,8,11,15,18,22,25) and those away from the ring β. Substitution at the β positions is more typical. For substituted phthalocyanines with an odd number of substituents on the isoindoles there are usually positional isomers, and these isomers are usually not specifically enumerated. PcF16 where all positions bear a fluorine and the Cu(II) complex, are commonly used in photovoltaic applications.
Porphyrins, metalloporphyrins, and their derivatives are relatively non-toxic and prime candidates for a host of molecular electronics applications. As a class of molecule, they possess distinctive, reversible oxidation and reduction chemistry that potentiates use as wires, switches, transistors, junctions, and photodiodes. The electronic and photophysical properties are governed by the bound metal ion, which can be nearly every element in the periodic table, exocyclic moieties on the pyrrole and/or on the meso positions, and the matrix surrounding the chromophore. Synthetic porphyrin chemistry is well developed and the Gouterman [41] and Michl [42] molecular orbital models accurately predict the electronic consequences of appending organic substituents and binding metal ions.
Appending exocyclic moieties designed for specific intermolecular interactions, e.g. ligands and H-bonds, is now routinely used to construct supramolecular materials. As materials, there continues to be a substantial amount of work on the design of new porphyrin derivatives that self-assemble into discrete nanoarchitectures or self-organize into a diverse menu of nanostructures [43–47] such as films [48, 49], crystals [38, 50–52], tubes [53–55], rods, wires [56], nanoparticle spheres [57, 58], and complex fractal-like or chiral patterns [59, 60]. A variety of nanofibers have also been reported [61–64]. Coordination chemistry is most often used because the metal ion binding geometry can be exploited as a design element and coordination bonds are robust [65], but electrostatic [26, 54] and hydrogen bond [52, 66] interactions are also used in formation of porphyrinic materials. Reversible coordination of metal ions dictates the conformation of porphyrins linked to the 5,5’ positions of a 2,2’-bipyridyl bridge thereby reversibly switching the electronic communication between the chromophores [67]. Geometric complementarities between pyridyl substituted porphyrins and metal ion bind facilitates formation of many structures ranging from linear tapes to squares and boxes.[68] Coordination chemistry can also be used to stitch together different chromophores into 3×3 and other arrays [69–73], and complementary coordination chemistry between covalently linking porphyrins yields photonic and/or conducting wires [35, 74–78]. In most cases, self-assembly and self-organization are accomplished under mild conditions. There is also a large amount of ongoing work on photo initiated electron transfer reactions in molecules and assemblies containing porphyrins [79–81]. In materials, the photonic properties of a given porphyrin are also highly dependent on the surrounding matrix of porphyrins, wherein the number, connectivity, relative orientation, and dynamics of both the molecules and the intermolecular interactions dictate the function of the chromophore and the material. Because of their adjustable photonic properties, robustness to decomposition, and ease of synthesis, porphyrins and porphyrinoid materials continue to be thoroughly investigated as components of organic light emitting devices, sensors, solar cells, and photocatalysts.
1.2. Chelated Metal ions
In addition to serving as a design element in the formation of supramolecular materials via endocyclic and exocyclic metal ion coordination, it should be noted that metal ions bound by these macrocyles further potentiate molecular electronics applications. The degree of electron delocalization between the chelated metal ion and the porphyrins depends on the relative energies and mixing of the orbitals, and has been extensively studied by EPR and other spectroscopies for both paramagnetic metal centers and ligand centered radical ions. For example, diamagnetic closed shell Zn(II) complexes, diamagnetic open shell Ni(II), and paramagnetic Mn(III), Fe(II), Co (II) complexes have significantly different photophysical properties and redox chemistry. The availability and energies of the metal orbitals can also be varied, e.g. closed shell Ti(IV), Zr(IV), and Hf(IV) complexes. The relative orbital energies, and thus the electronic populations of the central metal ion, are also dictated by the coordination chemistry via ligand field effects. The forced square planar geometry of the macrocycles and the degree of axial ligation can have profound effects on the relative energies and the population of the d orbitals of complexed open shell metalloporphyrins, such as for the Ni(II) porphyrins described below [82, 83]. In non-luminescent Ni(II) porphyrins, the deactivation of the excited state proceeds through a metal centered d,d (dx2-y2, dz2) excited state. Coordination of pyridine at both axial positions raises the dz2 orbital energy, thereby altering the photophysical properties, such that the population of the excited d,d state transiently forces the deligation of the pyridines [84]. It should also be noted that ground state [85] and excited state [82] molecular dynamics can have profound impacts on the function of these molecules in devices. The role of counter ions in complexes with trivalent and tetravalent metal ions can be equally important to the photonic properties, and thus their ability to serve as components of devices [86]. Similar orbital and metal ion binding considerations apply to phthalocyanines [87]. The role of metal ions in the conductance of porphyrin wires has been studied theoretically [77].
1.3 Synthesis
Meso tetraarylporphyrin synthesis is generally accomplished by Rothemund-Adler-type reactions [88, 89] and MacDonald-Lindsey-type reactions [90–92]. The former synthesis mixes an aldehyde with pyrrole and when two or more aldehydes are used a mixture is produced (Figure 1) [93], while the latter multistep strategy can yield specific target macrocycles. A recent report on the synthesis of meso-tetraarylporphyrins under aerobic oxidation conditions produced tetraphenylporphyrin in over 60% yields [94]. The meso aryl substituents are orthogonal to the macrocycle because of steric interactions with the pyrrole βH, so the porphyrin faces are somewhat blocked. When the meso positions are unsubstituted and there are eight alkyl substituents on the pyrroles, most commonly octaethylporphyrin, the macrocycle can lay flat onto a substrate. There are likewise many routes to phthalocyanines. The most common methods are to heat phthalonitriles or phthalimides in high boiling solvents, sometimes in the presence of metal ion template. Non-symmetrically substituted starting materials result in isomers.[20, 22]
2. Measurements of Charge Transport in Molecular Assemblies
The specific electronic properties of porphyrins govern their use as active electronic components and must therefore be fully characterized before the molecule can be implemented into a device. In order to characterize the electrical properties of molecules, a range of techniques have been developed. The electronic properties of single molecules or small ensembles of molecules have been measured through a number of different electrical test beds. These include electrical and mechanical break junctions [95–99], cross-wire junctions [100], nanopores [101], mercury drop contacts [102], cyclic voltammetry (CV) [103, 104], conducting-probe atomic force microscopy (CP-AFM) [105, 106], and scanning tunneling microscopy (STM) [99, 107, 108]. Among these, STM and CP-AFM can provide complementary surface analyses, as they enable investigation of individual molecules on surfaces on the atomic scale. In addition to structural details, the STM images themselves can further elucidate information regarding local tunneling probabilities as well as the local electronic density of molecular orbitals (e.g. HOMO and LUMO) participating in the tunneling process. Highly π-conjugated molecules, such as porphyrins are of particular interest since the π-electron delocalization inside such molecules typically results in lower injection barriers and more efficient tunneling or electron transfer. Specific electronic properties of porphyrins such as tunneling efficiency and the transport pathway are of significant interest in designing molecules for use as electrical components. These measurement techniques have been recently reviewed, and the reader is directed to these articles for details [109, 110].
3. Porphyrinoids on Surfaces
Porphyrinoids can be organized onto surfaces in two general ways: (1) self-assembled monolayers (SAMs) wherein the active molecules are covalently attached to the surface via well-established surface chemistries (Figure 3), and (2) self-organization into 2-dimensional arrays and/or layers by adsorption onto surfaces (Figure 4) [111–118]. In both cases the surface chemistry, energetics, and structure play key roles in the final structure and organization of the photonic materials [103, 119, 120]. For SAMs, there are four essential parts of the molecule: the electronically active dye, the linker, the tether, and the surface-reactive functional group. Each part plays a role in the molecular electronic properties.
Figure 3.
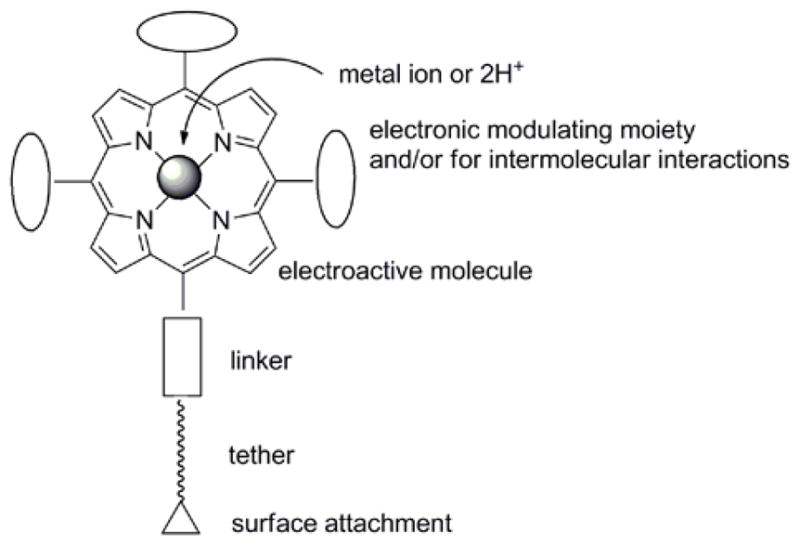
Parts of a porphyrin molecule for surface attachment include the reactive surface moiety, a tether, and the linking group to the porphyrin. Metal ions and exocyclic moieties modulate electronic properties.
Figure 4.

Large planar macrocycles such as porphyrins and phthalocyanines tend to adsorb cofacially on surfaces such as highly ordered pyrolytic graphite (HOPG), Au(III), NaCl, and other atomically smooth substrates. Here Ni(TPP) and PcF16 are deposited on HOPG as well as a 2:1 mixture. Reproduced from ref. [117] with permission of the copyright holders.
3.1 Adsorption
The self-organization of porphyrins on surfaces has been extensively studied by Hipps and co-workers who have characterized the electronic properties of porphyrins and related compounds such as phthalocyanines adsorbed as monolayers on surfaces [113, 115, 116, 118, 121, 122]. These studies conclude that the orbital energies of the macrocycles including the metal ions, molecular states, and the surface states determine the coupling, and thus tunneling probability (Figure 5). They have also conducted a variety of studies on the nanomanipulation and self-organization of porphyrins on HOPG as well as metal substrates such as Au. Studies which combine a range of surface analytical tools, such as x-ray photoelectron spectroscopy, inelastic tunneling spectroscopy, reflection absorption IR spectroscopy, ultra-violet photoelectron spectroscopy as well as orbital-mediated tunneling spectroscopy, along with STM have provided a significant amount of information regarding the physical and electronic properties of porphyrins on surfaces. This arena of research has shown that porphyrins can be valuable components of electronic devices since they possess desired properties such the ability to be deposited on surfaces without a change in composition or oxidation state and their frontier orbital energies lie close to the Fermi level of the Au substrates.
Figure 5.
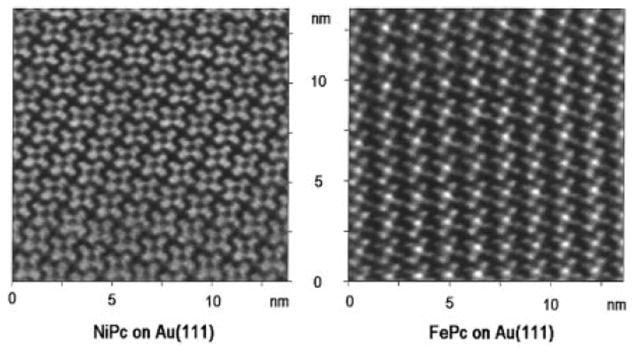
Assemblies of Ni(II) and Fe(II) phthalocyanine on Au(111). While both systems contain a central metal atom with the same valance, the Fe d6 system has greater orbital density near the Fermi level leading to an observed increased tunneling probability than that of the Ni d8 containing species. Reproduced from ref. [112] with permission of the copyright holders. These authors also examined d7 and d9 phthalocyanines [111].
Various strategies toward multiporphyrin arrays have been reported [123–125]. Recently, Hecht and coworkers reported that the formation of 2-dimensional arrays of porphyrins on gold surfaces can be accomplished by evaporative deposition or casting of tetra(4-bromophenyl)porphyrin onto gold surfaces, which results in a reasonably well ordered supramolecular array of up to ca. 100 nm. These arrays are pre-positioned to form covalent 2-dimensional networks upon heating, or if deposited above a critical temperature [126]. Similar to the coordination arrays, the substitution pattern on the porphyrin can dictate the nanoarchitecture of the resulting covalent array, for example the 5,15-derivative leads to chains (Figure 6), but the formation of high fidelity 2-dimensional structures over large areas is yet to be achieved [126]. More than just thickness, research groups are able to control the orientation of the molecules comprising the SAM, and there are many reports on the effect of reaction conditions on the ordering of molecules in a monolayer. For example, Ha et al. demonstrated the effect of annealing the first adsorbed layer on the orientation of the second layer [127]. Hydrogen bonding interactions and halogen-halogen interactions alter the thermodynamic properties and therefore binding modes of the adsorbed molecules on surfaces [128, 129].
Figure 6.
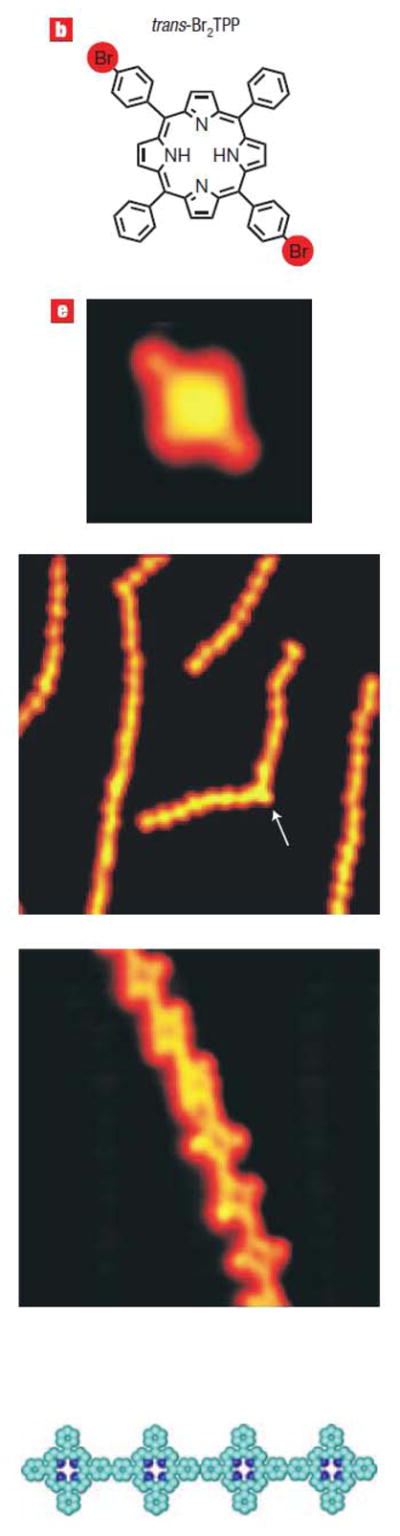
Formation of supramolecular arrays of halogenated aryl porphyrins allows covalent bond assembly of 1- and 2- dimensional structures on a gold surface upon heating. In this case the formation of the 1-dimensional tapes is shown. The arrow indicates overlapping of two linear chains rather than a covalent bond. Reproduced from ref. [126] with permission of the copyright holders.
3.2 Surface Attachments
3.2.1 Thiols on gold
The use of porphyrins in molecular electronics largely came about from the formation of liquid crystalline films and attachment of the molecules to surfaces in the form of the now ubiquitous self-assembled monolayer (SAM) [127, 128, 130]. It was noted early on that thiols create well ordered structures on gold substrates, and similarly a variety of porphyrins have been bound to gold as SAMs [131–137]. Numerous patterns and arrangements of single molecules and supramolecular assemblies have been reported [129]. Many of the early SAMs were created as Langmuir Blodgett films, and later were fabricated by dipping the substrate into a solution containing the thiol for extended periods of time. Extensive techniques have been developed to fabricate and characterize these assemblies, especially ultrahigh vacuum techniques that allows layers to be patterned with precision approaching nanometer control [138]. Assembly approaches can be further extended by the use of nanopatterning, via techniques such as scanning probe lithography [120, 123, 130]. In our own work, we have recently shown that porphyrin assemblies may be patterned on Au surfaces using nanografting. In nanografting, an AFM tip is used to displace surface bound matrix molecules (typically an alkanethiol) in a background solution of the molecule of interest. During this process, the molecule of interest, in this example a porphyrin appended with a thiol, then bonds within the open surface region created by the removal of the matrix molecules [139]. Using scanning probe lithography, thiol tethered porphyrin assemblies with features down to ca. 10 nm in dimension (Figure 7) can be fabricated to create well defined nanostructures on surfaces.
Figure 7.

A 3×3 array of nanografted islands of a zinc porphyrinthiol (top) patterned in a background matrix of dodecanethiol. The feature size illustrated here is ca. 20 nm (FWHM) as determined from the topographic image (left). The porphyrins are found to be protruding above the dodecanethiol matrix by ca. 0.6 nm. The friction image (right) more clearly shows the patterned array. The scalebars in the lower right are 50 nm. (Batteas and coworkers, unpublished results).
There are several modes of porphyrin attachments to gold surfaces depending on the location and size of the linker moieties and attachment groups. For example, Perrine et al, attached thiols directly to the porphyrin macrocycle on either opposing pyrrole β positions or on opposing meso positions, therefore attached to different macrocycle molecular orbitals [140]. This geometry allows the cofacial deposition of the porphyrin with the π-system remarkably close to the Au surface with reasonable reliability (Figure 8). Porphyrin oligomers terminated on either end with thiols allow them to bridge between gold electrodes, and where I–V plots show that the fused systems show markedly different electric properties than similar oligomers directly connected via meso positions (Figure 9) [141]. Cycling of these electrodes however, indicated that the porphyrins may be aggregating due to mobility or lability of the Au-S bonds. Similar constructs with multimers of acetylene-bridged zinc porphyrins using phenylacetylene tethers on each end bridged between gold electrodes showed that the conductance is not linearly dependent on the distance (Figure 9) [36]. A theoretical approach indicated that a porphyrin bearing eight thiols, two on each pyrrole, may bridge between four gold electrodes to serve as a photo gated current router, but construction of this type of device will be problematic [142]. The free base and the Co(II) tetra phenylporphyrins with thiols on the 4-positions on opposing meso positions (5,15 in Figure 1) can be attached to gold nanowires. In this case the metalloporphyrin served as a memory bit wherein the charge was located as the Co(II/III) couple, which then alters the conductance of the nanowire. The free base exhibited no memory effects [40]. In all of the constructs with thiols the facile formation of the disulfides, in the presence of oxygen or from electrode generated redox processes, can complicate the formation of the material and interpretation of the data. Also, the mobility and lability of the S-Au bond must be considered.
Figure 8.

Perrine et al. demonstrated cofacial deposition of porphyrins on Au surfaces with thiols directly attached to the porphyrin. Reproduced from ref [140] with permission of the copyright holders.
Figure 9.

Top: Electrical properties of directly linked porphyrin wires across Au nanoelectrodes with spacing of less than 5 nm were prepared using an electromigration-induced break-junction technique Reproduced from ref. [141] with permission of the copyright holders. Bottom: an acetylene-linked porphyrin construct has been studied to look at distance dependence on the conductivity. Reproduced from ref. [36] with permission of the copyright holders.
Surface Attachment Chemistry
| Structure/surface | Comments, and surface bond energies [6] | Recent examples |
|---|---|---|
| Por-tether-S-/gold | Migration and lability of S-Au bond can be used to form islands of electroactive molecules and can present issues for preparing useful devices. Surface bond energy ~1.9eV | [131–137] |
| Por-tether-CH=CH-/Si(100) Por-tether-CH2-/Si(100) |
This bond is less labile, surface bond energy ~3.7 eV. | [143] |
| Por-tether-O-/Si(100) | Surface bond energy ~3.7 eV | [144] |
| Adsorption | Surface bond energy <0.5 eV | [113, 114, 116, 117, 121, 123] |
3.2.2. Other attachment chemistry
The use of a single alkylthiol tether can also be used to implant these molecules into a background matrix, allowing for the investigation of single molecules as well as assemblies of these molecules to be investigated by techniques such as scanning tunneling microscopy (e.g. Figure 7). Beyond metal surfaces, attachment to semi-conducting and oxide surface can be accomplished via a range of addition reactions. These have different surface bond energies (Table 1). In each of these cases the first key issue surrounding this approach to device fabrication includes understanding the attachment chemistry required to organize the molecular components on surfaces and the role of these bonds in electronic coupling. The implementation of strategies which link molecular components directly to semiconductors such as Si have a significant advantage in light of the extensive technologies and fabrication methods already built up around Si in the existing semiconductor industry. To this end, several groups are reshaping our views of traditional organic chemistry by establishing reaction mechanisms of small organics on semiconductor surfaces such as Si [145–147]. Recent work has also shown that porphyrin films can be attached to Si surfaces via Si-O linkages [144], and that these molecular monolayers are very robust under elevated temperatures, maintaining their electroactivity, thus making them reasonable candidates for device fabrication [148]. Other linking chemistries, including the use of chlorosilanes [149] and phosphates [150] can be used for the formation of self-assembled monolayers (SAMs) on oxide surfaces. Additionally, direct attachment to H-terminated Si can be accomplished through the use of alkyne and hydroxyl groups to form Si-C and Si-O bonds [151, 152]. Absent from this body of work; however, are the details of how the film quality (i.e. defect density and local aggregate dimension) impact performance, as well as how the specifics of the electronic structure, packing geometry and coupling group exert influences on the charge transport behavior of the system.
3.3 Monolayers on Other Surfaces: Information Storage
Matching the appended attachment group to the surface chemistry allows formation of monolayers on a variety of surfaces. Alkynes add to Si(100) surfaces in hydrosilylation reactions. For example, an alkyne-terminated tether attached to silicon can subsequently react with a porphyrin bearing an azido moiety on a pyrrole in a click-type 1,3-Huisgen cycloaddition [143]. Similarly, vinyl groups can be added to Si(100) surfaces [153, 154]. Porphyrin and phthalocyanine based molecular memory devices are among the most promising in the arena of molecular electronics because these macrocycles are generally robust enough to withstand the elevated temperatures and reaction conditions needed to fabricate monolayers on silicon. Adsorbed or bound to a conducting or semiconducting surface, domains of porphyrins are excellent candidates for memory storage due to their distinct yet easily manipulated and often photoactive electronic states. By exploiting the reversible redox states of a porphyrin an “on-off switch” is created to function as the unit by which information is stored. The porphyrin layer also stores charge in this manner thereby serving as a molecular capacitor and mimicking the inorganic transistor/capacitor system currently used for memory storage. Because applications for molecular memory demand a high charge density and a small footprint to minimize feature size, molecules with multiple reversible redox states may potentiate a new generation of devices.
Lindsey, Bocian and coworkers presented some of the best and most thorough approaches to designing and creating functional and potentially economically viable molecular memory devices that are stable to ~ 400 °C and can undergo ~1012 cycles [104, 155]. These devices, and others like them, are well characterized by numerous electrochemical and microscopic techniques [154]. Molecules with as many as four discrete redox states maintain a small footprint because of their unique binding mode to the surface (Figure 10) [156, 157]. Carcel et al. created a series of molecules to investigate: (a) electron transfer kinetics between the surface and the adsorbed molecule by varying the moiety used for attachment, (b) a series of porphyrins designed to investigate the effects of charge density, and (c) a set to investigate methods of chip patterning. Here it was found that electron transfer rates increase as packing density in the SAM decrease [158].
Figure 10.

(A) The dynamics of a surface attached trimer is one of the factors that determine the surface density, where SAG is the surface attachment group. (B) The heteroleptic Por-Eu-Pc-Eu-Pc serves as a multi-bit information storage molecule. Reproduced from ref [155] with permission of the copyright holders.
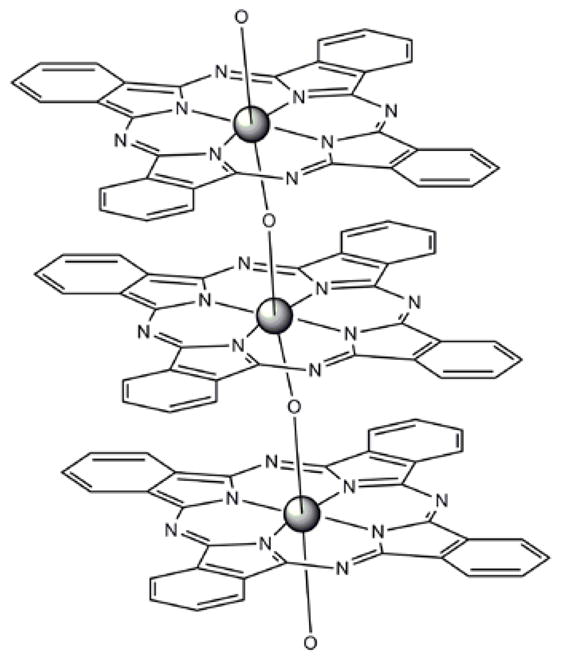
Work by Lindsey and Bocian in the past decade has shown that the reversible redox activity of porphyrin complexes can be exploited for molecular information storage applications where the oxidation of the porphyrin is the write cycle and the reduction is the read cycle [159, 160]. Several porphyrin complexes that can serve as molecular charge storage elements were designed to form robust monolayers and to be compatible with conventional semiconductor lithography. Careful investigations of the attachment chemistry and topology, tether, and linker moieties have yielded insights to the functional role of each part of the molecule. The electronic functional properties of monolayers of a diverse set of porphyrinoids were investigated to decipher the molecular architecture in terms of all four parts of the molecule (Figure 2). These studies included: monopodal, bipodal, and tripodal attachment geometries together with variations in the binding chemistry to silicon using oxygen, sulfur, and selenium [132, 134, 153–158]. These molecules and their derivatives have also found potential uses in the field of molecular memory devices. In terms of the electronically active porphyrinoid portion of the molecule, a single first row transition metal porphyrin complex usually has 1–2 reversible redox processes. Lanthanides and other metal ions with large ionic radii form sandwich complexes with porphyrins and phthalocyanines because they protrude significantly out of the mean plane of the macrocycle. Higher order, and mixed porphyrin-phthalocyanine sandwich complexes can also be formed in reasonable yields, and these tend to have more reversible redox states than the monomers (Figure 10). Attachment of a tether to these large multiporphyrinoid systems allows construction of a monolayer wherein each molecule has a minimal footprint to maximize the surface density of the electronically active units, and the distinct oxidation states of each unit allows them to be used as multi-bit devices [154].
3.4. Nano Islands
Our groups are interested in designing components for electronics using a porphyrin core system as shown in Figure 7. Specifically, we have designed a free-base porphyrin macrocycle bearing three 4-pyridyl moieties and one pentafluorophenyl substituent in the meso positions as a core platform for the rapid, high yield attachment of tethers that can be tailored to both the surface chemistry and other design criteria. In this case the pentafluorophenyl moiety serves as a “universal” linker to which a variety of nucleophiles can be appended via replacement of the 4-fluoro group. The pyridyl motifs allow design of intermolecular interactions to yield hierarchically organized monolayers or can provide a convenient attachment point for additional molecules via metal-ligand coordination chemistry. Initial studies used a terminal dithioalkane to yield a derivative for immobilization onto a gold surface via strong sulfur-gold chemisorption. Moreover, by controlling the extent of fluorination of this linker group, this phenyl ring, can be used as an internal barrier to control the tunneling between the macrocycle and the thiol tether. This barrier can be modulated by systematic variation of the number and position of the fluoro groups on the ring, thus affording some control of the relative orbital energies of this phenyl group and the degree of steric interactions between the 2,6-positions on this phenyl group with the pyrrole β-hydrogens. Thus, control of these interactions provides a means to dictate the electronic coupling between the macrocycle and the tether.
To optimize our ability to investigate these molecules either individually or as small monolayer aggregates (about 6 nm in dimension), we have characterized SAMs of these porphyrin based molecules with thiol linkers mixed into a pre-assembled dodecanethiol matrix [137]. These insertion based experiments afford the means of orienting molecules at the surface by covering the surface with a protective capping group first (in this case a simple alkanethiol). Assemblies of such molecules on clean open metal surfaces show that they will frequently lay down on the surface as described above, driven by the strong interaction between the π system and the metal Au terraces. In characterizing the free-base and zinc porphyrin derivatives, it was found that the molecule inserts on edges of the substrate and into defects in the pre-assembled dodecanethiol SAM. The free-base analogue tends to insert as single molecules and small monolayer domains dispersed within the background SAM. The small islands of the free-base porphyrin molecules undergo stochastic switching as observed during STM measurements in the mixed monolayer. This is likely associated with conformational changes within the monolayer. The I–V spectra are shown in Figure 11. The zinc metalated derivative has a much higher proclivity for aggregation mediated by both coordination chemistry and π-π interactions, thereby creating larger domains that are approximately 10 nm in width on average. These large domains of the metalloporphyrin afford interesting electronic properties, such as Coulomb blockades, that are not seen in the small clusters or with the free-base analogue (see below).
Figure 11.
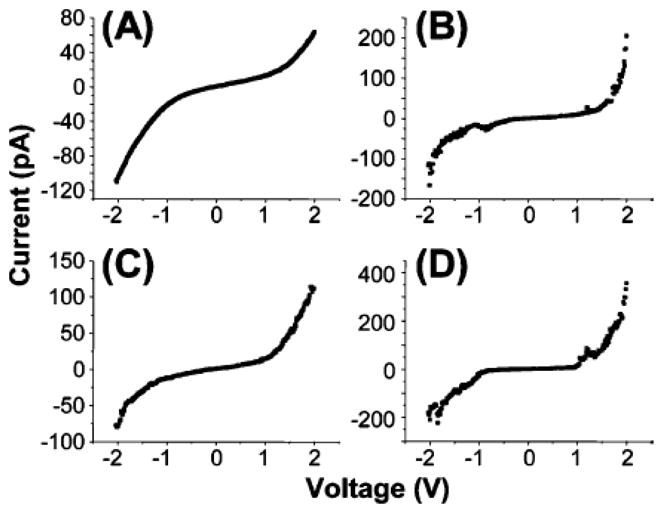
I–V spectra (averaged from 50 curves each) for the (A) dodecanethiol matrix, (B) small (~2 nm) porphyrin domains and (C, D) large (>6 nm) porphyrin domains. Reproduced from ref. [137] with permission of the copyright holders.
3.5. Other Devices
In many molecular electronic devices, efficient charge separation and charge stability is essential. Photo initiated charge transfer reactions of porphyrins have been extensively studied and many form long lived charge separate states. The charge stability combined with excellent charge carrier mobilities make these systems prime candidates for molecular electronic applications [161–163]. Photo initiated charge injection from porphyrins into semiconducting surfaces has been studied in the context of solar energy devices and the interfacial electron transfer rates are generally less than a ps [17, 79]. Much work is being done in the design of new molecules capable of forming long lived charge separated states based on the distinct electronic states of porphyrinoids that enable functions such as current routers, gates and field effect transistors [39, 135, 142, 164–168]. Linear gates and T gates have been fabricated and can be controlled by porphyrin redox chemistry as well as photochemistry [169, 170]. Recent studies have also demonstrated that porphyrins on surfaces display stochastic switching [137]. Recently we have found that by forming nanoscale assemblies of zinc metalloporphyrins similar to those in Figure 7, stable Coulomb islands can be formed as evidenced by the appearance of a Coulomb staircase in the I–V measurements (unpublished results).
Derivatives of the generic molecule shown in Figure 3 have shown promise to serve as molecular capacitors and single electron tunneling devices in which small islands are assembled within electrode gaps and their charge state tuned using a gate voltage. In one example, we investigated the transport properties of zinc metalloporphyrin islands inserted into a dodecanethiol matrix on Au(111). Using scanning tunneling microscopy (STM), it was shown that the zinc porphyrin islands exhibited bias dependent switching at high surface coverage, where the conductance of the molecules increased when sweeping above the threshold voltage, and the islands returned to a lower conductance state when the tip was swept to the negative bias. This switching behavior at high coverage was found to result from the formation of Coulomb islands driven both by enhanced π-π aggregation of the macrocycles and by increased oxidation potential of the zinc metalloporphyrins. Low temperature measurements (~4 K) from crossed-wire junctions verified the appearance of a Coulomb staircase and blockade for only aggregates of the zinc porphyrins. In other examples, free base porphyrins in nanogap electrodes (formed as break junctions) have been reported to act as Coulomb islands [171–173]. Wakayama et al. observed Coulomb blockade staircases in the I–V measurements of these systems and also showed that the threshold voltages were sensitive to changes in the intermolecular interactions [174–176]. Overall, it has been shown that intermolecular interactions such as aggregation play a significant role in the electronic properties of porphyrin molecules. These aggregates readily show the ability to stabilize charge and this affords a means of creating bias switchable devices.
4. Space charge
Materials composed of small molecular aggregates have been shown to exhibit the ability to store charge. In analogy to the semiconductor counterparts, the development of charge in the molecules of a SAM bound to a surface saturates before every molecule becomes ionized because of space charge limitations, and densely packed monolayers of porphyrinoids are an excellent example [159, 177, 178]. Space charge limited conductance of supramolecular porphyrin wires in lipid bilayers was previously noted (Figure 12) [23–26]. Thus the optimum surface or device density of active molecules represents a balance between efficient use of molecules because of space charge limits and the need for redundancy to assure performance. Space charge scales with q/4 πε0ε1 where q is the charge ε0 is the vacuum permittivity (8.85 × 10−12 J−1 C2m−1), and ε1 is the dielectric constant of the medium. Perhaps secondary SAM molecules in the matrix that can diminish the space charge effects by raising the dielectric constant (~3 for an alkane 20–30 for a polyethyleneglycol) or serve as built-in counter ions may allow greater surface densities of the active molecules.
Figure 12.

The conductance of tetraphenylboride (TPhB−) through a lipid bilayer saturates with increasing concentration because of space charge limits (●). An implicit equation describes the space charge limited current, solid line: ρTPhB− = 0.602C β exp(−qVTPhB) where 0.602 converts units of concentration mol liter−1, to ions per nm3, C is the concentration of the lipophilic ion, β is the partition coefficient, q is the molecular charge, and V is the potential of the ion inside the membrane based on electrostatic calculations. Cancelation of the space charge of the lipophilic anion by photo-formation of a porphyrin cation and self-assembly into an ion chain can increase the conductance by >20-fold (○), were the dashed line is the calculated non-space charge limited conductance. This photogated device is an early example of an all-organic molecular electronic. Reproduced from ref. [26] with permission of the copyright holders.
5. Past, Present, and Future
Previously, in the pre-scanning probe era, there was excellent work on the conductivity of crystalline and polycrystalline materials that were in many ways the forerunners of today’s efforts in molecular electronics. The pioneering work of Marks and coworkers on crystalline and polycrystalline halogen-doped [M(Pc)O]Ix, materials in the late 1970s (Figure 13) [179–181] is noteworthy. Electrical conductivities of the best performing wires, where M=Si, Ge, and Sn with x=1–2, were ~0.1 Ω cm−1. In view of the new analytical tools and new approaches to forming nanowires, it would be interesting to revisit these systems to assess the transport mechanism(s) and their photonic properties on the nano scale.
Figure 13.
Conductive polycrystalline wires of Marks and coworkers reported in the late 1970s. Redrawn from ref. [178–179].
Currently, molecular electronics based on porphyrinoid systems shows great promise for the future. The molecular design and formation of functional devices has been aided by new fabrication strategies, measurement tools, and theoretical models. Several key results should be noted. All parts of the molecule play critical roles in the molecular electronic properties: redox active moiety, linker, tether, and attachment group. The matrix surrounding the probed molecule has a profound effect on the activity of the molecule whether the matrix consists of passive alkanes, active molecules of the same kind, or active molecules of other types. The detailed electronic properties of a single molecule can be quite different than nanoscale domains or islands of these molecules. Space charge and redox potentials can limit the charge density on a given surface and number of charges on given molecules, respectively. The hierarchical organization of molecules, wherein the supramolecular structure varies with scale, has the potential to modulate function. For example, the intermolecular interactions between free-base porphyrins are different than the corresponding zinc metalloporphyrins in nanodomains of these molecules in SAMs. The precise architecture mediated by supramolecular interactions ranging from π-π interactions to coordination bonds dictates the electronic communication between the molecules in these nanodomains, thus have profound effects on the observed molecular electronic properties.
In the future, developing better theoretical and experimental probes of how both the ensemble and matrix effects molecular electronic properties will facilitate design of enabling technologies. For example, predictably designing specific switching properties and thresholds are keystone issues to be developed. The matrix surrounding the active molecules is an essential design element that has heretofore not been specifically studied. The role of molecular dynamics on multiple time scales (vibrational and conformational) on the molecular electronic properties is beginning to be appreciated both in terms of the initial charge transport efficiency and rate, and in terms of while the molecule is in the ‘on’ or ‘off’ redox state. Yet, there is a paucity of studies that systematically vary temperature with observed transport properties of porphyrinoid SAMs or those using matrix molecules with varying stiffness. We have not specifically discussed the commercial viability of the dyes, but if the molecule is to be deployed on a large scale, easily scalable and economical synthetic strategies will be needed eventually.
Much of the work described above is on large areas of SAMs of porphyrinoids wherein little molecular level or nanoscale level measurements have been made. Though the surface may appear to be uniformly covered with the active molecules by electrochemical and spectroscopic methods, it is yet to be determined how uniform the molecular electronic properties are on the nanoscale. Are cadres of supramolecularly organized substructures present in the SAM? If so, do they have different transport properties as we observed in the case of the porphyrins in the SAM matrix? Many of these systems are demonstrated to be remarkably robust, but can defective molecules be replaced to restore the functional parameters the device? What is the right balance between molecular economy and needed redundancy for a given function?
Indeed, as Feynman noted there has been a lot of public relations concerning the potential of molecular electronics, but as we develop a better understanding of Nature (e.g. the physics of molecular conduction), the long-held promise of molecular electronics may be moving from bench to successful technology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Abruña HD, Ratner MA, Zee RDv, González CA, Kagan CR, Stewart DR, Walker AV, Batteas JD, Chidsey CED, Seideman T. Ballston, Virginia: 2007. National Science Foundation pp. [Google Scholar]; (b) Mirkin CA, Ratner MA. Annual Rev Phys Chem. 1992;43:719. [Google Scholar]; (c) Annal New York Acad Sci. 2002;960 [Google Scholar]
- 2.Hille B. Ion Channels of Excitable Membranes. 3. Sinauer Associates; Sunderland: 2001. [Google Scholar]
- 3.Jin Y, Friedman N, Sheves M, He T, Cahen D. Proc Natl Acad Sci USA. 2006;103:8601. doi: 10.1073/pnas.0511234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong FT. Biosensors and Biocomputers. Springer; New York: 1990. Molecular Electronics. [Google Scholar]
- 5.Stoddart JF. Chem Soc Rev. 2009;38:1802. doi: 10.1039/b819333a. [DOI] [PubMed] [Google Scholar]
- 6.McCreery RL, Bergren AJ. Adv Mater. 2009;21:4303. doi: 10.1002/adma.200802850. [DOI] [PubMed] [Google Scholar]
- 7.Pognon G, Boudon C, Schenk KJ, Bonin M, Bach B, Weiss J. J Am Chem Soc. 2006;128:3488. doi: 10.1021/ja058132l. [DOI] [PubMed] [Google Scholar]
- 8.Suslick KS, Rakow NA, Kosal ME, Chou JH. J Porphyrins Phthalocyanines. 2000;4:407. [Google Scholar]
- 9.Fages F, Wytko JA, Weiss J. C R Chim. 2008;11:1241. [Google Scholar]
- 10.Kubatkin S, Danilov A, Hjort M, Cornil J, Bredas JL, Stuhr-Hansen N, Hedegard P, Bjornholm T. Nature. 2003;425:698. doi: 10.1038/nature02010. [DOI] [PubMed] [Google Scholar]
- 11.Lafferentz L, Ample F, Yu H, Hecht S, Joachim C, Grill L. Science. 2009;323:1193. doi: 10.1126/science.1168255. [DOI] [PubMed] [Google Scholar]
- 12.Lindsey JS. New J Chem. 1991;15:153. [Google Scholar]
- 13.Burrell AK, Officer DL, Plieger PG, Reid DC. Chem Rev. 2001;101:2751. doi: 10.1021/cr0000426. [DOI] [PubMed] [Google Scholar]
- 14.Burrell AK, Wasielewski MR. J Porphyrins Phthalocyanines. 2000;4:401. [Google Scholar]
- 15.Wu W, Liu Y, Wang Y, Xi H, Gao X, Di C, Yu G, Xu W, Zhu D. Adv Funct Mater. 2008;18:810. [Google Scholar]
- 16.Boyle NM, Rochford J, Pryce MT. Coord Chem Rev. 2010;254:77. [Google Scholar]
- 17.Campbell WM, Burrell AK, Officer DL, Jolley KW. Coord Chem Rev. 2004;248:1363. [Google Scholar]
- 18.Shea PB, Pattison LR, Kawano M, Chena C, Chend J, Petroff P, Martin DC, Yamadac H, Onoc N, Kanicki J. Synth Met. 2007;157:190. [Google Scholar]
- 19.Liang W, Shores MP, Bockrath M, Long JR, Park H. Nature. 2002;417:725. doi: 10.1038/nature00790. [DOI] [PubMed] [Google Scholar]
- 20.Kadish K, Smith KM, Guiard R. Academic Press. Vol. 2000. New York: 2003. [Google Scholar]
- 21.Smith KM. Porphyrins and Metaloporphyrins. Elsevier; Amsterdam: 1972. [Google Scholar]
- 22.Leznoff C, Lever A. Wiley VCH. New York: 1993. [Google Scholar]
- 23.Drain CM, Christensen B, Mauzerall DC. Proc Natl Acad Sci USA. 1989;86:6959. doi: 10.1073/pnas.86.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drain CM, Mauzerall D. Bioelectrochem Bioenerg. 1990;24:263. [Google Scholar]
- 25.Drain CM, Mauzerall DC. Biophys J. 1992;63:1556. doi: 10.1016/S0006-3495(92)81739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mauzerall DC, Drain CM. Biophys J. 1992;63:1544. doi: 10.1016/S0006-3495(92)81738-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drain CM. Proc Natl Acad Sci USA. 2002;99:5178. doi: 10.1073/pnas.062635099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu C-y, Pan H-l, Fox MA, Bard AJ. Science. 1993;261:897. doi: 10.1126/science.261.5123.897. [DOI] [PubMed] [Google Scholar]
- 29.Liu C-y, Pan H-l, Fox MA, Bard AJ. Chem Mater. 1997;9:1422. [Google Scholar]
- 30.Bai Y, Liu X, Chen L, Khizar-ul-Haq, Khan MA, Zhu WQ, Jiang XY, Zhang ZL. Microelectronics J. 2007;38:1185. [Google Scholar]
- 31.Grätzel M. J Photochem Photobiol C: Photochem Rev. 2003;4:145. [Google Scholar]
- 32.Guldi DM. Chem Soc Rev. 2002;31:22. doi: 10.1039/b106962b. [DOI] [PubMed] [Google Scholar]
- 33.Sgobba V, Giancane G, Conoci S, Casilli S, Ricciardi G, Guldi DM, Prato M, Valli L. J Am Chem Soc. 2007;129:3148. doi: 10.1021/ja0655789. [DOI] [PubMed] [Google Scholar]
- 34.Yang F, Forrest SR. ACS Nano. 2008;2:1022. doi: 10.1021/nn700447t. [DOI] [PubMed] [Google Scholar]
- 35.Ozawa H, Kawao M, Tanaka H, Ogawa T. Chem Lett. 2009;38:542. [Google Scholar]
- 36.Sedghi G, Sawada K, Esdaile LJ, Hoffmann M, Anderson HL, Bethell D, Haiss W, Higgins SJ, Nichols RJ. J Am Chem Soc. 2008;130:8582. doi: 10.1021/ja802281c. [DOI] [PubMed] [Google Scholar]
- 37.Koepf M, Trabolsi A, Elhabiri M, Wytko JA, Paul D, Albrecht-Gary AM, Weiss J. Org Lett. 2005;7:1279. doi: 10.1021/ol050033p. [DOI] [PubMed] [Google Scholar]
- 38.Iengo E, Zangrando E, Minatel R, Alessio E. J Am Chem Soc. 2002;124:1003. doi: 10.1021/ja016162s. [DOI] [PubMed] [Google Scholar]
- 39.Ambroise A, Kirmaier C, Wagner R, Loewe R, Bocian D, Holten D, Lindsey J. J Org Chem. 2002;67:3811. doi: 10.1021/jo025561i. [DOI] [PubMed] [Google Scholar]
- 40.Li C, Ly J, Lei B, Fan W, Zhang D, Han J, Meyyappan M, Thompson M, Zhou C. J Phys Chem B. 2004;108:9646. [Google Scholar]
- 41.Gouterman M. Review of the Porphyrin 4 Orbital Model. In: Dolphin D, editor. The Porphyrins. Academic Press; New York: 1978. p. 1. [Google Scholar]
- 42.Waluk J, Michl J. J Org Chem. 1991;56:2729. [Google Scholar]
- 43.Drain CM, Batteas JD, Smeureanu G, Patel S. Encyclopedia of Nanoscience and Nanotechnology. Marcel Dekker; New York: 2004. Self-Assembled Porphyrinic Materials on Surfaces; p. 3481. [Google Scholar]
- 44.Drain CM, Bazzan G, Milic T, Vinodu M, Goeltz JC. Isr J Chem. 2005;45:255. [Google Scholar]
- 45.Drain CM, Chen X. Self-Assembled Porphyrinic Nanoarchitectures. In: Nalwa HS, editor. Encyclopedia of Nanoscience & Nanotechnology. American Scientific Press; New York: 2004. p. 593. [Google Scholar]
- 46.Drain CM, Goldberg I, Sylvain I, Falber A. Top Curr Chem. 2005;245:55. [Google Scholar]
- 47.Medforth CJ, Wang Z, Martin KE, Song Y, Jacobsen JL, Shelnutt JA. Chem Commun. 2009:7261. doi: 10.1039/b914432c. [DOI] [PubMed] [Google Scholar]
- 48.Nishiyama F, Yokoyama T, Kamikado T, Yokoyama S, Mashiko S. Appl Phys Lett. 2006;88:253113. [Google Scholar]
- 49.Bazzan G, Smith W, Francesconi L, Drain CM. Langmuir. 2007;24:3244. doi: 10.1021/la7031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SJ, Mulfort KL, O’Donnell JL, Zuo X, Goshe AJ, Wesson PJ, Nguyen ST, Hupp JT, Tiede DM. Chem Commun. 2006:4581. doi: 10.1039/b610025b. [DOI] [PubMed] [Google Scholar]
- 51.Liu B, Qian DJ, Huang HX, Wakayama T, Hara S, Huang W, Nakamura C, Miyake J. Langmuir. 2005;21:5079. doi: 10.1021/la050064t. [DOI] [PubMed] [Google Scholar]
- 52.Muniappan S, Lipstman S, George S, Goldberg I. Inorg Chem. 2007;46:5544. doi: 10.1021/ic0701099. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z, Lybarger LE, Wang W, Medforth CJ, Miller JE, Shelnutt JA. Nanotechnology. 2008;19:395604. doi: 10.1088/0957-4484/19/39/395604. [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Medforth CJ, Shelnutt JA. J Am Chem Soc. 2004;126:15954. doi: 10.1021/ja045068j. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Medforth CJ, Shelnutt JA. J Am Chem Soc. 2004;126:16720. doi: 10.1021/ja044148k. [DOI] [PubMed] [Google Scholar]
- 56.Lee SJ, Hupp JT, Nguyen ST. J Am Chem Soc. 2008;130:9632. doi: 10.1021/ja801733t. [DOI] [PubMed] [Google Scholar]
- 57.Drain CM, Smeureanu G, Patel S, Gong X, Garno J, Arijeloye J. New J Chem. 2006;30:1834. [Google Scholar]
- 58.Gong X, Milic T, Xu C, Batteas JD, Drain CM. J Am Chem Soc. 2002;124:14290. doi: 10.1021/ja027405z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Chen P, Ma Y, He S, Liu M. Appl Mater Interfaces. 2009;1:2036. doi: 10.1021/am900399w. [DOI] [PubMed] [Google Scholar]
- 60.Ozawa H, Tanaka H, Kawao M, Unoa S, Nakazato K. Chem Commun. 2009:7411. doi: 10.1039/b915832d. [DOI] [PubMed] [Google Scholar]
- 61.Radivojevic I, Likhtina I, Shi X, Singh S, Drain CM. Chem Commun. 2010:1643. doi: 10.1039/b919468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haino T, Fujii T, Watanabe A, Takayanagi U. Proc Natl Acad Sci USA. 2009;106:10477. doi: 10.1073/pnas.0809602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu G, Zhang X, Cai X, Jiang J. J Mater Chem. 2009;19:2417. [Google Scholar]
- 64.Sooambar C, Troiani V, Bruno C, Marcaccio M, Paolucci F, Listorti A, Belbakra A, Armaroli N, Magistrato A, Zorzi RD, Geremia S, Bonifazi D. Org Biomol Chem. 2009;7:2402. doi: 10.1039/b820210a. [DOI] [PubMed] [Google Scholar]
- 65.Scandola F, Chiorboli C, Prodi A, Iengo E, Alessio E. Coord Chem Rev. 2006;250:1471. doi: 10.1002/cphc.200600065. [DOI] [PubMed] [Google Scholar]
- 66.Drain CM, Shi X, Milic T, Nifiatis F. Chem Commun. 2001:287. [Google Scholar]
- 67.Cheng KF, Drain CM, Grohmann K. Inorg Chem. 2003;42:2075. doi: 10.1021/ic025985v. [DOI] [PubMed] [Google Scholar]
- 68.Drain CM, Lehn J. M Chem Commun. 1994:2313. [Google Scholar]
- 69.Drain CM, Batteas JD, Flynn GW, Milic T, Chi N, Yablon DG, Sommers H. Proc Natl Acad Sci USA. 2002;99(Suppl 2):6498. doi: 10.1073/pnas.012521899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drain CM, Nifiatis F, Vasenko A, Batteas JD. Angew Chem. 1998;37:2344. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2344::AID-ANIE2344>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 71.Cheng KF, Thai NA, Grohmann K, Teague LC, Drain CM. Inorg Chem. 2006;45:6928. doi: 10.1021/ic060448m. [DOI] [PubMed] [Google Scholar]
- 72.Cheng KF, Thai NA, Teague LC, Grohmanna K, Drain CM. Chem Commun. 2005:4678. doi: 10.1039/b503439f. [DOI] [PubMed] [Google Scholar]
- 73.Koepf M, Trabolsi A, Elhabiri M, Wytko JA, Paul D, Albrecht-Gary AM, Weiss J. Organic Letters. 2005;7:1279. doi: 10.1021/ol050033p. [DOI] [PubMed] [Google Scholar]
- 74.Kobuke Y. Struct Bond. 2006;121:49. [Google Scholar]
- 75.(a) Reimers JR, Lu TX, Crossley MJ, Hush NS. Chem Phys Lett. 1996;256:353. [Google Scholar]; (b) Hush NS, Reimers JR, Hall LE, Johnson LA, Crossley MJ. Optimization and Chemical Control of Porphyrin-Based Molecular Wires and Switches. In: Aviram A, Ratner M, editors. Molecular Electronics: Science and Technology. New York Academy of Sciences; New York: 1998. p. 1. [Google Scholar]
- 76.Tagami K, Tsukada M, Matsumoto T, Kawai T. Phys Rev B. 2003;67:245324. [Google Scholar]
- 77.Long MQ, Wang L, Chen KQ. Mod Phys Lett B. 2008;22:661. [Google Scholar]
- 78.Tagami K, Tsukada M. Thin Solid Films. 2004;464–465:429. [Google Scholar]
- 79.Wasielewski MR. J Org Chem. 2006;71:5051. doi: 10.1021/jo060225d. [DOI] [PubMed] [Google Scholar]
- 80.Hayes RT, Wasielewski MR, Gosztola D. J Am Chem Soc. 2000;122:5563. [Google Scholar]
- 81.Wang Y, Wang X, Ghosh SK, Lu HP. J Am Chem Soc. 2009;131:1479. doi: 10.1021/ja806988d. [DOI] [PubMed] [Google Scholar]
- 82.Drain CM, Gentemann S, Roberts JA, Nelson NY, Medforth CJ, Jia S, Simpson MC, Smith KM, Fajer J, Shelnutt JA, Holten D. J Am Chem Soc. 1998;120:3781. [Google Scholar]
- 83.Drain CM, Kirmaier C, Medforth CJ, Nurco DJ, Smith KM, Holten D. J Phys Chem. 1996;100:11984. [Google Scholar]
- 84.Retsek JL, Drain CM, Kirmaier C, Nurco DJ, Medforth CJ, Smith KM, Sazanovich IV, Chirvony VS, Fajer J, Holten D. J Am Chem Soc. 2003;125:9787. [Google Scholar]
- 85.Kottas GS, Clarke LI, Horinek D, Michl J. Chem Rev. 2005;105:1281. doi: 10.1021/cr0300993. [DOI] [PubMed] [Google Scholar]
- 86.Jiao J, Schmidt I, Taniguchi M, Lindsey JS, Bocian DF. Langmuir. 2008;24:12047. doi: 10.1021/la8019843. [DOI] [PubMed] [Google Scholar]
- 87.Xiao J, Dowben P. J Mater Chem. 2009;19:2172. [Google Scholar]
- 88.Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. J Org Chem. 1967;32:476. [Google Scholar]
- 89.Lindsey JS, Schreiman IC, Hsu HC, Kearney PC, Marguerettaz AM. J Org Chem. 1987;52:827. [Google Scholar]
- 90.Lindsey JS. New J Chem. 1991;15:153. [Google Scholar]
- 91.Lindsey JS. Synthesis of meso substituted porphyrins. In: Kadish K, Smith KM, Guiard R, editors. The Porphyrin Handbook. Academic Press; New York: 2000. p. 45. [Google Scholar]
- 92.Loewe RS, Ambroise A, Muthukumaran K, Padmaja K, Lysenko AB, Mathur G, Li Q, Bocian DF, Misra V, Lindsey JS. J Org Chem. 2004;69:1453. doi: 10.1021/jo034946d. [DOI] [PubMed] [Google Scholar]
- 93.Drain CM, Singh S. The Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine. World Scientific Publisher; Singapore: 2010. Combinatorial Chemistry of Porphyrins; p. 485. [Google Scholar]
- 94.Sharghi H, Nejad AH. Tetrahedron. 2004;60:1863. [Google Scholar]
- 95.Reed MA, Zhou C, Muller CJ, Burgin TP, Tour JM. Science. 1997;278:252. [Google Scholar]
- 96.Li X, He J, Hihath J, Xu B, Lindsay SM, Tao N. J Am Chem Soc. 2006;128:2135. doi: 10.1021/ja057316x. [DOI] [PubMed] [Google Scholar]
- 97.Park J, Pasupathy AN, Goldsmith JI, Chang C, Yaish Y, Petta JR, Rinkoski M, Sethna JP, Abruna HD, McEuen PL, Ralph DC. Nature. 2002;417:722. doi: 10.1038/nature00791. [DOI] [PubMed] [Google Scholar]
- 98.Venkataraman L, Klare JE, Nuckolls C, Hybertsen MS, Steigerwald ML. Nature. 2006;442:904. doi: 10.1038/nature05037. [DOI] [PubMed] [Google Scholar]
- 99.Dunbar TD, Cygan MT, Bumm LA, McCarty GS, Burgin TP, Reinerth WA, Jones L, Jackiw JJ, Tour JM, Weiss PS, Allara DL. J Phys Chem B. 2000;104:4880. [Google Scholar]
- 100.Kushmerick JG, Naciri J, Yang JC, Shashidhar R. Nano Lett. 2003;3:897. [Google Scholar]
- 101.Chen J, Reed MA. Chem Phys. 2002;281:127. [Google Scholar]
- 102.Weiss EA, Chiechi RC, Kaufman GK, Kriebel JK, Li Z, Duati M, Rampi MA, Whitesides GM. J Am Chem Soc. 2007;129:4336. doi: 10.1021/ja0677261. [DOI] [PubMed] [Google Scholar]
- 103.Yoshimoto S, Itaya K. J Porphyrins Phthalocyanines. 2007;11:313. [Google Scholar]
- 104.Liu Z, Yasseri AA, Lindsey JS, Bocian DF. Science. 2003;302:1543. doi: 10.1126/science.1090677. [DOI] [PubMed] [Google Scholar]
- 105.Morita T, Lindsay S. J Am Chem Soc. 2007;129:7262. doi: 10.1021/ja072040+. [DOI] [PubMed] [Google Scholar]
- 106.Wold DJ, Frisbie CD. J Am Chem Soc. 2001;123:5549. doi: 10.1021/ja0101532. [DOI] [PubMed] [Google Scholar]
- 107.Donhauser ZJ, Mantooth BA, Kelly KF, Bumm LA, Monnell JD, Stapleton JJ, Price DW, Rawlett AM, Allara DL, Tour JM, Weiss PS. Science. 2001;292:2303. doi: 10.1126/science.1060294. [DOI] [PubMed] [Google Scholar]
- 108.Samori P, Rabe JP. J Phys: Condens Matter. 2002;14:9955. [Google Scholar]
- 109.Chen F, Tao NJ. Acc Chem Res. 2009;42:429. doi: 10.1021/ar800199a. [DOI] [PubMed] [Google Scholar]
- 110.Chen F, Hihath J, Huang Z, Li X, Tao NJ. Annu Rev Phys Chem. 2007;58:535. doi: 10.1146/annurev.physchem.58.032806.104523. [DOI] [PubMed] [Google Scholar]
- 111.Lu X, Hipps KW, Wang XD, Mazur U. J Am Chem Soc. 1996;118:7197. [Google Scholar]
- 112.Lu X, Hipps KW. J Phys Chem B. 1997;101:5391. [Google Scholar]
- 113.Ogunrinde A, Hipps KW, Scudiero L. Langmuir. 2006;22:5697. doi: 10.1021/la060233p. [DOI] [PubMed] [Google Scholar]
- 114.Scudiero L, Hipps KW, Barlow DE. J Phys Chem B. 2003;107:2903. [Google Scholar]
- 115.Deng W, Hipps KW. J Phys Chem B. 2003;107:10736. [Google Scholar]
- 116.Scudiero L, Barlow DE, Hipps KW. J Phys Chem B. 2002;106:996. [Google Scholar]
- 117.Hipps KW, Scudiero L, Barlow DE, Cooke MP. J Am Chem Soc. 2002;124:2126. doi: 10.1021/ja017561q. [DOI] [PubMed] [Google Scholar]
- 118.Scudiero L, Barlow DE, Hipps KW. J Phys Chem B. 2000;104:11899. [Google Scholar]
- 119.Nikiforov MP, Zerweck U, Milde P, Loppacher C, Park T-H, Uyeda HT, Therien MJ, Eng L, Bonnell D. Nano Letters. 2007;8:110. doi: 10.1021/nl072175d. [DOI] [PubMed] [Google Scholar]
- 120.Visser J, Katsonis N, Vicario J, Feringa BL. Langmuir. 2009;25:5980. doi: 10.1021/la804196r. [DOI] [PubMed] [Google Scholar]
- 121.Scudiero L, Hipps KW. J Phys Chem C. 2007;111:17516. [Google Scholar]
- 122.Scudiero L, Barlow DE, Mazur U, Hipps KW. J Am Chem Soc. 2001;123:4073. doi: 10.1021/ja0100726. [DOI] [PubMed] [Google Scholar]
- 123.Garno JC, Xu C, Bazzan G, Batteas JD, Drain CM. Designing Supramolecular Porphyrin Arrays for Surface Assembly and Patterning of Optoelectronic Materials. In: Schubert US, Newkome GR, Manners I, editors. Metal-Containing and Metallosupramolecular Polymers and Materials. American Chemical Society; 2006. p. 168. [Google Scholar]
- 124.Drain CM, Varotto A, Radivojevic I. Chem Rev. 2009;109:1630. doi: 10.1021/cr8002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beletskaya I, Tyurin VS, Tsivadze AY, Guilard R, Stern C. Chem Rev. 2009;109:1659. doi: 10.1021/cr800247a. [DOI] [PubMed] [Google Scholar]
- 126.Grill L, Dyer M, Lafferentz L, Persson M, Peters MV, Hecht S. Nature Nanotechnology. 2007;2:687. doi: 10.1038/nnano.2007.346. [DOI] [PubMed] [Google Scholar]
- 127.Ha S, Kaafarani B, Barlow S, Marder S, Kahn A. J Phys Chem C. 2007;111:10493. [Google Scholar]
- 128.Hipps K, Scudiero L, Barlow D, Cooke M. J Am Chem Soc. 2002;124:2126. doi: 10.1021/ja017561q. [DOI] [PubMed] [Google Scholar]
- 129.Barlow S, Raval R. Surf Sci Rep. 2003;50:201. [Google Scholar]
- 130.Smith R, Lewis P, Weiss P. Progr Surf Sci. 2004;75:1. [Google Scholar]
- 131.Li JZ, Gryko D, Dabke RB, Diers JR, Bocian DF, Kuhr WG, Lindsey JS. J Org Chem. 2000;65:7379. doi: 10.1021/jo000490d. [DOI] [PubMed] [Google Scholar]
- 132.Balakumar A, Lysenko AB, Carcel C, Malinovskii VL, Gryko DT, Schweikart KH, Loewe RS, Yasseri AA, Liu ZM, Bocian DF, Lindsey JS. J Org Chem. 2004;69:1435. doi: 10.1021/jo034944t. [DOI] [PubMed] [Google Scholar]
- 133.Wei LY, Padmaja K, Youngblood WJ, Lysenko AB, Lindsey JS, Bocian DF. J Org Chem. 2004;69:1461. doi: 10.1021/jo0349476. [DOI] [PubMed] [Google Scholar]
- 134.Yasseri AA, Syomin D, Malinovskii VL, Loewe RS, Lindsey JS, Zaera F, Bocian DF. J Am Chem Soc. 2004;126:11944. doi: 10.1021/ja047723t. [DOI] [PubMed] [Google Scholar]
- 135.Kang B, Aratani N, Lim JK, Kim D, Osuka A, Yoo K-H. Chem Phys Lett. 2005;412:303. [Google Scholar]
- 136.Vaughan OPH, Turner M, Williams FJ, Hille A, Sanders JKM, Lambert RM. J Am Chem Soc. 2006;128:9578. doi: 10.1021/ja061247k. [DOI] [PubMed] [Google Scholar]
- 137.Chan YH, Schuckman AE, Perez LM, Vinodu M, Drain CM, Batteas JD. J Phys Chem C. 2008;112:6110. [Google Scholar]
- 138.Forrest S. Chem Rev. 1997;97:1793. doi: 10.1021/cr941014o. [DOI] [PubMed] [Google Scholar]
- 139.Liu M, Amro NA, Liu G-y. Annu Rev Phys Chem. 2008;59:367. doi: 10.1146/annurev.physchem.58.032806.104542. [DOI] [PubMed] [Google Scholar]
- 140.Perrine TM, Smith RG, Marsh C, Dunietz BD. J Chem Phys. 2008;128:154706. doi: 10.1063/1.2897425. [DOI] [PubMed] [Google Scholar]
- 141.Kang BK, Aratani N, Lim JK, Kim D, Osuka A, Yoo K-H. Mat Sci Eng C. 2006;26:1023. [Google Scholar]
- 142.Thanopulos I, Paspalakis E, Yannopapas V. Nanotechnology. 2008;19:445202. doi: 10.1088/0957-4484/19/44/445202. [DOI] [PubMed] [Google Scholar]
- 143.Liu H, Duclairoir F, Fleury B, Dubois L, Chenavier Y, Marchon JC. Dalton Trans. 2009:3793. doi: 10.1039/b901309a. [DOI] [PubMed] [Google Scholar]
- 144.Roth KM, Yasseri AA, Liu Z, Dabke RB, Malinovskii V, Schwelkart KH, Yu L, Tiznado H, Zaera F, Lindsey JS, Kuhr WG, Bocian DF. J Am Chem Soc. 2003;125:505. doi: 10.1021/ja021169a. [DOI] [PubMed] [Google Scholar]
- 145.Teague LC, Boland JJ. J Phys Chem B. 2003;107:3820. [Google Scholar]
- 146.Teague LC, Chen D, Boland JJ. J Phys Chem B. 2004;108:7827. [Google Scholar]
- 147.Teague LC, Boland JJ. Thin Solid Films. 2004;464–465:1. [Google Scholar]
- 148.Liu Z, Yasseri AA, Lindsey JS, Bocian DF. Science. 2003;302:1543. doi: 10.1126/science.1090677. [DOI] [PubMed] [Google Scholar]
- 149.Yerushalmi R, Scherz A, van der Boom ME. J Am Chem Soc. 2004;126:2700. doi: 10.1021/ja038918o. [DOI] [PubMed] [Google Scholar]
- 150.Muthukumaran K, Loewe RS, Ambroise A, Tamaru S-i, Li Q, Mathur G, Bocian DF, Misra V, Lindsey JS. J Org Chem. 2003;69:1444. doi: 10.1021/jo034945l. [DOI] [PubMed] [Google Scholar]
- 151.Hurley PT, Ribbe AE, Buriak JM. J Am Chem Soc. 2003;125:11334. doi: 10.1021/ja035857l. [DOI] [PubMed] [Google Scholar]
- 152.Hacker CA, Anderson KA, Richter LJ, Richter CA. Langmuir. 2005;21:882. doi: 10.1021/la048841x. [DOI] [PubMed] [Google Scholar]
- 153.Schmidt I, Jiao J, Bocian DF, Lindsey JS. J Nanosci Nanotechnol. 2008;8:4813. doi: 10.1166/jnn.2008.ic85. [DOI] [PubMed] [Google Scholar]
- 154.Anariba F, Tiznado H, Diers JR, Schmidt I, Muresan AZ, Lindsey JS, Zaera F, Bocian DF. J Phys Chem C. 2008;112:9474. [Google Scholar]
- 155.Holten D, Bocian DF, Lindsey JS. Acc Chem Res. 2002;35:57. doi: 10.1021/ar970264z. [DOI] [PubMed] [Google Scholar]
- 156.Padmaja K, Youngblood W, Wei L, Bocian D, Lindsey J. Inorg Chem. 2006;45:5479. doi: 10.1021/ic060387s. [DOI] [PubMed] [Google Scholar]
- 157.Ng D, Jiang J. Chem Soc Rev. 1997;26:433. [Google Scholar]
- 158.Carcel C, Laha J, Loewe R, Thamyongkit P, Schweikart K, Misra V, Bocian D, Lindsey J. J Org Chem. 2004;69:6739. doi: 10.1021/jo0498260. [DOI] [PubMed] [Google Scholar]
- 159.Roth KM, Lindsey JS, Bocian DF, Kuhr WG. Langmuir. 2002;18:4030. [Google Scholar]
- 160.Roth KM, Dontha N, Dabke RB, Gryko DT, Clausen C, Lindsey JS, Bocian DF, Kuhr WG. J Vac Sci Tech B. 2000;18:2359. [Google Scholar]
- 161.Wiberg J, Guo L, Pettersson K, Nilsson D, Ljungdahl T, Martensson J, Albinsson B. J Am Chem Soc. 2006;129:155. doi: 10.1021/ja066346c. [DOI] [PubMed] [Google Scholar]
- 162.Ghirotti M, Chiorboli C, You C, Wurthner F, Scandola F. J Phys Chem A. 2008;112:3376. doi: 10.1021/jp7109516. [DOI] [PubMed] [Google Scholar]
- 163.D’Souza F, Chitta R, Gadde S, Islam D, Schumacher A, Zandler M, Araki Y, Ito O. J Phys Chem B. 2006;110:25240. doi: 10.1021/jp064504g. [DOI] [PubMed] [Google Scholar]
- 164.Albinsson B, Martensson J. J Photochem Photobio C. 2008;9:138. [Google Scholar]
- 165.Ozawa H, Kawao M, Tanaka H, Ogawa T. Chem Lett. 2009;38 [Google Scholar]
- 166.Sedghi G, Sawada K, Esdaile L, Hoffmann M, Anderson H, Bethell D, Haiss W, Higgins S, Nichols R. J Am Chem Soc. 2008;130 doi: 10.1021/ja802281c. [DOI] [PubMed] [Google Scholar]
- 167.Miyachi M, Ohta M, Nakai M, Kubota Y, Yamanoi Y, Yonezawa T, Nishihara H. Chem Lett. 2008;37:404. [Google Scholar]
- 168.Madru R, Guillaud G, Al Sadoun M, Maitrot M, André JJ, Simon J, Even R. Chem Phys Lett. 1988;145:343. [Google Scholar]
- 169.Ambroise A, Wagner RW, Rao PD, Riggs JA, Hascoat P, Diers JR, Seth J, Lammi RK, Bocian DF, Holten D, Lindsey JS. Chem Mater. 2001;13:1023. [Google Scholar]
- 170.Wagner R, Lindsey J, Seth J, Palaniappan V, Bocian D. J Am Chem Soc. 1996;118:3996. [Google Scholar]
- 171.Noguchi Y, Kubota T, Mashiko S, Wakayama Y. J Appl Phys. 2005;97:073513. [Google Scholar]
- 172.Noguchi Y, Nagase T, Kubota T, Kamikado T, Mashiko S. Thin Solid Films. 2006;499:90. [Google Scholar]
- 173.Noguchi Y, Ueda R, Kubota T, Kamikado T, Yokoyama S, Nagase T. Thin Solid Films. 2008;516:2762. [Google Scholar]
- 174.Wakayama Y, Kubota T, Suzuki H, Kamikado T, Mashiko S. J Appl Phys. 2003;94:4711. [Google Scholar]
- 175.Wakayama Y, Kubota T, Suzuki H, Kamikado T, Mashiko S. Nanotechnology. 2004;15:1446. [Google Scholar]
- 176.Wakayama Y, Ogawa K, Kubota T, Suzuki H, Kamikado T, Mashiko S. Appl Phys Lett. 2004;85:329. [Google Scholar]
- 177.Roth K, Gryko D, Clausen C, Li J, Lindsey J, Kuhr W, Bocian D. J Phys Chem B. 2002;106:8639. [Google Scholar]
- 178.Turek P, Petit P, Andre JJ, Simon J, Even R, Boudjema B, Guillaud G, Maitrot M. J Am Chem Soc. 1987;109:5119. [Google Scholar]
- 179.Schoch KF, Kundalkar BR, Marks TJ. J Am Chem Soc. 1979;101:7071. [Google Scholar]
- 180.Diel BN, Inabe T, Lyding JW, Schoch KF, Kannewurf CR, Marks TJ. J Am Chem Soc. 1983;105:1551. [Google Scholar]
- 181.Dirk CW, Inabe T, Schoch KF, Marks TJ. J Am Chem Soc. 1983;105:1539. [Google Scholar]