

Abstract
Maternal care alters epigenetic programming of glucocorticoid receptor (GR) gene expression in the hippocampus, and increased postnatal maternal licking/grooming (LG) behavior enhances nerve growth factor-inducible protein A (NGFI-A) transcription factor binding to the exon 17 GR promoter within the hippocampus of the offspring. We tested the hypothesis that NGFI-A binding to the exon 17 GR promoter sequence marks this sequence for histone acetylation and DNA demethylation and that such epigenetic alterations subsequently influence NGFI-A binding and GR transcription. We report that (1) NGFI-A binding to its consensus sequence is inhibited by DNA methylation, (2) NGFI-A induces the activity of exon 17 GR promoter in a transient reporter assay, (3) DNA methylation inhibits exon 17 GR promoter activity, and (4) whereas NGFI-A interaction with the methylated exon 17 GR promoter is reduced, NGFI-A overexpression induces histone acetylation, DNA demethylation, and activation of the exon 17 GR promoter in transient transfection assays. Site-directed mutagenesis assays demonstrate that NGFI-A binding to the exon 17 GR promoter is required for such epigenetic reprogramming. In vivo, enhanced maternal LG is associated with increased NGFI-A binding to the exon 17 GR promoter in the hippocampus of pups, and NGFI-A-bound exon 17 GR promoter is unmethylated compared with unbound exon 17 GR promoter. Knockdown experiments of NGFI-A in hippocampal primary cell culture show that NGFI-A is required for serotonin-induced DNA demethylation and increased exon 17 GR promoter expression. The data are consistent with the hypothesis that NGFI-A participates in epigenetic programming of GR expression.
Keywords: maternal behavior, rat, hippocampus, transcription factors, glucocorticoid receptor, DNA methylation
Introduction
In rodents and primates, there are maternal effects on defensive responses in the offspring, including the development of hypothalamic–pituitary–adrenal (HPA) responses to stress (Higley et al., 1991; Agrawal, 2001; Meaney, 2001). In the rat, the adult offspring of mothers that exhibit increased levels of pup licking/grooming (i.e., high LG mothers) over the first week of life show increased hippocampal glucocorticoid receptor (GR) expression, enhanced glucocorticoid feedback sensitivity, decreased hypothalamic corticotrophin-releasing factor expression, and more modest HPA stress responses compared with animals reared by low LG mothers (Liu et al., 1997; Francis et al., 1999). Cross-fostering studies (Francis et al., 1999; Caldji et al., 2003) suggest direct effects of maternal care on both gene expression and stress responses.
Increased maternal LG is associated with demethylation of the 5′ CpG dinucleotide within a nerve growth factor-inducible protein A (NGFI-A) transcription factor response element located within the exon 17 GR promoter (Weaver et al., 2004). The difference in the methylation status of this CpG site between the offspring of high and low LG mothers emerges over the first week of life, is reversed with cross-fostering, persists into adulthood, and is associated with altered histone acetylation and NGFI-A binding to the GR promoter (Weaver et al., 2004). This epigenomic programming of the exon 17 GR promoter by maternal care might serve as a model for a novel mechanism through which the social environment programs adaptation at the level of the genome (Meaney and Szyf, 2005).
In vivo and in vitro studies suggest that maternal LG or postnatal handling, which increases maternal LG, increases GR gene expression in the offspring through a thyroid hormone-dependent increase in serotonin (5-HT) activity at 5-HT7 receptors and the subsequent activation of cAMP and cAMP-dependent protein kinase A (PKA) (Meaney et al., 1987, 2000; Laplante et al., 2002). Both the in vitro effects of 5-HT and the in vivo effects of maternal behavior on GR mRNA expression are accompanied by an increased hippocampal expression of NGFI-A transcription factor. The noncoding exon 1 region of the hippocampal GR includes a promoter region, exon 17, that contains a binding site for NGFI-A (McCormick et al., 2000). Splice variants of the GR mRNA transcripts containing the exon 17 sequence are found predominantly in the brain, and hippocampal expression of exon 17 GR mRNAs transcripts is increased with manipulations such as postnatal handling (McCormick et al., 2000) that increase maternal LG (McCormick et al., 2000). Postnatal handling increases hippocampal expression of both NGFI-A and GR, and such effects are eliminated by thyroid hormone synthesis inhibitors or a 5-HT2/7 receptor antagonist (Meaney et al., 1987, 2000; Mitchell et al., 1990, 1992). In these studies we examined (1) whether differential methylation of the NGFI-A binding site alters NGFI-A binding and GR expression, and (2) the potential role of NGFI-A in establishing the difference in the methylation status of the 5′ CpG within the NGFI-A response element.
Materials and Methods
Animals and maternal behavior.
The animals used were outbred Long–Evans, hooded rats born in our colony and housed in 46 × 18 × 30 cm Plexiglas cages that permitted a clear view of all activity within the cage. Food and water were provided ad libitum. The colony was maintained on a 12 h light/dark schedule with lights on at 8:00 A.M. All procedures were performed according to guidelines developed by the Canadian Council on Animal Care and protocols approved by the McGill University Animal Care Committee. At the time of weaning on day 22 of life, the offspring were housed in same-sex, same-litter groups of two animals per cage.
Maternal behavior was scored as described previously in detail (Champagne et al., 2003). Maternal behavior was observed for five, 75 min periods/d for the first 6 postpartum days at three periods during the light (9:00 A.M., 1:00 P.M., and 5:00 P.M.) and two during the dark (6:00 A.M. and 9:00 P.M.) phases of the light/dark cycle. The behavior of each mother was scored every 3 min (25 observations/period × 5 periods per day = 125 observations per mother per day) for the following behaviors: mother off pups, mother carrying pup, mother licking/grooming any pup, and mother nursing pups in an arched-back posture, a “blanket” posture in which the mother lays over the pups, or a passive posture in which the mother is lying either on her back or side while the pups nurse (for a detailed description, see Champagne et al., 2003). The frequency of maternal licking/grooming across a large number of mothers is normally distributed (Champagne et al., 2003). Hence, high and low LG mothers represent two ends of a continuum rather than distinct populations. High LG mothers were defined as females whose frequency scores for licking/grooming were >1 SD above the cohort mean. Low LG mothers were defined as females whose frequency scores for licking/grooming were >1 SD below the cohort mean.
Reverse transcription-PCR analysis.
Whole brains were removed by rapid decapitation <1 min after the animal's removal from the home cage. The hippocampal tissue was dissected, snap frozen on dry ice, and stored at −80°C. Total hippocampal RNA was isolated with the Trizol reagent method (Invitrogen, Burlington, Ontario, Canada), and the overall quality and yield of the RNA preparation was determined using an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA). cDNA was synthesized in a 20 μl reaction volume containing 2 μg of total RNA, 40 U of Moloney murine leukemia virus reverse transcriptase (MBI Fermentas, Hanover, MD), 5 μm random primer (Roche Molecular Biochemicals, Indianapolis, IN), a 1 mm concentration of each of the four deoxynucleotide triphosphates, and 40 U of RNase inhibitor (Roche Molecular Biochemicals). The mRNA was denatured (5 min, 70°C), the random primers were annealed (10 min, 25°C), and mRNA was reverse transcribed (1 h, 37°C). The reverse transcriptase was heat inactivated (10 min, 72°C), and the products were stored at −20°C. The rat hippocampal exon 17 GR region (GenBank accession number AJ271870) was subjected to PCR amplification (forward primer, 5′-TCCCAGGCCAGTTAATATTTGC-3′; reverse primer, 5′-TTGAACTCTTGGGGTTCTCTGG-3′). To control for equal loading, the rat hippocampal β-actin exon region (GenBank accession number V01217) was also subjected to PCR amplification (forward primer, 5′-GTTGCTAGCCAGGCTGTGCT-3′; reverse primer, 5′-CGGATGTCCACGTCACACTT-3′). The GR exon 17 and β-actin amplification were performed in parallel, using a 25 μl reaction mixture containing 1.5 μl of synthesized cDNA product, 2.5 μl of 10× PCR buffer, 1.5 mm MgCl2, 0.2 mm dNTPs, 1 U of Taq polymerase (all from MBI Fermentas), and 0.5 μm of each primer. The thermocycler protocol involved an initial denaturation cycle (5 min, 95°C), 20–30 cycles of denaturation (30 s, 95°C), annealing (30 s, 60°C), and extension (30 s, 72°C), followed by a final extension cycle (5 min, 72°C) terminating at 4°C. Samples were removed every two cycles between 20 and 30 cycles to determine the linear range of the PCR amplification. Products were separated on an agarose gel (2%) to visualize bands corresponding to the GR exon 17 (514 bp) or β-actin (470 bp) cDNA fragments. Nucleic acids were transferred by Southern blot (14 h, 22°C) to positively charged nylon transfer membrane (Hybond-N+; Amersham Biosciences, Arlington Heights, IL). An oligonucleotide (20 bp) specific for the GR exon 17 sequence (GenBank accession number AJ271870; forward, 5′-TCCCAGGCCAGTTAATATTTGC-3′) was synthesized, as well as an oligonucleotide (21 bp) specific for the β-actin sequence (GenBank accession number V01217; forward, 5′-GTTGCTAGCCAGGCTGTGCT-3′). The oligonucleotides were radiolabeled [1 μl of T4 polynucleotide kinase (PNK); Promega, Madison, WI] with 5 μl of [γ-32P]ATP (Amersham Biosciences) (2 h, 37°C) and then hybridized to the membranes, which were exposed to PhosphorImager screens (Type BAS-III Imaging Plate; Fuji, Tokyo, Japan) overnight at 22°C. The screens were scanned by PhosphorImager (Storm840; Molecular Dynamics, Sunnyvale, CA) running Storm840 software (Molecular Dynamics). Relative optical density (ROD) readings were determined using a computer-assisted densitometry program (ImageQuant; Molecular Dynamics). To control for equal loading between samples, the signal of the GR exon 17 region was divided by the signal from the β-actin region amplified from the same sample.
Quantitative real-time reverse transcription-PCR analysis.
The same samples as described above were used to amplify the rat hippocampal exon 17 GR region (forward primer, 5′-CCTCCCAGGCCAGTTAATATTTGC-3′; reverse primer, 5′-AAGGAGAATCCTCTGCTGCT-3′). To control for equal loading, the rat neuronal-specific β-III tubulin exon region (GenBank accession number NM_139254) was also subjected to PCR amplification (forward primer, 5′-TGCGTGTGTACAGGTGAATGC-3′; reverse primer, 5′-AGGCTGCATAGTCATTTCCAAG-3′). The GR exon 17 and β-III tubulin amplification were performed in parallel, using a 25 μl reaction mixture containing 1.0 μl of synthesized cDNA product, 12.5 μl of SuperArray master mix (SuperArray Bioscience Corporation, Frederick, MD), and 0.5 μm of each primer. The real-time thermocycler protocol (LIGHTCYCLER 3.5; Roche Molecular Biochemicals) involved an initial HotStart TaqDNA polymerase activation cycle (10 min, 95°C, with a temperature transition rate set at 20°C/s), 45 cycles of denaturation (10 s, 95°C, with a temperature transition rate set at 20°C/s), annealing (10 s, 60°C, with a temperature transition rate set at 20°C/s), and elongation (10 s, 72°C, with a temperature transition rate set at 20°C/s). A single fluorescence reading was acquired at the end of each elongation step. Arithmetic background subtraction was used, and the fluorescence channel was set to F1. To determine the cycle threshold (Ct) for both exon 17 GR region and β-III tubulin, a four-point calibration curve of increasing amounts of cDNA (0.5, 1, 2, and 4 μl) as well as a no-template negative control was performed by using separate tubes for each reaction (for each dilution and for each gene). The relative amount of both gene transcripts in each sample was determined by plotting the Ct value for each gene on the y-axis and the log of the amount of cDNA used for each reaction on the x-axis. To calculate fold change in gene expression, the relative amount of the exon 17 GR region was divided by the relative amount of β-III tubulin for each sample. The specificity of the amplified PCR products was assessed by performing a melting curve analysis cycle after the PCR amplification (30 s, 95°C, with a temperature transition rate set at 20°C/s; 30 s, 60°C, with a temperature transition rate set at 20°C/s; and 95°C, with a temperature transition rate set at 0.2°C/s) that terminated with a cooling step (30 s, 40°C, with a temperature transition rate set at 20°C/s). The fluorescence of the SYBR Green I dye bound to double-stranded amplified product declines sharply as the fragment is denatured. The melting temperature (Tm) of this fragment was visualized by plotting the first negative derivative (df/dT) of the melting curve on the y-axis and temperature (degrees Celsius) on the x-axis. No primer–dimers were detected that interfered with the quantification of the PCR products. The amplified products were also separated on an agarose gel (2%) and poststained with ethidium bromide to visualize bands corresponding to exon 17 GR region or β-III tubulin cDNA fragments, which were then photographed with an Eagle Eye apparatus (Speed Light/BT Sciencetech-LT1000).
Electrophoresis mobility shift assay.
NGFI-A electrophoresis mobility shift assay (EMSA) was performed using a NGFI-A cDNA (Milbrandt, 1987) in a cytomegalovirus–neo expression/shuffle vector (pJOM464; Invitrogen), provided as a kind gift from Prof. Jeffrey Milbrandt (Washington University, St. Louis, MO). The NGFI-A coding sequence was subcloned into a TOPO–His vector (pCRT7/CT TOPO; Invitrogen) following the guide of the manufacturer (pCRT7 TOPO TA Expression kits; Invitrogen). Primers (forward, 365-GACCATGGACAACTACCCCAAA-384, Tm of 66°C; reverse: 2553-GCAAATTTCAATTGTCCTAGG-2532, Tm of 58°C) were designed (GenBank accession number M18416). The thermocycler protocol involved an initial denaturation cycle (3 min, 98°C), 35 cycles of denaturation (30 s, 98°C), annealing (30 s, 55–65°C), and extension (1 min 30 s, 72°C), followed by a final extension cycle (10 min, 72°C) terminating at 4°C. The ligated plasmid vector containing the NGFI-A coding region was transformed into TOP10F′ Escherichia coli cells. Colonies were selected and analyzed for insert and correct orientation by sequencing (data not shown). Bacterial cells [BL21(DE3)] transfected with the NGFI-A expression vector were allowed to grow (14 h, 37°C), and an aliquot was removed and grown in LB ampicillin (100 mg/ml) until the OD600 of the solution reached 0.6 (3 h, 37°C). Isopropylthio-β-d-galactoside (IPTG) (Invitrogen) was added to the bacteria to a final concentration of 1 mm IPTG and incubated further (6 h, 37°C). The bacteria were subsequently centrifuged (4000 rpm, 25 min, 4°C), and the pelleted bacteria were resuspended in 10 ml of lysis buffer containing SDS (1%), EDTA (1 mm), Tris (50 mm), pH 8, and one protease inhibitor cocktail tablet (Complete, Mini; Roche Molecular Biochemicals). The bacteria were freeze–thawed three times, sonicated (Vibra Cell; Sonics & Materials Inc., Fisher Scientific, Houston, TX) on ice (10 s pulse at 40% every 20 s for 15 min), and centrifuged (10,000 rpm, 25 min, 4°C), and the supernatant containing the recombinant protein was extracted. The recombinant protein was purified by metal-ion affinity chromatography (Schmitt et al., 1993). In brief, 1 ml of glass–wool was packed into a 3 ml syringe and washed through with 1 vol of iminodiacetic acid immobilized on Sepharose 6β/fast flow elution/epoxy (IAA; Sigma, St. Louis, MO). The beads were packed with 6 vol of ddH2O and labeled with 3 vol of NiCl2 (200 mm). The column was equilibrated with 5 vol of ddH2O until no NiCl2 was eluted. The medium containing the bacteria (10 ml) was loaded onto the column. The medium that flowed through the column was collected and reloaded onto the column four times. The column was washed with 25 ml of lysis buffer (20 mm Tris HCl, 50 mm NaCl, 0.05% Tween 20, and 10% glycine) and then again with 10 and 20 mm imidazole in lysis buffer (25 ml of each) to remove nonspecifically bound proteins. Elution was performed with 250 mm imidazole in lysis buffer (4 ml) that was subsequently resuspended in 5× binding buffer (20% glycine, 5 mm MgCl2, 2.5 mm EDTA, 2.5 mm DTT, 250 mm NaCl, and 50 mm Tris-Cl, pH 7.6) to a final volume of 15 ml. The elution containing the protein was filtered (Amicon, Beverly, MA) (Ultra-15 Centrifugal Ultracel Low Binding Regenerated Cellulose Filter; Millipore, Bedford, MA) over four 15 min centrifugations (4000 rpm, 4°C), resuspending the protein in 5× binding buffer (to a final volume of 15 ml) between spins. Total protein was recovered after a final extended centrifugation (4000 rpm, 25 min, 4°C). Aliquots were taken to determine the levels of total protein. Differentially methylated oligonucleotide sequences (27 bp) of the NGFI-A consensus binding site (GenBank accession number AJ271870) were used: (1) nonmethylated (1881-GAGCTGGGCGGGGGCGGGAGGGAGCCT-1907); (2) methylated in the 5′ CpG dinucleotide (1881-GAGCTGGGMCGGGGGCGGGAGGGAGCCT-1907); (3) methylated in the 3′ CpG dinucleotide (1881-GAGCTGGGCGGGGGMCGGGAGGGAGCCT-1907); or (4) methylated in both CpG dinucleotide sites (1881-GAGCTGGGMCGGGGGMCGGGAGGGAGCCT-1907). The single-stranded DNA oligonucleotides were denatured (10 min, 100°C) with NaCl (150 mm) and Tris, pH 7.5, and annealed (3 h, 20–23°C) to form double-stranded DNA (dsDNA) (500 mg/25 μl). The oligonucleotides were end labeled using a fill-in reaction with a DNA polymerase- (Pol-) I Klenow fragment and [γ-32P]dCTP. In brief, oligonucleotides were radiolabeled (1 ml of T4 PNK; Promega) with 2 μl of [γ-32P]ATP (Amersham Biosciences) (1 h, 37°C). The [γ32-P]ATP-labeled dsDNA oligonucleotide was dissolved in loading buffer (250 mm Tris-Cl, pH 7.6, and 40% glycine) and separated on a (5%) nondenaturing acrylamide gel (200 vol, 2 h, 4°C). Labeled dsDNA oligonucleotides were eluted (14 h, 37°C) from the gel in 1× TNE (5 mm EDTA, 15 mm Tris, pH 7.6, and 180 mm NaCl). Following standard phenol-chloroform DNA extraction methodology, the oligonucleotides were centrifuged (13,200 rpm, 5 min) and further precipitated [95% ethanol (2 v/v), 10% Na acetate (1:10 v/v), and 10 ng/ml tRNA]. The pelleted [32P-γ]ATP-labeled dsDNA oligonucleotide was resuspended in ddH2O (50,000 cpm/μl). The binding reaction was conducted by incubating the end-labeled oligonucleotide probes (50,000 cpm) with purified NGFI-A protein (36 μm) and of poly[d(I-C)] (1 μg) in 5× binding buffer in a final volume of 50 μl (30 min, 20–23°C). For competition experiments, a 100- to 10,000-fold molar excess of competitor DNA is incubated in the mixture before adding the purified protein. DNA–protein complexes were resolved on a nondenaturing (5%) polyacrylamide gel in 0.5× TBE (89 mm Tris, 89 mm boric acid, and 2 mm EDTA) buffer (200 vol, 2 h, 4°C). Gels were dried and subjected to autoradiography.
Human embryonic kidney HEK 293 cell cultures and transient transfections.
In preparation of the GR promoter–luciferase plasmid, the rat exon 17 GR promoter region (GenBank accession number AJ271870) was subjected to PCR amplification (forward, 1557-AGACGCTGCGGGGGTG-1572; reverse, 2555-CGACCTGGCCTGGGAG-1940). The thermocycler protocol involved an initial denaturation cycle (30 s, 95°C), 35 cycles of denaturation (30 s, 95°C), annealing (30 s, 56°C), and extension (30 s, 72°C), followed by a final extension cycle (5 min, 72°C) terminating at 4°C. The amplified DNA fragment was cloned into a pCR2.1 plasmid (Original TA cloning kit; Invitrogen) and then subcloned into a pGL2 plasmid. Accordingly, the pCR2.1 plasmid was digested with HindIII and then EcoRI, and the pGL2 plasmid was digested with BamH1 and then HindIII. The released exon 17 fragment was then ligated 5′ to 3′ at HindIII and BamHI sites or 3′ to 5′ at Xba and HindIII sites within the pGL2 plasmid. In vitro mutation of the exon 17 GR promoter was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) and following the guide of the manufacturer. Mutagenesis primers were designed for cytosine to adenine conversion within the 5′ CpG (site 16) dinucleotide (forward, 5′-CTCGGAGCTGGGAGGGGGGG GGAGG-3′; reverse, 5′-CCTCCCGCCCCCTCCCAGCTCCGAG-3′) and 3′ CpG (site 17) dinucleotide (forward, 5′-GCTGGGCGGGGGAGGGAGGGAGCCT-3′; reverse, 5′-AGGCTCCCTCCCTCCCCCGCCCAGC-3′) in the NGFI-A response element on the exon 17 GR promoter (GenBank accession number AJ271870). For in vitro methylation, the exon 17 GR promoter–pGL2 plasmid (10 μg) was incubated (2 h, 37°C) with SssI CpG DNA methyltransferase (20 U; New England Biolabs, Beverly, MA) in a buffer containing S-adenosylmethionine (160 μl). This procedure was repeated twice or until full protection from HpaII digestion was observed. In preparation of the NGFI-A expression plasmid, the NGFI-A coding sequence was subcloned into a TOPO–His vector following the guide of the manufacturer (pEF6/V5-His TOPO TA Expression kit; Invitrogen). Primers (forward, 365-GACCATGGACAACTACCCCAAA-384, Tm of 66°C; reverse, 2553-GCAAATTTCAATTGTCCTAGG-2532, Tm of 58°C) were designed (GenBank accession number M18416). The thermocycler protocol involved an initial denaturation cycle (3 min, 98°C), 35 cycles of denaturation (30 s, 98°C), annealing (30 s, 55–65°C), and extension (1 min 30 s, 72°C), followed by a final extension cycle (10 min, 72°C) terminating at 4°C. To generate empty control plasmids, the pEF6/V5–His TOPO construct was digested with BstXI to remove the multiple cloning site and then self-ligated. In cotransfection studies, human embryonic kidney HEK 293 cells were plated at a density of 6 × 104 in six-well dishes and transiently cotransfected with a total amount of 1.5 μg of plasmid DNA (1.0 μg of exon 17 GR promoter–pGL2 and/or 0.5 μg of NGFIA expression plasmid) using the calcium phosphate method as described previously (Rouleau et al., 1992). HEK 293 cells were maintained as a monolayer in DMEM (Invitrogen) containing fetal calf serum (10%; Colorado Serum Company, Denver, CO). The cells were harvested 72 h after transfection and lysed, and luciferase activity was assayed using the Luciferase Assay System (Promega) according to the protocol of the manufacturer. Transfections were repeated a minimum of three times using different cultures of HEK 293 cells.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays (Crane-Robinson et al., 1999) were performed using the ChIP assay kit protocol (Upstate Biotechnology, Lake Placid, NY). Hippocampi were dissected from each rat brain, and chromatin was immunoprecipitated using one of the following: rabbit polyclonal antibody to acetyl-histone H3 (Upstate Cell Signaling Solutions, Beverly, MA), rabbit polyclonal antibody to cAMP response element binding protein (CREB) binding protein (CBP)-N-terminal (Upstate Biotechnology), rabbit polyclonal antibody to NGFI-A, or normal rabbit IgG non-immune antibody (both from Santa Cruz Biotechnology, Santa Cruz, CA). One-tenth of the lysate was kept to quantify the amount of DNA present in different samples before immunoprecipitation (“input”). The rat hippocampal GR exon 17 promoter region (GenBank accession number AJ271870) of the uncrosslinked DNA was subjected to PCR amplification (forward primer, 1750-TGTGACACACTTCGCGCA-1767; reverse primer, 1943-GGAGGGAAACCGAGTTTC-1926). PCR reactions were done with the FailSafe PCR system protocol using FailSafe PCR 2× PreMix D (Epicenter; InterScience, Markham, Ontario, Canada). The thermocycler protocol involved an initial denaturation cycle (5 min, 95°C), 34 cycles of denaturation (1 min, 95°C), annealing (1 min, 56°C), and extension (1 min, 72°C), followed by a final extension cycle (10 min, 72°C) terminating at 4°C. To control for unequal loading of acetyl-histone histone 3–lysine 9 (H3–K9) immunoprecipitate, the rat hippocampal β-actin promoter-α region (GenBank accession number V01217) of the uncrosslinked DNA was subjected to PCR amplification (forward primer, 10-TCAACTCACTTCTCTCTACT-29; reverse primer, 161-GCAAGGCTTTAACGGAAAAT-180). PCR reactions were done with the same protocol but using FailSafe PCR 2× PreMix L (Epicenter; InterScience) with the same thermocycler protocol as described previously. To control for purity of the NGFI-A immunoprecipitate, we PCR amplified the rat hippocampal exon 1b estrogen receptor (ER)-α promoter region, which does not contain binding sites for NGFI-A (GenBank accession number X98236) of the uncrosslinked DNA (forward primer, 1836-GAAGAAACTCCCCTCAGCAT-1855; reverse primer, 2346-GAAATCAAAACACCGATCCT-2327), this time using FailSafe PCR 2× PreMix A (Epicenter; InterScience). The thermocycler protocol involved an initial denaturation cycle (5 min, 95°C), 34 cycles of denaturation (1 min, 95°C), annealing (1 min, 60°C), and extension (1 min, 72°C), followed by a final extension cycle (10 min, 72°C) terminating at 4°C. PCR reactions on DNA purified from non-immunoprecipitated samples and immunoprecipitated samples were repeated exhaustively using varying amounts of template and cycles to ensure that results were within the linear range of the PCR. Products were separated on an agarose gel (2%) to visualize bands corresponding to the exon 17 GR promoter (194 bp), β-actin promoter-α (171 bp), or exon 1b ER-α promoter (493 bp) DNA fragments. Nucleic acids were transferred by Southern blot (14 h, 22°C) to positively charged nylon transfer membrane (Hybond-N+; Amersham). An oligonucleotide (20 bp) specific for the exon 17 GR promoter sequence (GenBank accession number AJ271870; forward, 1881-TCCCGAGCGGTTCCAAGCCT-1907) was synthesized, as well as an oligonucleotide (21 bp) specific for the β-actin promoter-α sequence (GenBank accession number V01217; forward, 95-GTAAAAAAATGCTGCACTGTC-115) and an oligonucleotide (20 bp) specific for the exon 1b ER-α promoter sequence (GenBank accession number X98236; forward, 1942-AGAAAGCACTGGACATTTCT-1961). The oligonucleotides were radiolabeled (1 μl of T4 PNK; Promega) with 5 μl of [γ-32P]ATP (Amersham Biosciences) (2 h, 37°C) and then hybridized to the membranes that were then subjected to autoradiography. ROD readings were determined using a computer-assisted densitometry program (MCID Systems; Imaging Research, St. Catharines, Ontario, Canada). To calculate the final signal for each sample, the ROD value of the band within the antibody lane (A) was divided by the ROD value of the band within the input lane (I). To control for equal loading between samples, the final signal of the exon 17 GR promoter, amplified from the acetyl-histone H3–K9 immunoprecipitations, was divided by the final signal from the β-actin promoter-α amplified from the same precipitate.
Sodium bisulfite mapping.
Sodium bisulfite mapping was performed as described previously (Frommer et al., 1992; Clark et al., 1994). The rat GR exon 17 genomic region (GenBank accession number AJ271870) of the sodium bisulfite-treated hippocampal DNA (50 ng/ml) was subjected to PCR amplification using outside primers (forward, 1646-TTTTTTAGGTTTTTTTAGAGGG-1667; reverse, 1930-ATTTCTTTAATTTCTCTTCTCC-1908). The thermocycler protocol involved an initial denaturation cycle (5 min, 95°C), 34 cycles of denaturation (1 min, 95°C), annealing (2 min 30 s, 56°C), and extension (1 min, 72°C), followed by a final extension cycle (5 min, 72°C) terminating at 4°C. The PCR product (285 bp) was used as a template for subsequent PCR reactions using nested primers (forward, 1738-TTTTTTTGTTAGTGTGATATATTT-1761; reverse, 1914-TTCTCCCAAACTCCCTCC-1897). The nested PCR product (177 bp) was then subcloned (Original TA cloning kit; Invitrogen) and transformed, and 10 different clones per plate were mini-prepped. Ten plasmids containing the ligated exon 17 GR promoter DNA fragment were sequenced per animal (T7 sequencing kit, USB; Amersham Biosciences) starting from procedure C in the protocol of the manufacturer. The sequencing reactions were resolved on a denaturing polyacrylamide gel (6%) and visualized by autoradiography.
Hippocampal cell cultures.
Hippocampal cell cultures from embryonic day 20 Long–Evans rat fetuses were prepared (Banker and Cowan, 1977; Mitchell et al., 1992; Bhatnagar and Meaney, 1995) in minimum essential medium (Invitrogen) containing fetal calf serum (10%), penicillin (0.1%), and streptomycin (0.1%), and supplemented with HEPES (15 mm), potassium chloride (20 mm), and glucose (55 mm), pH 7.4. Hippocampal tissue was mechanically dissociated in HBSS buffered (pH 7.4) with HEPES (15 mm), washed, and digested with trypsin (2.5 mg/ml; Invitrogen), and the digestion was stopped with fetal calf serum (10%). The cells were seeded at a density of 3 × 106 cells (60 mm) or 8 × 106 (100 mm) cells on poly-d-lysine (Boehringer Mannheim, Mannheim, Germany)-coated Petri dishes. Two days after seeding, uridine (20 mm) and 5-fluorodeoxyuridine (20 mm) were added to the medium to prevent proliferation of glial cells. Cell cultures were maintained at 37°C in a humid atmosphere with 5% carbon dioxide.
Glucocorticoid receptor assays.
For measurement of glucocorticoid receptor binding capacity [3H]RU 28362 (11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one) (specific activity 77.5 Ci/mmol; NEN, Boston, MA) was used as radioligand in an incubation range of 0.l to l5.0 nm for saturation experiments and at a saturating 10 nm concentration in single-point assays (Mitchell et al., 1990). Nonspecific binding was determined using a 200-fold excess of nonlabeled dexamethasone. After incubation (20 h, 4°C), labeled [3H]RU 28362 was separated from free steroid using LH-20 Sephadex chromatography and quantified with scintillation counting.
Antisense oligonucleotide transfection.
Chemically modified phosphorothioate antisense (AS) (170-GCGGGGTGCAGGGGCACACT-151) and scrambled antisense (SAS) (170-TCACACGGGGACGTGGGGCG-151) oligonucleotide (Biognostik, Göttingen, Germany) were designed and based on the rat NGFI-A genomic region (GenBank accession number M18416). The optimum sequence of 20 bases for generating specific oligonucleotides was searched by analyzing the 5′ upstream regions of the open reading frames of the NGFI-A gene and exactly correspond to the sequences reported by Kukita et al. (1997). Scrambled sequence with the same base ratio as the antisense were confirmed to have no homology to any sequences reported to date in the GenBank DNA database. The cultures received a single treatment of either 5-HT (100 nm), 8-bromo-cAMP (10 mm), or 5-HT (100 nm), followed by NGFI-A antisense (1 μm) oligonucleotides and were harvested 4 d later.
Western blotting.
Whole-cell extracts were harvested in TEDGM (10 mM Tris, 1.5 mM EDTA, 1 mM dithiothreitol, 10% glycerol, and 25 mM sodium molybdate), and the protein concentrations for each sample were determined according to the method of Bradford (Bradford, 1976). Equal quantities of protein (35 μg) were mixed with an equal volume of sample buffer (0.125 m Tris-HCl, 20% glycerol, 4% SDS, 0.005% bromophenol blue, and 5% β-mercaptoethanol) and subjected to denaturing and reducing electrophoresis on 10% Tris-glycine polyacrylamide gels. Proteins were then electrophoretically transferred onto nitrocellulose membranes and air dried. The membranes were blocked (1 h, 22°C) with Carnation dried milk (5 or 10%) in Tris-buffered saline with Tween 20 (TBS-T) (Tris, NaCl, and 0.1% Tween-20, pH 7.6), washed briefly in TBS-T, and incubated (14 h, 4°C) with either anti-rat α-GR (1:4000; PA1–510; Affinity BioReagents, Neshanic Station, NJ), V5 (1:1000; R960-25; Invitrogen Canada, Burlington, Ontario, Canada) or NGFI-A (1:100; sc-189; Santa Cruz Biotechnology) primary antibodies diluted in TBS-T containing 0.5 or 5% milk. Membranes were washed with TBS-T (20 min, 22°C) and then incubated (1 h, 22°C) with horseradish peroxidase-conjugated sheep anti-mouse or anti-rabbit IgG antibody (1:20,000 or 1:5000, respectively; Amersham Biosciences). After four washes for 15 min, membranes were detected using a chemiluminescence kit (Renaissance kit; DuPont, Billerica, MA) and exposed to Hyperfilm (Amersham Biosciences). Membranes were then stripped and reprobed using either an α-tubulin or β-actin antibody (1:5000; Biodesign International, Saco, ME) to control for equal loading.
Results
Maternal effects on hippocampal expression of the exon 17 GR promoter
The exon 17 GR promoter region contains an NGFI-A response element, and postnatal handling as well as increased maternal LG are associated with increased hippocampal NGFI-A expression in pups (Meaney et al., 2000). We therefore examined whether differences in activity of the exon 17 GR promoter are established in early postnatal life, a time that corresponds to the period when differences in maternal behavior of high and low LG mothers are apparent and remain stable into adulthood. Reverse transcription-PCR and quantitative reverse transcription-PCR analysis was performed using purified hippocampal mRNA from day 6 (neonatal) and day 90 (adult) offspring of high and low LG dams (Fig. 1 a–c). There was a significant maternal effect on levels of GR mRNA transcripts containing the exon 17 sequence in hippocampi from both neonatal (t (1,14) = 3.4; p < 0.002) and adult (t (1,14) = 3.6; p < 0.001) animals. Additionally, these changes in mRNA transcripts were translated into increases in GR protein levels as assayed through Western blot analysis (Fig. 1 d,e). These findings suggest that levels of GR mRNA and protein associated with transcriptional activity from the exon 17 promoter are increased in the neonatal and adult offspring of high LG mothers compared with those of low LG dams. This effect is apparent in early life and maintained into adulthood. Global abundance of total mRNA and protein in the hippocampus was not apparently affected, as indicated by comparable levels of β-actin and α-tubulin immunoreactivity, respectively (Fig. 1 a,d).
Figure 1.

Maternal effect on hippocampal exon 17 GR mRNA expression in young [postnatal day 6 (P6)] and adult [postnatal day 90 (P90)] animals (n = 5 animals per group). a, Representative Southern blots of the amplified exon 17 region from hippocampal tissue (514 bp band). b, Mean ± SEM ROD of hippocampal GR expression (**p < 0.002; **p < 0.001). c, Quantitative reverse transcription-PCR analysis of the amplified exon 17 region from hippocampal tissue relative to the levels of neuronal-specific β-III tubulin (*p < 0.01). d, Representative Western blot of GR expression in rat hippocampal tissue at postnatal days 6 and 90. e, Mean ± SEM ROD of hippocampal GR expression (P6, **p < 0.002; P90, **p < 0.001). IR, Immunoreactivity.
NGFI-A binding to its response element is inhibited by DNA methylation
Maternal care influences hippocampal GR expression and exon 17 activity in early life as well as in adulthood (Liu et al., 1997; Weaver et al., 2004, 2005). Weaver et al. (2004, 2005) reported that maternal care alters the methylation status of the 5′ CpG dinucleotide of the NGFI-A response element within the exon 17 promoter. This 5′ CpG dinucleotide is hypermethylated in the adult offspring of low LG mothers compared with the adult offspring of high LG dams (Weaver et al., 2004, 2005), and hippocampal tissue from adult animals showed approximately threefold greater levels of NGFI-A association with the exon 17 GR promoter in the adult offspring of high compared with low LG mothers (Weaver et al., 2004). Constitutive expression of NGFI-A in adults appears unaffected by maternal care (I. C. G. Weaver, M. Szyf, and M. J. Meaney, unpublished observations). We hypothesized that the increased NGFI-A binding to the exon 17 GR promoter is attributable to differences in the cytosine methylation state of the exon 17 GR promoter. To test this idea, we used an EMSA to determine the in vitro binding of purified recombinant NGFI-A protein (Milbrandt, 1987) to its response element (Fig. 2 a) while varying the methylation state of the 5′ and 3′ CpG dinucleotides of the NGFI-A response element. We predicted that methylation of the 5′ CpG should reduce NGFI-A binding to its response element.
Figure 2.

DNA methylation inhibits protein–DNA complex formation between purified recombinant NGFI-A and oligonucleotide containing its recognition sequence in vitro. a, The exon 17 GR promoter sequence with the NGFI-A response element (RE) (encircled). Beneath is a bead-on-string representation of a synthesized radiolabeled oligonucleotide probe, highlighting the two CpG dinucleotides (ovals represent the cytosines within the 5′ CpG and 3′ CpG dinucleotides) within the NGFI-A response element. b, EMSA analysis of protein–DNA complex formation between recombinant purified NGFI-A protein and radiolabeled oligonucleotide (shown in a) bearing the NGFI-A response element containing differentially methylated cytosines within the 5′ CpG and 3′ CpG dinucleotides. Nonmethylated cytosines are represented by gray ovals, methylated cytosines are shown as black ovals, and white ovals are mutated CpG dinucleotides with an adenosine replacing the cytosine. The presence of increasing amounts of purified NGFI-A protein (9, 18, or 36 pm) is indicated by the black triangle. The black arrow indicates the shift in labeled oligonucleotide mobility. Lanes 1, 5, 9, 13, and 17 are free labeled oligonucleotides with the modifications indicated by the shading of the ovals. These oligonucleotides were labeled with increasing amount of NGFI-A as indicated by the black triangles. c, EMSA analysis of competition of protein–DNA complex formation between NGFI-A protein and a radiolabeled wild-type oligonucleotide probe containing the NGFI-A response element (shown in a) by an excess of nonlabeled oligonucleotides containing differentially methylated cytosines within the 5′ and 3′ CpG dinucleotides or mutations of the NGFI-A response element as indicated by the shaded ovals. The presence of increasing amounts of purified NGFI-A protein (9, 18, or 36 pm) is indicated by the gray triangle in lanes 2–4. The presence of increasing amount of nonlabeled oligonucleotide competitor (1:100-fold, 1:500-fold, or 1:1000-fold) is indicated by the black triangle. The black arrow indicates the shift in labeled oligonucleotide mobility. Lane 1, The free labeled oligonucleotide.
NGFI-A formed a protein–DNA complex with the nonmethylated oligonucleotide (Fig. 2 b, lanes 2–4), although the protein was unable to form a complex with either a fully methylated sequence or a sequence that was methylated at the 5′ CpG site (Fig. 2 b, lanes 10–12, 14–16). NGFI-A binding was apparent with the sequence methylated only at the 3′ CpG site (Fig. 2 b, lanes 6–8). The specificity of the protein–DNA interaction is indicated by the fact that the recombinant protein fails to form a complex with the mutated NGFI-A response element (Fig. 2 b, lanes 18–20). The results indicate that methylation of the cytosine within the 5′ CpG dinucleotide reduced NGFI-A protein binding to a level comparable with that of methylation at both CpG sites. Methylation of the cytosine within the 3′ CpG dinucleotide only partially reduced NGFI-A protein binding (Fig. 2 a,b).
The difference in NGFI-A binding was confirmed by competition experiments in which NGFI-A recombinant protein was incubated with a labeled, nonmethylated oligonucleotide (Fig. 2 a) in the presence of increasing concentrations of the nonlabeled differentially methylated oligonucleotides. The results (Fig. 2 c) reveal marked, concentration-dependent competition for NGFI-A binding with either the nonmethylated oligonucleotide sequence (lanes 5–7) or with the sequence methylated in the 3′, but not the 5′, CpG (lanes 8–10). Methylation of the 5′ CpG eliminated the ability of the oligonucleotide sequences containing the NGFI-A response element to compete for NGFI-A binding (lanes 11–16). These findings support the hypothesis that methylation of the cytosine in the 5′ CpG dinucleotide within the NGFI-A response element of the exon 17 GR promoter inhibits NGFI-A protein binding, potentially explaining the reduced NGFI-A–exon 17 promoter association and GR gene transcription in the offspring of the low LG mothers.
NGFI-A binds and transactivates transcription from exon 17 GR promoter
We next examined (1) whether NGFI-A induces exon 17 GR promoter activity and (2) whether such effects are regulated by CpG methylation. We resorted to a transient transfection assay in HEK 293 cells to analyze the interactions between NGFI-A and GR exon 17 promoter in isolation from other tissue-specific factors. HEK 293 cells were used for this assay because they are transfected at high efficiency, which allows for effective cotransfection analyses of a number of expression vectors concurrently. The goal of this experimental paradigm was to determine the effects of definite concentrations of ectopic NGFI-A on defined exogenously introduced GR exon 17 promoter constructs. Moreover, because these plasmids do not replicate in HEK 293 cells during a transient transfection assay (Cervoni and Szyf, 2001), the plasmid does not lose its methylation pattern by passive demethylation through replication. Thus, any demethylation measured in this system most likely reflects active demethylation.
We transiently transfected plasmids containing the exon 17 GR promoter previously characterized by McCormick et al. (2000) fused to a luciferase reporter gene (Fig. 3 a) in either the absence or presence of an NGFI-A overexpression vector (Fig. 3 b,c) into HEK 293 cells. Luciferase activity was significantly (p < 0.001) increased in cells cotransfected with the nonmethylated exon 17 GR promoter in the presence of ectopically expressed NGFI-A (Fig. 3 d). There was dramatically reduced luciferase activity in cells transfected with the in vitro methylated exon 17 sequence (Fig. 3 d). Nevertheless, NGFI-A significantly (p < 0.001) enhanced luciferase activity through the methylated exon 17 GR promoter, although absolute levels were substantially reduced compared with those with the nonmethylated promoter construct (p < 0.01) (Fig. 3 d). These findings suggest that (1) NGFI-A can activate gene expression through the exon 17 GR promoter and (2) methylation of the exon 17 GR promoter region dampens NGFI-A-induced transcriptional activity from this promoter sequence.
Figure 3.

DNA methylation affects NGFI-A binding to and activation of an exon 17 GR promoter–luciferase reporter construct. a, Physical map of the exon 17 GR promoter–luciferase reporter construct. b, Physical map of the pEF6/V5–His–NGFI-A expression vector. c, Western blot analysis of NGFI-A expression in HEK 293 cells 72 h after transfection with the pEF6/NGFI-A/V5–His plasmid. The top shows the level of V5 immunoreactivity with the ectopically expressed NGFI-A protein. The middle shows the endogenous and exogenous levels of NGFI-A immunoreactivity. Because of the strong affinity of the NGFI-A antibody for the ectopically expressed NGFI-A protein, the signal for the endogenous NGFI-A protein could not be detected on the same film. Therefore, although the samples were run on and transferred from the same gel, the nitrocellulose membrane was cut so that one piece contained the untransfected and V5 empty vector transfected samples, and the other piece contained the sample transfected with V5 vector expressing NGFI-A. To detect a signal for the endogenous NGFI-A protein, the membrane was exposed to film for 10 min, whereas to detect the exogenous NGFI-A protein, the membrane was exposed to film for <20 s. The bottom shows the level of β-actin immunoreactivity to control for equal loading. d, Effects of ectopic NGFI-A expression on exon 17 GR promoter function. Mean ± SEM of luciferase expression (**p < 0.001) from the nonmethylated and methylated exon 17 GR promoter–luciferase reporter plasmid cotransfected without or with an NGFI-A expression plasmid (n = 4 samples per group). NGFI-A binding to either the nonmethylated or methylated exon 17 GR promoter–luciferase reporter plasmid cotransfected without or with an NGFI-A expression plasmid in HEK 293 cells was determined by ChIP analysis (n = 4 samples per group). Lanes were loaded with non-immunoprecipitated input (I), NGFI-A primary antibody immunoprecipitated (A), or non-immune IgG antibody immunoprecipitated (N) extracts. e, Representative Southern blots of the amplified exon 17 GR promoter region from NGFI-A immunoprecipitated (IP) HEK 293 cells (194 bp band). f, Mean ± SEM ROD of exon 17 sequence amplified from NGFI-A immunoprecipitated cell extract normalized to the input values (*p < 0.05; **p < 0.001; n = 4 samples per group).
We then performed ChIP analysis using an NGFI-A primary antibody on the cotransfected HEK 293 cells described above to examine the effect of cytosine methylation on NGFI-A binding in the living cell. NGFI-A overexpression significantly increased NGFI-A association with the nonmethylated exon 17 sequence (p < 0.001). NGFI-A association with the methylated exon 17 sequence was reduced by comparison with that with the nonmethylated sequence, although a significant (p < 0.05) increase in association was apparent (Fig. 3 e,f). These findings suggest that methylation of the exon 17 sequence alters NGFI-A association, although NGFI-A association was not completely abolished with the use of the methylated exon 17 construct.
Transcriptional activation from exon 17 GR promoter requires NGFI-A binding
We performed site-directed mutagenesis of the 5′ and 3′ CpG sites in the NGFI-A response element (Fig. 4 a) to examine whether activation of exon 17 GR promoter by NGFI-A requires direct interaction of NGFI-A with its response element within this promoter. NGFI-A overexpression significantly increased NGFI-A association with the exon 17 promoter in cells cotransfected with nonmutated (p < 0.001) or, to a lesser extent, the 5′ CpG dinucleotide mutated (p < 0.02) exon 17 GR promoter sequences (Fig. 4 b). There was no significant effect (p > 0.05) of NGFI-A overexpression on NGFI-A binding in cells cotransfected with 3′ CpG dinucleotide mutated exon 17 GR promoter, suggesting that the 3′ CpG sequence is critical for NGFI-A binding.
Figure 4.

Effects of mutation and methylation of the NGFI-A response element on exon 17 GR promoter function. a, In vitro mutation of the exon 17 GR promoter was performed using primers designed for cytosine to adenine conversion within the 5′ CpG and 3′ CpG dinucleotide in the NGFI-A response element. b, NGFI-A binding of the nonmethylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type (WT) nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected in the absence or presence of an NGFI-A expression plasmid (n = 4 samples per group). Mean ± SEM ROD of exon 17 sequence amplified from NGFI-A immunoprecipitated (IP) cell extract (*p < 0.02; **p < 0.001). Lanes were loaded with non-immunoprecipitated input (I), NGFI-A primary antibody immunoprecipitated (A), or non-immune IgG antibody immunoprecipitated (N) extracts. c, NGFI-A binding, to the methylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected in the absence or presence of an NGFI-A expression plasmid (n = 4 samples per group). Mean ± SEM ROD of exon 17 sequence amplified from NGFI-A immunoprecipitated cell extract (*p < 0.05; ***p < 0.0001). d, Mean ± SEM of luciferase expression (*p < 0.05; **p < 0.001) from the nonmethylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected in the absence or presence of an NGFI-A expression plasmid (n = 4 samples per group). e, Mean ± SEM of luciferase expression (**p < 0.001; ***p < 0.0002) from the methylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected without or with an NGFI-A expression plasmid (n = 4 samples per group).
We then examined the effect of mutagenesis on NGFI-A binding to the methylated exon 17 GR promoter transfected into HEK 293 cells (Fig. 4 c). NGFI-A overexpression significantly increased NGFI-A association with the exon 17 promoter in cells cotransfected with methylated and nonmutated (p < 0.05) or methylated and 5′ CpG dinucleotide mutated (p < 0.0001) exon 17 GR promoter sequences. We saw no evidence of NGFI-A association with the methylated exon 17 promoter construct mutated at the 3′ CpG dinucleotide. Thus, mutation of the 3′ CpG dinucleotide appears to be crucial for the association of NGFI-A to both methylated and unmethylated exon 17 promoters. Interestingly, NGFI-A association was increased in the methylated promoter bearing a 5′ ApG dinucleotide compared with the wild-type condition bearing a methylCpG at this position (Fig. 5 c). Because mutation of this CpG site to ApG renders it constitutively nonmethylated, this suggests that methylation of this cytosine inhibits NGFI-A binding. An alternative explanation for the increased binding of the ApG mutation is that the CpG sequence itself is repressive. However, this interpretation is not supported by the data showing that mutation of the nonmethylated CpG to ApG does not increase binding when the rest of the sequence is methylated (Fig. 5 c), suggesting that the methylation of this cytosine rather than the sequence has the main repressive effects.
Figure 5.

NGFI-A binding recruits CBP and results in H3–K9 acetylation associated of both nonmethylated and methylated exon 17 GR promoter. a, b, CBP association and histone H3–K9 acetylation of the nonmethylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type (WT) nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected without or with an NGFI-A expression plasmid (n = 4 samples per treatment). a, Mean ± SEM ROD of exon 17 sequence amplified from CBP immunoprecipitated (IP) cell extract (*p < 0.04; **p < 0.002). b, Mean ± SEM ROD of exon 17 sequence amplified from histone H3–K9 acetylation immunoprecipitated cell extract (*p < 0.05; **p < 0.001). c, d, CBP association and histone H3–K9 acetylation of the methylated exon 17 GR promoter–luciferase reporter plasmid containing either a wild-type nonmutated, 5′ CpG mutated or 3′ CpG mutated NGFI-A response element, cotransfected without or with an NGFI-A expression plasmid (n = 4 samples per treatment). c, Mean ± SEM ROD of exon 17 sequence amplified from CBP immunoprecipitated cell extract (*p < 0.05). d, Mean ± SEM ROD of exon 17 sequence amplified from histone H3–K9 acetylation immunoprecipitated cell extract (*p < 0.05; **p < 0.001). Lanes were loaded with non-immunoprecipitated input (I), NGFI-A primary antibody immunoprecipitated (A), or non-immune IgG antibody immunoprecipitated (N) extracts.
We then measured the impact of these mutations on the ability of NGFI-A to induce expression from the nonmethylated exon 17 GR promoter (Fig. 4 d). The effect of NGFI-A was significantly (p < 0.03) reduced by the mutation at the 5′ CpG dinucleotide site that partially inhibits NGFI-A binding but was completely abolished by the 3′ CpG mutation that blocked NGFI-A binding (Fig. 4 d). Thus, the 5′ CpG mutation of the cytosine reduces, but does not completely abolish, binding or the effects of NGFI-A.
Finally, we measured the impact of these mutations on the ability of NGFI-A to induce expression from the methylated exon 17 GR promoter (Fig. 4 e). Because conversion of the 5′ CpG dinucleotide to ApG renders it constitutively resistant to methylation, the fact that it remains sensitive to NGFI-A allows us to examine the specific role of methylation at this site. Despite the overall reduction in transcriptional activity in the methylated exon 17 GR promoter (note the differences in the scales for the ordinates in Fig. 4 d,e), NGFI-A overexpression significantly (p < 0.001) increased luciferase gene expression in cells cotransfected with the methylated, wild-type and, interestingly, to a significantly greater extent (p < 0.0002) with the methylated and 5′ CpG dinucleotide mutated exon 17 GR promoter (Fig. 4 e). The conversion of the 5′ CpG dinucleotide to ApG renders the site constitutively unmethylated and sensitive to NGFI-A, thus explaining the enhanced activity compared with the methylated wild-type construct (Fig. 4 e). This finding suggests that the 5′ CpG dinucleotide within the NGFI-A response element functions as a “DNA methylation sensor,” and its methylation status is critical for the silencing of gene expression by cytosine methylation. There was no significant effect of NGFI-A overexpression on luciferase activity in cells cotransfected with 3′ CpG dinucleotide mutated and methylated exon 17 GR promoter (p > 0.05). Because this mutation abolishes NGFI-A binding, this finding demonstrates that activation of the methylated exon 17 GR promoter requires direct interaction of NGFI-A with this promoter.
NGFI-A binding recruits CBP and histone acetylation to the exon 17 GR promoter sequence
NGFI-A directly interacts with CBP (Silverman et al., 1998). CBP is a histone acetyltransferase (HAT) that activates chromatin by catalyzing histone acetylation (Ogryzko et al., 1996). We used a ChIP analysis with either a CBP primary antibody or an acetylated H3–K9 histone primary antibody to test the hypothesis that NGFI-A recruits CBP and histone acetylation to the nonmethylated exon 17 GR promoter (Fig. 5 a,b). NGFI-A overexpression significantly increased CBP association (p < 0.002) with and H3–K9 acetylation (p < 0.001) of the nonmethylated, wild-type exon 17 sequence. Mutation of the 5′ CpG dinucleotide reduced CBP association and H3–K9 acetylation in response to NGFI-A overexpression, although significant increases (p < 0.04 and p < 0.05, respectively) were observed (Fig. 5 a,b). There was no significant effect of NGFI-A overexpression on CBP association or H3–K9 acetylation in cells cotransfected with 3′ CpG dinucleotide mutated exon 17 GR promoter. Because the 3′ CpG mutation is critical for NGFI-A binding to this promoter (Fig. 4 b), these findings suggest that NGFI-A interaction with the exon 17 GR promoter is required for CBP recruitment and histone acetylation.
We then performed the same analysis using the methylated version of the exon 17 GR promoter (Fig. 5 c,d). Although methylation reduced CBP binding, NGFI-A overexpression nevertheless significantly increased CBP association (p < 0.05) with and H3–K9 acetylation of (p < 0.05) the wild-type exon 17 sequence. In cells transfected with the methylated exon 17 GR promoter mutated at the 5′ CpG dinucleotide, NGFI-A expression also increased both CBP association (p < 0.05) with and H3–K9 acetylation of (p < 0.001) the GR exon 17 promoter. However, mutation of the 3′ CpG dinucleotide completely eliminated both CBP association with and H3–K9 acetylation of the methylated exon 17 sequence. Thus, mutation of the 3′ CpG dinucleotide seems crucial for the association of NGFI-A, CBP, and histone H3–K9 acetylation with both the unmethylated and methylated exon 17 GR promoter sequence. These results suggest that interaction of NGFI-A with the methylated exon 17 GR promoter can recruit CBP and result in histone acetylation of the exon 17 GR promoter.
NGFI-A association demethylates the methylated exon 17 GR promoter sequence
Methylation decreases NGFI-A binding to and transcriptional activity through the exon 17 promoter. Nevertheless, the previous studies reveal some evidence for NGFI-A binding and NGFI-A-induced transcriptional activity even with the methylated exon 17 GR promoter–luciferase construct (Figs. 3, 4). There are at least two possible explanations. First, methylation may not completely inhibit NGFI-A binding. Alternatively, NGFI-A overexpression might result in the gradual demethylation of the previously methylated exon 17 construct, resulting in transcriptional activation. We (Cervoni and Szyf, 2001) provided evidence suggesting that histone acetylation can result in a replication-independent demethylation of DNA sites (Chen et al., 2003; Martinowich et al., 2003). We suggested that transcription factors direct demethylation activity to specific promoters by targeting HATs (Cervoni and Szyf, 2001). NGFI-A overexpression results in increased CBP association with and histone acetylation of a methylated exon 17 promoter construct so long as the 3′ CpG site remains intact (Fig. 5 c,d). We tested the hypothesis that interaction of NGFI-A with the methylated exon 17 sequence results in GR promoter demethylation. We used sodium bisulfite mapping to characterize the global methylation state of exon 17 GR promoter sequence in HEK 293 cells 72 h after transient transfection with the methylated version of the exon 17 GR promoter construct in either the absence or presence of ectopically expressed NGFI-A (data not shown). NGFI-A overexpression significantly (p < 0.01) decreased levels of cytosine methylation in cells cotransfected with methylated exon 17 GR promoter. Ectopic expression of NGFI-A resulted in ∼50% demethylation of the cotransfected exon 17 GR promoter. Because the exon 17 GR promoter–luciferase reporter is a plasmid that does not bear an origin of replication and does not replicate in HEK 293 cells during the period of transient transfection (Cervoni and Szyf, 2001), the results suggest that NGFI-A overexpression provokes active demethylation of the exon 17 GR promoter.
Mutation of the exon 17 GR promoter NGFI-A response element blocks NGFI-A-induced demethylation
We then tested whether direct interaction of NGFI-A with the exon 17 GR promoter is responsible for demethylation taking advantage of the site-specific mutants in the NGFI-A response element described above (Fig. 4). In the wild-type condition, NGFI-A overexpression significantly (p < 0.001) decreased levels of cytosine methylation at the 5′ CpG site (i.e., DNA demethylation) in cells cotransfected with methylated and nonmutated exon 17 GR promoter (Fig. 6 a). These findings replicate those of the previous study. In contrast, there was no significant effect of NGFI-A overexpression on levels of cytosine methylation of the 5′ CpG dinucleotide in cells cotransfected with methylated exon 17 GR promoter mutated at the 3′ CpG dinucleotide (Fig. 6 a). Because this site appears critical for NGFI-A binding, these results suggest that the NGFI-A-induced demethylation of the 5′ CpG dinucleotide requires a direct interaction of NGFI-A with the exon 17 GR promoter sequence. We propose that, although the affinity of NGFI-A for the methylated promoter is lower than to the unmethylated promoter, high abundance of the NGFI-A transcription factor will result in the association of the NGFI-A protein with the exon 17 GR promoter sequence resulting in demethylation of a methylated promoter at the 5′ CpG site. Interestingly, the demethylation effect is site specific. There was no effect of NGFI-A overexpression on the methylation status of the 3′ CpG dinucleotide (Fig. 6 b) in either the wild-type condition or the construct bearing the mutated 5′ CpG dinucleotide.
Figure 6.

NGFI-A binding to its response element triggers active demethylation of GR promoter. a, Mean ± SEM percentage methylation of the 5′ CpG dinucleotide for NGFI-A bound exon 17 GR and its respective mutants from the cotransfection shown in Figure 4 c (6–10 clones sequenced per sample; n = 4 samples per group) in the absence (control) and presence of an NGFI-A expression vector (**p < 0.001). b, Mean ± SEM percentage methylation of the 3′ CpG dinucleotide (6–10 clones sequenced per sample; n = 4 samples per group).
Maternal effect on NGFI-A and CBP binding, histone acetylation, and demethylation of the exon 17 GR promoter
We then tested whether our findings from in vitro experiments are consistent with the in vivo condition focusing on the first week of postnatal life, during which there occurs the demethylation of the 5′ CpG site (Weaver et al., 2004). We performed a ChIP analysis of CBP association, histone H3–K9 acetylation, and NGFI-A protein binding to the exon 17 GR promoter in the native chromatin environment in vivo in intact hippocampi from day 6 offspring of high and low LG mothers. The results indicated significantly greater binding of NGFI-A (p < 0.0001), increased CBP association (p < 0.05), and greater histone H3–K9 acetylation (p < 0.001) of the hippocampal exon 17 GR promoter in the neonatal offspring of high compared with low LG mothers (Fig. 7 a–d). Thus, maternal programming of the exon 17 GR promoter is associated with differences in NGFI-A binding, CBP association, and histone H3–K9 acetylation in the neonatal offspring of high and low LG mothers.
Figure 7.
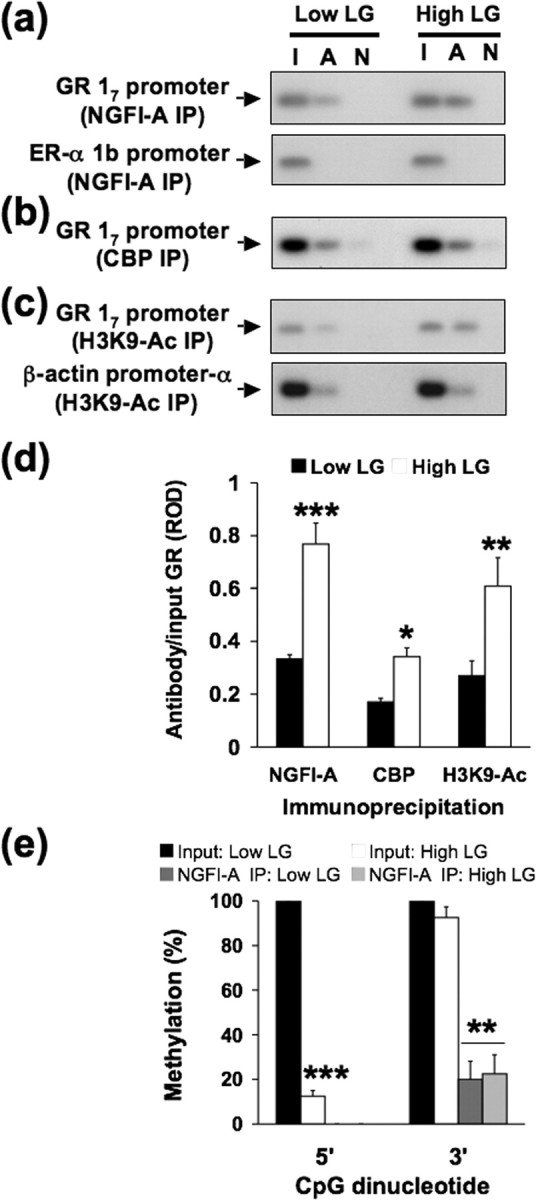
Chromatin immunoprecipitation of CBP, histone H3–K9 acetylation and NGFI-A binding to the exon 17 GR and DNA methylation analysis of NGFI-A-bound exon 17 GR sequence in hippocampal tissue of 6-d-old high and low LG offspring (n = 4 animals per group). a–c, Lanes were loaded with non-immunoprecipitated input (I), CBP, acetylated histone H3–K9 or NGFI-A primary antibody immunoprecipitated (A), or non-immune IgG antibody immunoprecipitated (N) hippocampal extracts. a, Representative Southern blots of the amplified exon 17 region of the GR from NGFI-A immunoprecipitated (IP) hippocampal tissue (194 bp band). Exon 1b estrogen receptor-α promoter region, which does not contain NGFI-A recognition elements (493 bp), amplified from the same NGFI-A immunoprecipitated hippocampal tissue was run as a control for specificity and showed no signal. b, Representative Southern blots of the amplified exon 17 region of the GR from CBP immunoprecipitated hippocampal tissue (194 bp band). c, Representative Southern blots of the amplified exon 17 region from acetyl-histone H3–K9 immunoprecipitated hippocampal tissue (194 bp band) and β-actin (171 bp band) control. d, ROD (mean ± SEM) of exon 17 sequence amplified from cAMP, acetyl-histone H3–K9, or NGFI-A immunoprecipitated hippocampal tissue of 6-d-old high and low LG offspring (n = 4 animals per group; *p < 0.05; **p < 0.001; ***p < 0.0001). e, Mean ± SEM percentage methylation per cytosine for the 5′ and 3′ CpG dinucleotides within the NGFI-A binding region of the exon 17 GR promoter bound to NGFI-A protein in 6-d-old offspring of high and low LG mothers (6–10 clones sequenced per animal; n = 4 animals per group; **p < 0.001; ***p < 0.0001).
Sodium bisulfite mapping was performed using the same samples of NGFI-A immunoprecipitated hippocampal DNA from the neonatal offspring of high and low LG mothers to establish the DNA methylation pattern of the NGFI-A-bound exon 17 GR promoter sequence (Fig. 7 e). The 5′ and 3′ CpG dinucleotides within the NGFI-A response element on the exon 17 GR promoter bound to NGFI-A were significantly (p < 0.0001 and p < 0.001, respectively) (Fig. 7 e) unmethylated compared with the non-immunoprecipitated (i.e., sequences not bound to NGFI-A) Input DNA in the offspring of high and low LG mothers. It is interesting to note that, although the exon 17 GR promoter is hypermethylated in the hippocampus of the offspring of low compared with high LG mothers (note the highly significant difference in the methylation state of the input DNA in Fig. 7 e), the methylation status of the exon 17 promoter sequences that are bound to NGFI-A in vivo are equally unmethylated in both groups. These findings underscore the close in vivo link between NGFI-A binding and methylation status of the NGFI-A response element of exon 17 GR promoter in the day 6 hippocampus.
Serotonin programs in vitro hippocampal GR expression through NGFI-A
We used primary hippocampal neuronal cultures as a model system to further determine the role of NGFI-A in demethylation of the NGFI-A response element within the exon 17 GR promoter. Postnatal handling and enhanced maternal LG increase 5-HT turnover and cAMP levels in neonatal rat hippocampus. Both 5-HT and cAMP increase GR expression in primary hippocampal cell cultures (Mitchell et al., 1990, 1992; Laplante et al., 2002), providing evidence for the direct effects of these maternally regulated signals on hippocampal GR expression. These cultures are primarily (∼95%) neuronal (Banker and Cowan, 1977), and indeed 5-HT has little or no effect on GR levels in glial cultures prepared from hippocampal samples (Mitchell et al., 1990). Hippocampal cell cultures from embryonic day 19–20 fetuses were prepared as described by Laplante et al. (2002), and, 2 d after seeding, mitotic inhibitors were added to the medium to prevent glial proliferation. The cultures were then treated with 5-HT (100 nm) for 96 h. The 5-HT was then removed from the medium, and the cultures were harvested at various time points after the removal of 5-HT for in vitro assays of GR binding (Mitchell et al., 1990). Cells were maintained in heat-inactivated medium to ensure the effective removal of 5-HT, which was confirmed using HPLC analysis. GR binding levels (Fig. 8 a) were significantly (p < 0.01) elevated at each time point after the removal of 5-HT from the medium. Because the medium was heat inactivated to ensure the effective removal of 5-HT, the effect of 5-HT appears to persist well beyond the termination of 5-HT treatment. These findings suggest that GR levels are stably increased after 5-HT and provide evidence for an in vitro model of GR programming in hippocampal neurons.
Figure 8.

Effects of 5-HT cAMP and NGFI-A on hippocampal GR promoter methylation and gene expression. a, Primary hippocampal cultures received a single treatment of 5-HT (100 nm) for 96 h, 5-HT was then removed from the medium, and the cultures were harvested at various time points after the removal of 5-HT for in vitro assays of GR binding (n = 4 samples per group; *p < 0.01). b, Mean ± SEM percentage methylation per cytosine for the 5′ and 3′ CpG dinucleotides within the NGFI-A binding region of the GR promoter from 5-HT and cAMP-treated hippocampal cell culture (6–10 clones sequenced per sample; n = 4 samples/group; **p < 0.001). c, Mean ± SEM percentage methylation per cytosine for the 5′ and 3′ CpG dinucleotides within the NGFI-A binding region of the GR promoter from 5-HT/NGFI-A missense and 5-HT/NGFI-A antisense-treated hippocampal cell culture (6–10 clones sequenced per sample; n = 4 samples per group; **p < 0.001). d, e, Western blot analysis of GR expression from nontreated, 5-HT/NGFI-A missense and 5-HT/NGFI-A antisense-treated hippocampal cell culture (6–10 clones sequenced per sample; n = 4 samples per group; **p < 0.001).
We then examined (1) whether activation of the 5-HT/cAMP cascade in hippocampal cell cultures might alter the methylation status of the NGFI-A response element of the exon 17 GR promoter and (2) whether such effects might be mediated by NGFI-A. Hippocampal cell cultures treated for 96 h with medium alone, 5-HT (100 nm), or 8-bromo-cAMP (10 mm) [concentrations based on previous studies (Mitchell et al., 1990, 1992; Laplante et al., 2002)]. The cells were harvested 4 d later to determine levels of GR promoter methylation. Although no significant differences were found for the 3′ CpG dinucleotide, the 5′ CpG dinucleotide of the NGFI-A response element was significantly demethylated in the 5-HT- and cAMP-treated cultures compared with the control cultures (p < 0.001) (Fig. 8 b). Bromodeoxyuridine (BrdU) labeling, which marks newly generated cells, revealed little or no cell replication in the cultures at the time of 5-HT treatment; indeed, in accordance with the procedures for the establishment of neuronal cell cultures, the cultures were treated with mitotic inhibitors (Banker and Cowan, 1977). Thus, this difference in methylation occurs in the absence of DNA replication.
We examined the hypothesis that the downstream effects of the 5-HT/cAMP cascade are mediated by NGFI-A using a chemically modified phosphorothioate antisense oligonucleotide (Biognostik) to downregulate NGFI-A gene expression (see methods for oligonucleotide sequence modified from Kukita et al., 1997). The cultures received a single treatment of 5-HT (100 nm), followed by either NGFI-A AS oligonucleotide (1 μm) or the SAS control. Preliminary studies showed that this concentration of the NGFI-A AS/SAS was effective in completely blocking the effect of 5-HT on NGFI-A expression without any evidence of toxicity in viability tests (data not shown). The cells were harvested 96 h later to determine exon 17 promoter methylation status and GR expression. NGFI-A AS treatment eliminated the effects of 5-HT treatment on methylation of the 5′ CpG dinucleotide within the NGFI-A binding region of the GR promoter (p < 0.001) (Fig. 8 c). Subsequent Western blot analysis showed that NGFI-A AS treatment also blocked the effect of 5-HT treatment on GR protein expression (p < 0.001) (Fig. 8 d,e). These findings are consistent with the idea that NGFI-A mediated the 5-HT-activated, replication-independent epigenetic modification of the exon 17 promoter and GR programming.
Discussion
GR transcription from the exon 17 promoter in the adult rat is altered as a function of maternal care in early life (McCormick et al., 2000) (Fig. 1). The exon 17 GR promoter contains an NGFI-A consensus sequence (McCormick et al., 2000), and ChIP assays reveal increased NGFI-A association with the exon 17 promoter in the adult offspring of high compared with low LG mothers (Weaver et al., 2004, 2005). The results of the present experiments support the idea that NGFI-A can activate gene transcription through the exon 17 promoter (Fig. 3). The differences in NGFI-A binding to the exon 17 GR promoter in adult animals occur despite comparable levels of hippocampal NGFI-A expression. Weaver et al. (2004) proposed that differential methylation of the 5′ CpG site of the NGFI-A response element within the exon 17 GR promoter is associated with differences in NGFI-A binding to the exon 17 promoter and GR expression. Indeed, cytosine methylation is associated with decreased binding of certain transcription factors to their cognate binding sites (Prendergast and Ziff, 1991). The results of the current studies support this hypothesis. Thus, gel mobility shift assays revealed that methylation of the 5′ CpG dinucleotide within the NGFI-A response element reduced NGFI-A binding (Fig. 2). Moreover, cotransfection of HEK 293 cells with an NGFI-A expression vector and plasmid containing the exon 17 GR promoter coupled to a luciferase reporter revealed increased NGFI-A binding and increased transcriptional activity from a nonmethylated exon 17 GR promoter construct (Fig. 3). Both the levels of NGFI-A binding and NGFI-A-induced transcriptional activation were reduced when the transfection was performed using a methylated exon 17 GR promoter construct.
The results of site-directed mutagenesis provide additional support for the importance of the methylation state of the 5′ CpG site within the NGFI-A consensus sequence of the exon 17 GR promoter. Thus, the effect of NGFI-A on transcription through a methylated exon 17 GR promoter–luciferase reporter construct was significantly increased with a point mutation at the 5′ cytosine (a cytosine to adenine mutation) that rendered it unmethylatable (Fig. 4 c,e). Interestingly, mutation at the 3′ CpG dinucleotide eliminated both NGFI-A binding and NGFI-A transcriptional activity (Fig. 4 c,e). Thus, the 3′, but not the 5′, CpG dinucleotide is apparently obligatory for NGFI-A binding. Instead, the 5′ CpG dinucleotide appears to function as a DNA methylation sensor; it is the methylation status of the cytosine that is critical in determining NGFI-A binding.
Studies using the methylated exon 17 GR promoter revealed decreased NGFI-A binding and NGFI-A-induced transcriptional activation. There remained, however, a residual level of NGFI-A activity in cells harvested after prolonged exposure to the NGFI-A overexpression vector. The results of studies described in Figure 6 suggest that this finding was likely attributable to an NGFI-A-induced demethylation of the promoter construct. Interestingly, this demethylation was dependent on NGFI-A association with the exon 17 GR promoter; the effect was completely eliminated by mutation of the 3′ CpG dinucleotide that abolishes NGFI-A binding (Fig. 6 b). Moreover, the effect of NGFI-A on the methylation status of the exon 17 GR promoter was specific to the 5′ CpG site. These findings may prove critical in our understanding of the effect of maternal care on the programming of the exon 17 GR promoter. On the day of birth, the 5′ CpG site is hypermethylated in the offspring of both high and low LG mothers. Over the first week of postnatal life, the period when differences in maternal care are observed, there is a demethylation of the 5′ CpG site. This period corresponds to a time of increased NGFI-A association with the exon 17 GR promoter in the offspring of the high LG mothers (Fig. 7). In support of this hypothesis, we found that NGFI-A-bound exon 17 GR promoter in day 6 pups is highly enriched for unmethylated DNA (Fig. 7 e) as expected if NGFI-A binding targets demethylation.
We propose that the initial weak interaction of NGFI-A with the methylated exon 17 GR promoter initiates a cascade of events leading to demethylation of the 5′ CpG site and stable epigenetic reprogramming of the promoter that is defined by an increased affinity for NGFI-A and stable activation. Transcription factors can target demethylation of specific sequences. Examples of transcription factor-induced demethylation include the role of nuclear factor-κB in demethylation of Ig-κ light chain gene enhancer, which requires the presence of a cis-acting response element for this transcription factor (Kirillov et al., 1996), and demethylation mediated by the maize suppressor–mutator transposon-encoded TnpA protein (Dahlman-Wright et al., 1991).
The demethylation of the 5′ CpG dinucleotide within the NGFI-A response element is associated with variations in maternal care occurs over the first week of life (Weaver et al., 2004) and is associated with increased CBP association with the exon 17 GR promoter (Fig. 7 d). CBP is an HAT (Ogryzko et al., 1996) that interacts directly with NGFI-A (Silverman et al., 1998). The results of the current studies shown in Figure 5 reveal that NGFI-A overexpression serves to recruit CBP to the exon 17 sequence (Fig. 5 c), increasing histone acetylation of the exon 17 sequence (Fig. 5 d). Neither of these effects is apparent when the exon 17 sequence contains an NGFI-A response element mutated in 3′ CpG site, which eliminates NGFI-A binding. Cervoni and Szyf (2001) showed that pharmacological stimulation of histone acetylation by the histone deacetylase inhibitor trichostatin A (TSA) induces replication-independent demethylation and proposed that transcription factors trigger demethylation by recruiting HATs to the promoter. Likewise, chronic TSA infusion in the offspring of low LG mothers increases histone acetylation of and NGFI-A binding to the exon 17 promoter and results in the demethylation of the 5′ CpG dinucleotide within the NGFI-A response element (Weaver et al., 2004). Such effects are unique to the 5′ CpG site.
These findings support a hypothesis that the tactile stimulation associated with increased maternal LG initiates a cascade of intracellular events that result in increased NGFI-A expression in pups, stably altered methylation status of the 5′ CpG of the NGFI-A response element within the exon 17 promoter, and enhanced GR expression in adulthood. Jutapakdeegul et al. (2003) report that enhanced tactile stimulation (pup stroking with a brush) over the first week of life results in increased hippocampal GR expression in adulthood. Previous studies suggest that such effects as well as those on NGFI-A expression are mediated by increased hippocampal 5-HT activity at 5-HT7 receptors that are positively coupled to cAMP (Mitchell et al., 1990, 1992; Meaney et al., 2000). In hippocampal cell cultures, 5-HT increases GR expression in hippocampal cell cultures (Laplante et al., 2002). This effect is mimicked by treatment with the stable cAMP analog 8-bromo-cAMP and blocked with either a 5-HT7 receptor antagonist or an inhibitor of PKA. Importantly, the effect of 5-HT on GR expression in hippocampal cell cultures is completely blocked by concurrent treatment with an antisense targeting NGFI-A mRNA (Fig. 8 c–e). We found that 5-HT treatment stably alters GR expression in a manner similar to that of the in vivo effects of maternal care. Levels of GR in hippocampal cultures treated with 5-HT remained elevated long after the removal of 5-HT from the culture medium (Fig. 8 a). Importantly, both 5-HT and cAMP treatments resulted in the demethylation of the NGFI-A response element of the exon 17 sequence, and these effects were selective for the 5′ CpG site. BrdU labeling, which marks newly generated cells, reveals little or no cell replication in the cultures at the time of 5-HT treatment; indeed, these cells are treated with mitotic inhibitors to prevent glial proliferation and maintain the essentially neuronal character of the cultures (Banker and Cowan, 1977). These findings reinforce the idea that the alterations in cytosine methylation occur independently of cell replication and in response to intracellular signals associated with variations in maternal care. In support of this hypothesis, we found that an NGFI-A AS blocks the effects of 5-HT on the methylation status of the 5′ CpG site of the NGFI-A response element within the exon 17 GR promoter as well as on GR expression (Fig. 8 c–e).
A critical question concerns the mechanism for this active process of demethylation. One possibility is that NGFI-A directly recruits a demethylase to the gene or recruits an HAT, which increases acetylation, thus increasing the accessibility to demethylase (Cervoni and Szyf, 2001; Cervoni et al., 2002). Interestingly, NGFI-A can actively target methylated DNA binding proteins to genomic targets (Carvin et al., 2003). Additional studies will be required to identify the demethylase involved in this process. The current studies suggest a critical role of NGFI-A in the epigenetic programming of the exon 17 GR promoter. NGFI-A resides downstream to a signaling cascade triggered by 5-HT, which is activated in response to maternal care.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research (CIHR), and from the National Institutes of Health (HD-051897) (M.J.M., M.S.), and the National Cancer Institute of Canada (M.S.). S.E.B. is supported by a Doctoral Research Award from the CIHR. M.J.M. is supported by a CIHR Senior Scientist Award, and the project was supported by a Distinguished Investigator Award (M.J.M.) from the National Alliance for Research on Schizophrenia and Affective Disorders.
References
- Agrawal AA. Phenotypic plasticity in the interactions and evolution of species. Science. 2001;294:321–326. doi: 10.1126/science.1060701. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Meaney MJ. Hypothalamic-pituitary-adrenal function in chronic intermittently cold-stressed neonatally handled and non handled rats. J Neuroendocrinol. 1995;7:97–108. doi: 10.1111/j.1365-2826.1995.tb00672.x. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Caldji C, Diorio J, Meaney MJ. Variations in maternal care alter GABA(A) receptor subunit expression in brain regions associated with fear. Neuropsychopharmacology. 2003;28:1950–1959. doi: 10.1038/sj.npp.1300237. [DOI] [PubMed] [Google Scholar]
- Carvin CD, Parr RD, Kladde MP. Site-selective in vivo targeting of cytosine-5 DNA methylation by zinc-finger proteins. Nucleic Acids Res. 2003;31:6493–6501. doi: 10.1093/nar/gkg853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervoni N, Szyf M. Demethylase activity is directed by histone acetylation. J Biol Chem. 2001;276:40778–40787. doi: 10.1074/jbc.M103921200. [DOI] [PubMed] [Google Scholar]
- Cervoni N, Detich N, Seo SB, Chakravarti D, Szyf M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem. 2002;277:25026–25031. doi: 10.1074/jbc.M202256200. [DOI] [PubMed] [Google Scholar]
- Champagne FA, Francis DD, Mar A, Meaney MJ. Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol Behav. 2003;79:359–371. doi: 10.1016/s0031-9384(03)00149-5. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane-Robinson C, Myers FA, Hebbes TR, Clayton AL, Thorne AW. Chromatin immunoprecipitation assays in acetylation mapping of higher eukaryotes. Methods Enzymol. 1999;304:533–547. doi: 10.1016/s0076-6879(99)04031-8. [DOI] [PubMed] [Google Scholar]
- Dahlman-Wright K, Wright A, Gustafsson JA, Carlstedt-Duke J. Interaction of the glucocorticoid receptor DNA-binding domain with DNA as a dimer is mediated by a short segment of five amino acids. J Biol Chem. 1991;266:3107–3112. [PubMed] [Google Scholar]
- Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–1158. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley JD, Hasert MF, Suomi SJ, Linnoila M. Nonhuman primate model of alcohol abuse: effects of early experience, personality, and stress on alcohol consumption. Proc Natl Acad Sci USA. 1991;88:7261–7265. doi: 10.1073/pnas.88.16.7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutapakdeegul N, Casalotti SO, Govitrapong P, Kotchabhakdi N. Postnatal touch stimulation acutely alters corticosterone levels and glucocorticoid receptor gene expression in the neonatal rat. Dev Neurosci. 2003;25:26–33. doi: 10.1159/000071465. [DOI] [PubMed] [Google Scholar]
- Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. A role for nuclear NF-kappaB in B-cell-specific demethylation of the Igkappa locus. Nat Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- Kukita T, Kukita A, Harada H, Iijima T. Regulation of osteoclastogenesis by antisense oligodeoxynucleotides specific to zinc finger nuclear transcription factors Egr-1 and WT1 in rat bone marrow culture system. Endocrinology. 1997;138:4384–4389. doi: 10.1210/endo.138.10.5466. [DOI] [PubMed] [Google Scholar]
- Laplante P, Diorio J, Meaney MJ. Serotonin regulates hippocampal glucocorticoid receptor expression via a 5-HT7 receptor. Brain Res Dev Brain Res. 2002;139:199–203. doi: 10.1016/s0165-3806(02)00550-3. [DOI] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic- pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McCormick JA, Lyons V, Jacobson MD, Noble J, Diorio J, Nyirenda M, Weaver S, Ester W, Yau JL, Meaney MJ, Seckl JR, Chapman KE. 5′-heterogeneity of glucocorticoid receptor messenger RNA is tissue specific: differential regulation of variant transcripts by early-life events. Mol Endocrinol. 2000;14:506–517. doi: 10.1210/mend.14.4.0438. [DOI] [PubMed] [Google Scholar]
- Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–1192. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005;28:456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH, Sapolsky RM. Thyroid hormones influence the development of hippocampal glucocorticoid receptors in the rat: a mechanism for the effects of postnatal handling on the development of the adrenocortical stress response. Neuroendocrinology. 1987;45:278–283. doi: 10.1159/000124741. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Diorio J, Francis D, Weaver S, Yau J, Chapman K, Seckl JR. Postnatal handling increases the expression of cAMP-inducible transcription factors in the rat hippocampus: the effects of thyroid hormones and serotonin. J Neurosci. 2000;20:3926–3935. doi: 10.1523/JNEUROSCI.20-10-03926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbrandt J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science. 1987;238:797–799. doi: 10.1126/science.3672127. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Rowe W, Boksa P, Meaney MJ. Serotonin regulates type II corticosteroid receptor binding in hippocampal cell cultures. J Neurosci. 1990;10:1745–1752. doi: 10.1523/JNEUROSCI.10-06-01745.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JB, Betito K, Rowe W, Boksa P, Meaney MJ. Serotonergic regulation of type II corticosteroid receptor binding in hippocampal cell cultures: evidence for the importance of serotonin-induced changes in cAMP levels. Neuroscience. 1992;48:631–639. doi: 10.1016/0306-4522(92)90407-s. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science. 1991;251:186–189. doi: 10.1126/science.1987636. [DOI] [PubMed] [Google Scholar]
- Rouleau J, Tanigawa G, Szyf M. The mouse DNA methyltransferase 5′-region. A unique housekeeping gene promoter. J Biol Chem. 1992;267:7368–7377. [PubMed] [Google Scholar]
- Schmitt J, Hess H, Stunnenberg HG. Affinity purification of histidine-tagged proteins. Mol Biol Rep. 1993;18:223–230. doi: 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- Silverman ES, Du J, Williams AJ, Wadgaonkar R, Drazen JM, Collins T. cAMP-response-element-binding-protein-binding protein (CBP) and p300 are transcriptional co-activators of early growth response factor-1 (Egr-1) Biochem J. 1998;336:183–189. doi: 10.1042/bj3360183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, Szyf M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]