

Abstract
The human mitochondrial DNA polymerase (pol γ) is nuclearly encoded and is solely responsible for the replication and repair of the mitochondrial genome. The progressive accumulation of mutations within the mitochondrial genome is thought to be related to aging, and mutations in the pol γ gene are responsible for numerous heritable disorders including progressive external opthalmoplegia, Alpers syndrome, and parkinsonism. Here we investigate the kinetic effect of H932Y, a mutation associated with opthalmoplegia. Mutations H932Y and H932A reduce the specificity constant governing correct nucleotide incorporation 150- and 70-fold, respectively, without significantly affecting fidelity of incorporation or the maximum rate of incorporation. However, this leads to only a 2-fold reduction in rate of incorporation at a physiological nucleotide concentration (∼100 μm). Surprisingly, incorporation of T:T or C:T mismatches catalyzed by either H932Y or H932A mutants was followed by slow pyrophosphate release (or fast pyrophosphate rebinding). Also, H932Y readily catalyzed incorporation of multiple mismatches, which may have a profound physiological impact over time. His-932 is thought to contact the β-phosphate of the incoming nucleotide, so it is perhaps surprising that H932Y appears to slow rather than accelerate pyrophosphate release.
Keywords: DNA Polymerase, Enzyme Kinetics, Enzyme Mechanisms, Mitochondrial DNA, Mutagenesis Mechanisms, Parkinson Disease
Introduction
Mutations in DNA polymerase γ (pol γ)2 have been linked to several mitochondrial disorders that include progressive external opthalmoplegia (PEO), Alpers syndrome, parkinsonism, male infertility, and SANDO (sensory ataxic neuropathy, dysarthria, and opthalmoparesis) among others (1). The majority of the mutations in pol γ are associated with PEO, a mitochondrial disorder associated with accumulation of mitochondrial DNA deletions and mutations. The main clinical manifestation of PEO is progressive weakness of the external eye muscles and is often accompanied by dysphagia and variable weakness of the limb and neck muscles. Neurological symptoms could include depression or avoidant personality (2). Greater than 60 pathogenic mutations in the pol γA gene are associated with PEO, including about 14 dominant mutations, 36 recessive mutations, and other mutations of sporadic or unknown origin (1).
Recently the crystal structure of the human pol γ was solved by Lee et al. (3) and has provided an insight into understanding how processivity is achieved by the holoenzyme. The structure provides a framework to examine the physiological basis of heritable mutations in pol γ. Pol γ is a heterotrimer in which one molecule of the catalytic subunit (pol γA) binds to a dimeric pol γB. Similar to most DNA polymerases, the structure of pol γA resembles a human right hand with “palm,” “fingers,” and “thumb” domains (4, 5). The palm domain is responsible for the catalysis of the phosphoryl transfer reaction, and the fingers domain is proposed to be involved in binding of the templating base and the incoming nucleotide and governs the fidelity of the DNA polymerase. The structures of the fingers domain differ between enzymes from different families, but a functionally conserved α-helix that binds the incoming nucleotide is located at a similar position within the fingers domain of different DNA polymerases. The thumb domain is mainly involved in the binding and translocation of the duplex DNA, playing an important role in replication processivity. Initial kinetic analysis using pre-steady state methods demonstrated that the catalytic subunit of the human mitochondrial DNA polymerase (pol γ) alone was insufficient for processive replication of the mitochondrial genome, with a relative weak binding affinity for DNA (∼39 nm) and a kpol of 8.7s−1 (6). The presence of the accessory subunit increased the affinity for the DNA as well as the polymerization rate (Kd, DNA∼10 nm, kpol∼45 s−1) without affecting the dissociation rate of the primer-template from the holoenzyme (6). The increase in kpol is the largest determinant of the increase in processivity defined as kpol/koff.
The crystal structure of the ternary complex of pol γ (with the DNA and nucleotide) is not yet known. Pol γ belongs to the Family A DNA polymerases represented by the extensively studied T7 DNA polymerase and Escherichia coli DNA polymerase I. The available structures of pol A family DNA polymerases revealed a large conformational change of the enzyme after nucleotide binding (7, 8). The open to closed conformational change suggests an induced-fit mechanism for nucleotide selectivity analogous to that seen for other members of the pol A family (9, 10). The high resolution crystal structures of the T7 DNA polymerase in a ternary complex with DNA and ddNTP as well as that of E. coli DNA polymerase I provide valuable insight to the probable functions of the pol γ active site residues. The four autosomal dominant PEO mutations, G923D, R943H, Y955C, and A957S in the active site of pol γA have been partially characterized using steady state kinetic measurements in an attempt to understand their role in enzyme catalysis (11). The homologous residues in the T7 DNA polymerase are a part of the O helix forming the nucleotide binding pocket within the finger domain, as illustrated in Fig. 1, which is derived from the T7 DNA polymerase structure but labeled with pol γ residue numbers. All of the non-bridging phosphate oxygen atoms interact either with a metal ion or with one or more of the conserved residues of the O-helix. Two oxygen atoms of the γ phosphate are contacted by Arg-943, Tyr-951 (helix O) and His-932 (helix N), both, contact the β phosphate, and Lys-947 contacts the α-phosphate.
FIGURE 1.

Catalytic residues on the fingers domain. Residues Lys-947, His-932, and Arg-943 provide hydrogen bonds to the α-, β-, and γ-phosphates of the incoming dNTP. In this figure the structure of T7 DNA polymerase, 1T7P.pdb, (5) was used to model the locations of the homologous residues in pol γ. The template strand is blue, the primer strand is green, the incoming dNTP is shown in magenta, and the catalytic residues are in yellow. Template positions are labeled relative to the templating nucleotide (T0).
The mutation of His-932 to a tyrosine is associated with autosomal recessive form of PEO and SANDO (sensory ataxic neuropathy, dysarthria, and opthalmoparesis). The H932Y mutation is found as a compound with T251I/P587L and with G1051R (12, 13). A preliminary pre-steady state kinetic study of nucleotide incorporation on H932Y mutation of human pol γ has quantified the effect of the mutation of the kinetics of correct nucleotide incorporation (14), but the role of His-932 in nucleotide selectivity has not been determined. Here we have evaluated the mechanistic role of the His-932 residue in the polymerase active site by systematically investigating H932Y and H932A mutations. In this study incorporation of the nucleotide (correct as well as mismatch) and dideoxynucleotide was examined by chemical quench assays, and the data were analyzed globally to obtain the pathway for the mutants. Our results present a quantitative and mechanistic understanding of the role of the His-932 residue in the catalysis of pol γ.
EXPERIMENTAL PROCEDURES
Cloning Expression and Purification of Pol γ B
The C-terminal His-tagged pol γB lacking the first 25 amino acids from the N terminus (the mitochondrial localization sequence) was cloned into pET43.1a vector and transformed in E. coli Rosetta 2 (DE3) from Novagen. A transformant selected from a single colony was used to inoculate a LB medium supplemented with ampicillin (50 μg/ml) and chloramphenicol (34 μg/ml). This starter culture was grown overnight at 37 °C and was subsequently used to inoculate a larger 1-liter LB medium. The expression of pol γB was induced with isopropyl 1 thio-β-d-galactopyranoside with a final concentration of 1 mm when the optical density at 600 nm reached 0.6. After 6 h of induction at 25 °C, the cells were harvested by centrifugation at 3500 rpm in a Beckman JLA8.1 rotor at 4 °C. The cell pellet was resuspended in pH 8 lysis buffer (20 mm Tris-HCl, 500 mm NaCl, 5 mm imidazole, 0.1% Triton X-100) supplemented with protease inhibitors (Roche Applied Science). The suspension was sonicated on an ice bath after incubation with lysozyme (0.3 mg/ml) for 20 min at 4 °C. The cell lysate was centrifuged at 15,000 rpm for 20 min, retaining the supernatant, and polyethyleneimine (PEI) was added to achieve a final concentration of 0.1% PEI and centrifuged for 30 min at 40,000 rpm in a Beckman 45Ti. Pol γB was purified further by loading the supernatant onto a nickel-nitrilotriacetic acid column (Qiagen) followed by S200 Superdex gel filtration column (Amersham Biosciences) as described previously (15). The purified fractions were pooled and dialyzed against a final storage buffer containing 50% glycerol, 50 mm Tris, 100 mm NaCl, 1 mm DTT, 2.5 mm EDTA. The dialyzed protein was then flash-frozen in liquid nitrogen and stored in aliquots at −80 °C. The protein concentration was determined by A280 using an extinction coefficient of 71,940 m−1cm−1 (16)
Cloning Expression and Purification of Pol γA
To observe misincorporation of an incorrect nucleotide by pol γ, it is necessary to use enzyme that is exonuclease-deficient and lacks proofreading activity (17, 18), so all experiments were performed with an exonuclease deficient mutant (D198A, E200A). The primary clone for the WT exonuclease-deficient C terminus His-tagged pol γA was maintained in pUC 19.1 and lacked the first 66 amino acids from the N terminus (including the mitochondrial localization sequence and the poly-glutamine tract). The mutations H932Y and H932A were made on the exonuclease-deficient wt pol γA (del 66) in pUC19.1 by site-directed mutagenesis. All the pol γA clones were transferred to a shuttle vector pBacPak9 (Clontech) by restriction digestion of the pUC 19.1 vector and subsequent ligation to pBacPak9. The pol γA clones in pBAcPak9 were then transferred into the baculovirus genome by recombination between the shuttle vector and the BacPak6 viral DNA (Clontech). Recombinant high viral titers were optimized for protein expression and used to infect SF9 cells at a multiplicity of infection greater than 10. Cells were harvested at 72 h post-infection at 1500 × g for 20 min at 4 °C, flash-frozen in liquid nitrogen, and stored at −80 °C until further use. The cell pellet was resuspended by stirring for 1 h in 6 volumes (mass to volume) of the lysis buffer composed of 0.32 m sucrose, 10 mm HEPES, pH 7.5, 0.5% Nonidet P-40, 3 mm CaCl2, 2 mm MgAc·4H2O, 0.1 mm EDTA, and protease inhibitor mixture from A. G. Scientific Inc. All steps were carried out in 4 °C unless otherwise specified. The nuclei were removed by centrifugation at 1500 × g for 10 min. The salt concentration of the supernatant was increased to 500 mm KCl by slowly adding 3 m KCl buffer (3 m KCl, 20 mm HEPES, pH 8, 5% glycerol). The supernatant was then stirred for 20 min on ice followed by centrifugation at 31,000 × g for 30 min in a Beckman 45-Ti rotor to remove the debris. The clear supernatant was removed and loaded onto a nickel-nitrilotriacetic acid column (Qiagen) followed by a Source S ion exchange column as described previously (3, 16). The Source S fractions containing pol γA were pooled, concentrated, and dialyzed into 50% glycerol buffer (50 mm Tris-HCl, pH7.5, 100 mm NaCl, 1 mm DTT, 2.5 mm EDTA). The dialysate was divided into aliquots, flash-frozen in liquid nitrogen, and stored in −80 °C until further use. The protein concentration was determined by A280 using an extinction coefficient of 243,790 m−1 cm−1 (16). The yield of the protein was typically ∼1–1.5 mg from 1 liter of culture. All experiments were performed with a reconstituted enzyme formed with a 5:1 molar excess of pol γB over pol γA. All references to pol γ are to the reconstituted holoenzyme.
Preparation of DNA
DNA oligomers were synthesized by Integrated DNA Technologies, Inc. and purified using 15% polyacrylamide/7 m urea denaturing gel electrophoresis. A 25-mer primer (5′-GCC TCG CAG CCG TCC AAC CAA CTC A-3′) and a 45-mer template (5′-GGA CGG CAT TGG ATC GAG GTT GAG TTG GTT GGA CGG CTG CGA GGC-3′) were adopted from previous studies to allow comparison between all the studies (6, 19). The dTMP-terminated primer was used for single nucleotide incorporation experiments, and it was 5′-32P-labeled using T4 polynucleotide kinase according to the manufacturer's instructions (Invitrogen). Heating at 95 °C for 5 min terminated the labeling reaction, and excess [γ-32P]ATP was removed using a Biospin 6 column (Bio-Rad). The primer and template were mixed at a 1:1 molar ratio, heated at 95 °C for 2 min, and allowed to cool slowly to room temperature to form the duplex as shown in Fig. 4.
FIGURE 4.

Misincorporation of TTP by H932Y exo− pol γ. Products from the incorporation of TTP (at 1000 μm) were resolved on a 15% polyacrylamide sequencing gel and revealed multiple incorporations past a T:T mismatch. The left axis displays the mismatch formed according to the templating base of the DNA substrate corresponding to each band. Above the figure we show the DNA sequence used. The details of the experiment and the analysis and fitting of these data are presented in Fig. 3B.
Nucleotide Incorporation Assays
Single nucleotide incorporation assays were performed with a RQF-3 rapid quench-flow apparatus (KinTeK Corp.) at 37 °C in a reaction buffer containing 50 mm Tris, pH 7.5, 100 mm NaCl, 12.5 mm MgCl2. For studying the incorporation of dATP and ddATP, one sample loop was loaded with an enzyme-DNA complex (formed with 300–350 nm enzyme, 1.2–1.4 μm small subunit, and 150–200 nm 5′-32P-labeled 25-mer/45-mer DNA duplex) in the reaction buffer without MgCl2. Another loop was loaded with nucleotide in the reaction buffer with 25 mm MgCl2. The reactions were started by rapidly mixing the two reactants (1:1) and then were quenched by mixing with 0.5 m EDTA (final concentration) after various time intervals. For the misincorporation experiments, preformed enzyme-DNA complex (200–300 nm enzyme, 0.8–1.2 μm small subunit, and 150 nm 5′-32P-labeled 25-mer/45-mer DNA duplex) in the reaction buffer without MgCl2 was used. For longer timescale misincorporation experiments (varying from 20 min to 1.5 h), manual hand mixing and quenching was done instead of using the rapid quench flow apparatus. In all assays a maximum of 5 mm dNTP (final concentration after mixing) was not exceeded to avoid complications due to nonspecific inhibition (17, 20).
The products from the assays described above were resolved on a 15% denaturing polyacrylamide-sequencing gel, and then the dried gel was exposed to a phosphor screen. The screen was scanned using a Storm 860 scanner (GE Healthcare), and the amount of product formation at each time point was analyzed using the ImageQuant software (GE Healthcare). The concentration of the product was calculated from the concentration of the starting isotope labeled DNA primer times the fraction of the materials in the product band.
Global Data Fitting
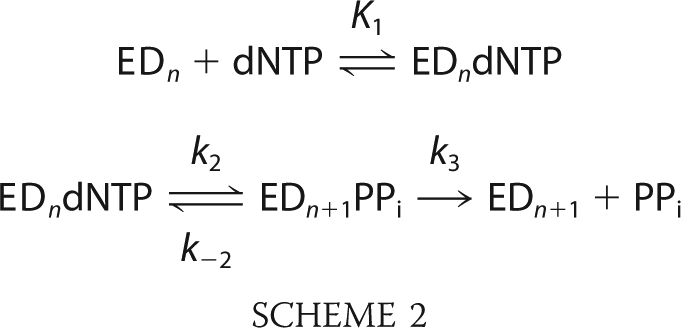
All of the data presented in this paper have been fit globally using the KinTek Explorer program (KinTek Corp.) based upon numerical integration of rate equations for the complete model. In fitting data to Schemes 1 or 2, we also included previously estimated rates for DNA binding and dissociation in constructing a comprehensive model. Concentrations of active enzyme were adjusted slightly (±10%) in deriving the best fit.
In addition to S.E. values, we have also presented upper and lower limits for each of our fitted parameters in the tables. These limits are derived based upon a threshold in the confidence contours equal to 10% increase in χ2, as described (21). This method of estimating errors is more robust than traditional S.E. analysis, which tends to underestimate errors and often fails to reveal parameters in the model that are under-constrained by the data.
In fitting data to Scheme 1 or Scheme 2, the equilibrium constant for the initial complex formation was estimated by assuming diffusion-limited nucleotide binding (k1 = 100 μm−1 s−1) and allowing the dissociation rate to vary during fitting to afford calculation of the equilibrium constant (K1 = k1/k−1). In some cases, during global fitting of data within a concentration series, a small background correction (less than 10% of the signal) was applied to individual traces based upon optimal fitting the data to the model.
Calculation of kcat and kcat/Km
Kinetic parameters kcat and kcat/Km were derived for Scheme 1 by the simple relationship kcat = kpol and Km = Kd,app. However, for the data fit to Scheme 2, the bulk parameters were calculated from the complete solution for the three-step model.
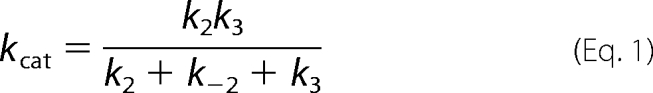 |
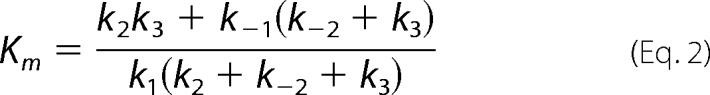 |
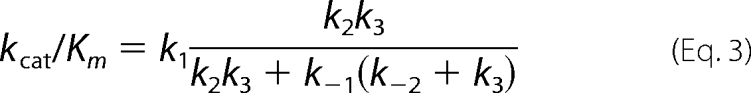 |
RESULTS
Kinetics of Single Nucleotide Incorporation Measured by Quench-flow Experiments
The kinetics of correct nucleotide incorporation were measured by rapid quench-flow methods. An enzyme-DNA complex was rapidly mixed with a solution containing the correct incoming nucleotide (dATP) at various concentrations. The experiments were performed under single turnover conditions in that the enzyme concentration exceeded the concentration of DNA. The nucleotide concentration dependence of the rate of polymerization provided estimates of an apparent nucleotide dissociation constant (Kd,app) and a maximum rate of nucleotide incorporation (kpol) according to Scheme 1 as described previously (17, 22). Fig. 2, A and B, show the time courses of the product (26-mer) formation at several nucleotide concentrations observed for the WT and the H932Y mutant, respectively. The data defining nucleotide incorporation were fit globally to the mechanism shown in Scheme 1 to obtain the maximum nucleotide incorporation rate (kpol) and the apparent nucleotide dissociation constant, Kd,app (EDn refers to the enzyme-DNA complex where the primer is n residues in length).
FIGURE 2.

Kinetics of incorporation of dATP and ddATP for the WT and H932Y mutant. For each concentration series, a preformed enzyme-DNA complex ([enzyme] > [DNA duplex]) was rapidly mixed with Mg2+ and various concentrations of dATP or ddATP. In each experiment the final concentrations of the enzyme and DNA after mixing were 150–175 and 75–100 nm, respectively. In globally fitting each data set, the concentration of active enzyme was adjusted to fit the amplitude. A, incorporation of dATP for WT exo− pol γ at various concentrations (0.2 (●), 0.5 (○), 1.5 (■), 3 (□), 5.5 (▴), 8.5 (△) μm) was globally fit to Scheme 1 yielding a kpol of 30 ± 2 s−1 and a Kd,app of 0.7 ± 0.14 μm. B, incorporation of dATP for H932Y exo− pol γ at various concentrations (2.5 (●), 7.5 (○), 20 (■), 40 (□), 100 (▴), 500 (△) μm from bottom to top) was globally fit to Scheme 1 yielding a kpol of 28.6 ± 2.9 s−1 and a Kd,app of 103 ± 15 μm. C, incorporation of ddATP for WT exo− pol γ at various concentrations (0.01 (●), 0.025 (○), 0.05 (■), 0.1 (□), and 5 (▴) μm) was globally fit to Scheme 1 yielding a kpol of 1.8 ± 0.2 s−1 and a Kd,app of 0.42 ± 0.06 μm. D, incorporation of ddATP for H932Y exo− polγ at various concentrations (0.5 (●), 2 (○), 5 (■), 15 (□), and 500 (▴) μm) was globally fit to Scheme 1 yielding a kpol of 1 ± 0.14 s−1 and a Kd,app of 61 ± 10 μm.
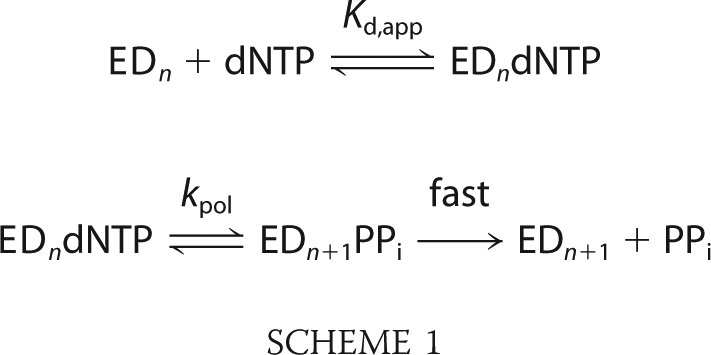 |
According to this simplified model, the polymerization rate is presumed to be governed by a single rate-limiting step (kpol), and the ground state binding occurs as a rapid equilibrium. After the incorporation of a correct nucleotide, pyrophosphate release and translocation appear to be fast (17, 22); therefore, the measured ratio of kpol/Kd,app defines kcat/Km, the specificity constant governing incorporation during processive polymerization. It should be noted that the apparent dissociation constant (Kd,app) cannot be interpreted as a simple substrate dissociation constant when binding occurs in multiple steps; rather, Kd,app more accurately reflects a Michaelis constant reflecting the relative rates of substrate binding and incorporation rather than the ground state nucleotide binding (9, 10). Regardless, kpol/Kd,app provides an accurate measurement of kcat/Km governing sequential nucleotide incorporation during processive synthesis. Kinetic parameters for the WT enzyme determined from the global fitting were in agreement with previously published data (6, 19). Interestingly, the H932Y mutant showed a maximum rate of polymerization similar to that of the WT enzyme, but kcat/Km was severely affected, (∼150-fold) as summarized in Table 1.
TABLE 1.
Effect of H932Y/A mutations on the specificity constant for incorporation of dATP and ddATP
Kinetic parameters governing correct (dATP) and dideoxynucleotide (ddATP) are summarized as derived from the data shown in Figs. 2 and 5. The S.E. estimates (±) were derived from the covariance matrix obtained by nonlinear regression. The upper and lower limits shown in parentheses were derived by confidence contour analysis (21).
| Enzyme/nucleotide | kpol | Kd,app | kpol/Kd,app | Discrimination |
|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 | ||
| WT | ||||
| dATP | 30 ± 2 | 0.7 ± 0.14 | 43 ± 9 | 10 |
| (29–33) | (0.62–0.84) | |||
| ddATP | 1.8 ± 0.2 | 0.42 ± 0.06 | 4.3 ± 0.8 | |
| (1.6–2) | (0.36–0.48) | |||
| H932Y | ||||
| dATP | 28.6 ± 2.9 | 103 ± 15 | 0.3 ± 0.05 | 15 |
| (27–30) | (97–110) | |||
| ddATP | 1 ± 0.14 | 61 ± 10 | 0.02 ± 0.003 | |
| (0.87–1.3) | (52–82) | |||
| H932A | ||||
| dATP | 23 ± 2.8 | 39 ± 6.4 | 0.6 ± 0.1 | 30 |
| (23–27) | (39–49) | |||
| ddATP | 0.7 ± 0.04 | 38 ± 2.6 | 0.02 ± 0.002 | |
| (0.67–0.75) | (35–42) | |||
Kinetics of Incorporation of Dideoxynucleotides
The incorporation of ddATP (the active metabolic product of the anti-HIV nucleoside analog didanosine, didexoyinosine) was measured by rapid quench flow methods similar to the measurements done for the incorporation of dATP. Fig. 2, C and D, show the time courses of the product formation observed for the WT and the H932Y mutation respectively with ddATP. The data defining nucleotide incorporation kinetics were then fit globally to the mechanism shown in Scheme 1 to obtain the maximum incorporation rate and the apparent dissociation constant, Kd,app (summarized in Table 1). It is important to note that for the WT enzyme, the lower concentration data do not reach the same end point because the nucleotide concentration was less than the enzyme concentration; this is taken into account during global fitting based upon numerical integration of the rate equations (23). The maximum rates of incorporation of ddATP by the mutants were close to the WT, but the kpol/Kd,app was reduced by >200-fold. The discrimination, defined as the ratio of kcat/Km for the correct nucleotide relative to that of the analog, was not significantly affected by mutating the H932. The decrease in kcat/Km for ddATP is mostly offset by a similar decrease in kcat/Km for dATP incorporation.
Kinetics of Nucleotide Misincorporation
Misincorporation experiments were performed to investigate the discrimination against mismatches exhibited by the H932Y mutant as compared with that of wild-type enzyme. We examined the misincorporation of TTP, dCTP, and dGTP opposite the template TMP. In each experiment the time course of the product formation (26-mer or greater) was quantified. In some cases multiple nucleotides were incorporated onto the primer strand such that products of up to 35 nucleotides long were observed; therefore, all of the extended primers were included in the total concentration of product, which was then plotted versus time. Misincorporation parameters for WT human pol γ have been published previously (18) and were used as a reference for our experiments but were repeated and reanalyzed with our updated global fitting methods. The kinetics of formation of a T:T mismatch and C:T mismatch by wild-type pol γ are shown in Fig. 3, A and B, respectively (G:T mismatch results are summarized in Tables 2 and 3). Each of these sets of data were globally fit to Scheme 1. The results are consistent with those previously reported and show that incorrect nucleotides have decreased rates of nucleotide binding and incorporation.
FIGURE 3.

Kinetics of misincorporation by WT enzyme and H932Y mutant. For each concentration dependence, a preformed enzyme-DNA complex ([enzyme] > [DNA duplex]) was rapidly mixed with Mg2+ and various concentrations of incorrect nucleotide. The time course of product formation was then fit globally. In each experiment, the final concentrations of the enzyme and DNA after mixing were 100–150 and 75 nm, respectively. In globally fitting each data set, the concentration of active enzyme was adjusted to fit the amplitude. A, formation of a T:T mismatch by WT exo− pol γ at each TTP concentration (1.5 (●), 5 (○), 15 (■), 50 (□), and 250 (▴) μm) was globally fit to the mechanism shown in Scheme 1, yielding an apparent Kd of 81.8 ± 10.9 μm and kpol of 0.01 ± 0.004 s−1. B, formation of a C:T mismatch by WT exo− pol γ at each dCTP concentration (15 (●), 50 (○), 250 (■), and 1000 (□) μm) was fit globally to the mechanism shown in Scheme 1, yielding an apparent Kd of 1030 ± 193 μm and kpol of 0.06 ± 0.018 s−1. C, formation of a T:T mismatch by H932Y exo− mutant at each TTP concentration (15 (●), 50 (○), 125 (■), 250 (□), 1000 (▴), and 5000 (△) μm) was fit globally (solid line) to the mechanism shown in Scheme 2, yielding an apparent Kd of 1630 ± 310 μm, k2 of 0.1 ± 0.02 s−1, k−2 of 0.01 ± 0.003 s−1, and k3 of 0.0004 ± 0.0005 s−1. The dashed line indicates an attempt to fit the data with the mechanism shown in Scheme 1, showing that it cannot account for the concentration dependence of the rate and amplitude. D, formation of a C:T mismatch by H932Y exo− mutant at various dCTP concentrations (175 (●), 375 (○), 750 (■), 1500 (□), and 4000 (▴) μm) were globally fit to the mechanism shown in Scheme 2, yielding an apparent Kd of 22200 ± 3420 μm, k2 of 0.02 ± 0.005 s−1, k−2 of 0.0004 ± 0.003 s−1, and k3 of 0.0003 ± 0.002 s−1. Traces for the formation of a G:T mismatch are not shown, but all time courses were fit to the mechanism shown in Scheme 1, and the resulting parameters are summarized in Table 2.
TABLE 2.
Effect of H932Y/A mutations on kinetic parameters governing discrimination
Kinetic parameters governing misincorporation opposite a template T catalyzed by H932Y and H932A are summarized as derived from the data shown in Figs. 3 and 5. The S.E. estimates (±) were derived from the covariance matrix obtained by nonlinear regression. The upper and lower limits shown in parentheses were derived by confidence contour analysis (21).
| Enzyme/nucleotide | 1/K1 | k2 | k−2 | k3 |
|---|---|---|---|---|
| μm | s−1 | s−1 | s−1 | |
| WT | ||||
| TTP | 81.8 ± 10.9 | 0.01 ± 0.004 | (0) | Fast |
| (40–90) | (0.006–0.01) | |||
| dCTP | 1,030 ± 193 | 0.06 ± 0.018 | (0) | Fast |
| (810–1,300) | (0.05–0.08) | |||
| dGTP | 1,300 ± 1,600 | 6.6 ± 7.4 | (0) | Fast |
| (836–4,100) | (4.5–20) | |||
| H932Y | ||||
| TTP | 1,630 ± 310 | 0.1 ± 0.02 | 0.01 ± 0.003 | 0.0004 ± 0.0005 |
| (1,300–1,920) | (0.08–0.1) | (0.01–0.014) | ( 0.0003–0.0006) | |
| dCTP | 22,200 ± 3,420 | 0.02 ± 0.005 | 0.0004 ± 0.003 | 0.0003 ± 0.002 |
| (17,800–6.7 × 1011) | (0.015–8.3 × 105) | (0.0002–0.002) | (2.8 × 10−8-0.001) | |
| dGTP | 24,000 ± 9,600 | 1.2 ± 0.4 | (0) | Fast |
| (18,300–1.5 × 105) | (1–7) | |||
| H932A | ||||
| TTP | 9,400 ± 2,700 | 0.47 ± 0.15 | 0.013 ± 0.0056 | 0.0003 ± 0.0004 |
| (6,600–26,500) | (0.39–1.86) | (0.01–0.02) | (2 × 10−5-0.0007) | |
| dCTP | 40,900 ± 9,470 | 0.038 ± 0.012 | 0.0009 ± 0.0006 | 0.0005 ± 0.001 |
| (26,000–1.9 × 105)a | (0.02–0.18) | (0.0005–0.0017) | (0.0002–0.0008) | |
| dGTP | 5,150 ± 1,500 | 0.27 ± 0.06 | (0) | Fast |
| (4,100–10,000) | (0.23–0.45) | |||
a No upper limit was found up to the value reported.
TABLE 3.
Effect of H932Y/A mutations on discrimination against mismatches
Kinetic parameters governing misincorporation opposite a template T catalyzed by H932Y and H932A are summarized as derived from the data shown in Figs. 3 and 5. The S.E. estimates (±) were derived by error propagation from the values shown in Table 2.
| Enzyme/nucleotide | kcat | Km | kcat/Km | Discrimination |
|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 | ||
| WT | ||||
| TTP | 0.01 ± 0.004 | 81.8 ± 10.9 | 0.0001 ± 0.00005 | 430,000 |
| dCTP | 0.06 ± 0.018 | 1030 ± 193 | 0.00006 ± 0.00002 | 717,000 |
| dGTP | 6.6 ± 7.4 | 1300 ± 1600 | 0.005 ± 0.008 | 8,600 |
| H932Y | ||||
| TTP | 0.0004 ± 0.0005 | 154 ± 60 | 0.000002 ± 0.000003 | 150,000 |
| dCTP | 0.0003 ± 0.002 | 750 ± 3875 | 0.0000004 ± 0.000003 | 750,000 |
| dGTP | 1.2 ± 0.4 | 24000 ± 9600 | 0.00005 ± 0.00003 | 6,000 |
| H932A | ||||
| TTP | 0.0003 ± 0.0004 | 260 ± 150 | 0.000001 ± 0.000002 | 600,000 |
| dCTP | 0.0005 ± 0.001 | 1450 ± 1330 | 0.0000003 ± 0.0000007 | 2,000,000 |
| dGTP | 0.27 ± 0.06 | 5150 ± 1500 | 0.00005 ± 0.00002 | 12,000 |
Analysis of the misincorporation catalyzed by H932Y showed some unexpected results. First, it was noted that misincorporation of TTP onto a template T was followed by additional misincorporation events (Fig. 4). According to the sequence of the template, the enzyme formed two sequential T:G mismatches, a correct T:A base pair and then a T:G and T:C mismatch. The sum of all products formed was used to quantify the kinetics of incorporation of the first T:T mismatch as shown in Fig. 3C. The kinetics of formation of the C:T mismatch are shown in Fig. 3D.
The data in Fig. 3C were initially fit to the model shown in Scheme 1, but it was immediately clear that the minimal model could not account for the trend observed in the data (Fig. 3C, dashed lines). Particularly for the T:T and C:T mismatches, it is clearly evident that the amplitude of product formation is concentration-dependent, whereas the model requires that the reaction goes to completion at each nucleotide concentration because the release of pyrophosphate drives the reaction forward even at subsaturating nucleotide concentrations. These data required a model in which the chemistry step is reversible and comes to an equilibrium that is linked to nucleotide binding. This interpretation requires that pyrophosphate release is slow or readily reversible (24, 25).
The data were fit to the model shown in Scheme 2, where k−2 > 0, and k3 is slow, ranging from 0.0002 to 0.0005 s−1. In this minimal model, the release of pyrophosphate is assumed to be irreversible, but further experiments are needed to address whether the release is slow or readily reversible.
 |
Data defining the kinetics of formation of a G:T mismatch by H932Y could be adequately fit to the simple model shown in Scheme 1 in that the reaction appears to go to the same end point at all dGTP concentrations. This could be due to faster pyrophosphate release or a slower rate constant for the reverse of chemistry (k−2) in forming the G:T mismatch. The kinetic parameters governing misincorporation are summarized in Tables 2 and 3.
Confidence Contour Analysis
Global data-fitting based upon numerical integration of rate equations is a powerful method in that parameters are derived by directly fitting to a complete model without simplifying assumptions, and the fitting requires that the model account for the concentration dependence of both the rate and amplitude of the reaction (23). However, fitting based upon computer simulation makes it too easy to include parameters in a model that are not well constrained by the data. To address whether the data are sufficient to define four kinetic parameters in fitting to Scheme 2, we performed a confidence contour analysis on the data from Fig. 3C to get the results shown in Fig. 5 (21). In this analysis, the extent to which each parameter can be varied is explored while allowing all other parameters to float in fitting the data. The results are then scored by the minimum χ2 value attainable, which is then plotted on a three-dimensional profile versus each pairwise combination of kinetic parameters. The results shown in Fig. 5 demonstrate that all four kinetic parameters are constrained by the data as indicated by the red central zone defining the area of good fit. The margin shown by the yellow band between the red and green zones defines a boundary representing a 10% increase in χ2, which is used to estimate upper and lower confidence intervals on each of the parameters. This analysis demonstrates that the concentration dependence of the rate and amplitude shown in Fig. 3C are sufficient to define the four kinetic parameters in Scheme 2. Confidence contour analysis was used to estimate confidence intervals for all of the experiments as summarized in the tables.
FIGURE 5.

Confidence contours for the global fit to four rate constants showing all six pairwise combinations of the four rate constants. The results were derived by globally fitting the data collected as shown in Fig. 3B. The central red zone shows the area of good fit, and the yellow band between the red and green zones shows the threshold representing a 10% increase in χ2, which is used to set upper and lower confidence limits on each of the kinetic parameters (21).
Kinetic Analysis of the H932A Mutant
To further explore the role of His-932 in catalysis, we examined the effect of an H932A mutation to get the results summarized in Fig. 6 and Tables 1–3. As shown in Fig. 6A, the H932A mutation showed a 72-fold reduction in the kcat/Km for correct incorporation (A:T) with respect to the WT, indicating that the tyrosine substitution had a more severe effect than the alanine substitution. The more severe effect of the H932Y substitution could be due to the additional steric hindrance imposed on the interactions important for nucleotide binding. The H932A mutant discriminated against the incorporation of ddATP 30-fold relative to dATP, which is only 2-fold greater than seen by H932Y (Fig. 6B, Table 1). Like H932Y, the kinetics of incorporation of T:T and C:T mismatches (Fig. 6, C and D) showed an amplitude concentration dependence and required fitting to the model invoking slow pyrophosphate release (Scheme 2). In each case, data fitting required a relatively slow rate of pyrophosphate release (k3) and slow reversal of chemistry (k−2), less than the rate of the forward chemistry step (k2) as summarized in Table 2. Reversal of chemistry and slow pyrophosphate release reduced the kcat/Km value governing net incorporation (Table 3). H932A showed a 2–3-fold increase in fidelity compared with H932Y.
FIGURE 6.

Kinetics of incorporation of for the H932A mutant. For each concentration series, a preformed enzyme-DNA complex ([enzyme] > [DNA duplex]) was rapidly mixed with Mg2+ and various concentrations of nucleotide. The time course of product formation was then fit globally (solid lines). In each experiment the final concentrations of the enzyme and DNA after mixing were 100–175 and 75–100 nm, respectively. In globally fitting each data set, the concentration of active enzyme was adjusted to fit the amplitude. A, incorporation of dATP for H932A exo− pol γ at various concentrations (0.2 (●), 0.5 (○), 2 (■), 4 (□), 7 (▴), 11 (△), 20 (▾), 30 (▿), and 55 (♦) μm) was globally fit to Scheme 1 yielding a kpol of 23 ± 2.8 s−1 and a Kd,app of 39 ± 6.4 μm. B, incorporation of ddATP for H932A exo− pol γ at various concentrations (0.5 (●), 2 (○), 5 (■), 15 (□), and 500 (▴) μm) was globally fit to Scheme 1 yielding a kpol of 0.7 ± 0.04 s−1 and a Kd,app of 38 ± 2.6 μm. C, formation of a T:T mismatch by H932A exo− pol γ at each TTP concentration (15 (●), 50 (○), 125 (■), 250 (□), and 5000 (▴) μm) was globally fit to the mechanism shown in Scheme 2, yielding an apparent Kd of 9400 ± 2700 μm, k2 of 0.47 ± 0.15 s−1, a k−2 of 0.013 ± 0.0056 s−1, and a k3 of 0.0003 ± 0.0004 s−1. D, formation of a C:T mismatch by H932A exo− pol γ at each dCTP concentration (175 (●), 375 (○), 750 (■), 1500 (□), and 5000 (▴) μm) was also globally fit to the mechanism shown in Scheme 2, yielding an apparent Kd of 40900 ± 9470 μm, k2 of 0.038 ± 0.012 s−1, k−2 of 0.0009 ± 0.0006 s−1, and k3 of 0.0005 ± 0.001. Traces for the formation of a G:T mismatch are not shown, but all time courses were fit to the mechanism shown in Scheme 1, and the resulting parameters are summarized in Tables 2 and 3.
DISCUSSION
Role of His-932 in dNTP Binding and Incorporation
The most conserved amino acid residue of the pre-motif B is an invariantly positively charged amino acid, usually a Lys or an Arg in family B DNA polymerases. In family A DNA polymerases, this residue is histidine with the only exception being SpO2 DNA polymerase, which has a tyrosine in that position (26). The active site structure of T7 DNA polymerase suggests that His-932 in pol γA is within H-bonding distance of the β phosphoryl oxygen of the incoming dNTP. Furthermore, the His-932 residue lies between Arg-943 and Tyr-951 (His-506, Arg-518, and Tyr-526 in T7 DNA polymerase, respectively), which are important for nucleotide selectivity and dideoxynucleotide discrimination, suggesting that mutation of His-932 would interfere with the interactions with the incoming nucleotide. Surprisingly, our data show that mutation of the His-932 does not significantly affect the kpol but severely affects kcat/Km, the effective nucleotide binding rate and the Km value for incoming nucleotide. Thus, His-932 is an important residue to enhance nucleotide incorporation, although it does not affect the rate of catalysis. In the absence of His-932, Tyr-951 could still H-bond to the β phosphate and, thus, along with metal ion B, contribute to stabilization of the transition state of the reaction (Fig. 1). This may explain why mutations H932Y/A do not significantly alter the maximum rate of polymerization, kpol. However H-bonding interaction between the His-932 and β phosphate appears to play a key role in the dynamics of nucleotide binding underlying the specificity constant (kcat/Km). Overall, the substitution of His-932 with a tyrosine had a more severe effect in slowing nucleotide binding than alanine, suggesting that the presence of the tyrosine residue may create additional steric effects or otherwise alter active site geometry.
One cannot simply interpret changes in the apparent Kd derived from the rapid quench studies because the value probably does not represent a true Kd for nucleotide binding but, rather, should be considered to represent a Km value governing incorporation. In studies on other pol A family DNA polymerases, nucleotide binding induces a rapid change in enzyme structure that governs the kinetics of correct base pair incorporation but comes to equilibrium preceding mismatch incorporation (9, 10, 27, 28). Therefore, calculations of the ratio of apparent Kd values for correct versus incorrect base incorporation are difficult to interpret, and our discussion of mechanism is focused on considering relative changes in kpol and kpol/Kd.
Residue Tyr-526 in T7 DNA polymerase (homologous to Tyr-951 in pol γ) has been shown to be essential for the incorporation of dideoxynucleoside triphosphates in that substitution with phenylalanine leads to significant discrimination against dideoxynucleotides (29). Tyrosine 951 is highly conserved in motif B of all the mitochondrial DNA polymerases and appears to facilitate the incorporation of nucleoside analogs. Because the His-932 residue is positioned close to Tyr-951 and both of these amino acid residues can H-bond to the β phosphate, we measured the incorporation of ddATP for the H932Y and H932A mutants and compared it to the wild type enzyme. Didanosine (dideoxyinosine) is used for the treatment of HIV infections but has significant toxic side effects due to its conversion to ddATP and subsequent incorporation in to human mitochondrial DNA (30, 31). Our results show that His-932 only contributes marginally (1.5–3-fold) to discrimination against ddA.
Mutations H932Y/A did not have any significant effect on the overall enzyme fidelity of incorporation measured in single turnover kinetic studies. The specificity constants for cognate and non-cognate base incorporation were reduced by the mutations by a similar fraction so the net effect on discrimination was minimal (Table 3). Interestingly for both mutations, H932Y/A, we observed that during incorporation of a T:T or C:T mismatch, the release of pyrophosphate was apparently slow with an average rate of ∼0.0003 s−1. This slow release of pyrophosphate and the reversible chemistry step was proposed to account for the nucleotide concentration dependence of the reaction amplitude (Figs. 3, C and D, and 5, C and D). Slow product release and reversal of chemistry drastically decreases the kcat/Km for the incorporation of a mismatch. This same effect has previously been seen in pol γ with the incorporation of 8-oxo-dGTP (24) and AZT-triphosphate (25), where kinetic studies demonstrated that pyrophosphate release was slow.
All of the data in this manuscript were fit globally based upon computer simulation (23). Fitting data directly to the model based upon numerical integration of rate equations has several advantages over the conventional data fitting (21). Conventional data fitting usually requires derivation of equations that are based upon simplifying assumptions that may not be valid. For example, pre-steady state burst data are usually fit to the sum of an exponential and a linear term, but the linear phase is not usually linear, and errors in the linear fit lead to errors in the rate and amplitude of the exponential phase. In contrast, we were able to fit a concentration series to a single, complete model without simplifying assumptions. Global data-fitting directly to the model requires that the concentration dependence of the rate and amplitude of the reaction are accounted for, and this allows the kinetic parameters to be better constrained by the data and eliminates errors committed when reaction amplitudes are ignored (32). Moreover, by using global fitting, more complex data can be analyzed rigorously. Conventional fitting of data to multiple exponential functions is unreliable, but fitting data directly to a model constrains the fitting of a series of experiments performed at multiple concentrations to conform to the underlying function.
Much of the data presented in this manuscript would have been difficult to interpret using conventional data fitting methods. For example, in Fig. 2C, concentrations of nucleotide covered a range below and above the enzyme concentration; therefore, conventional data fitting would yield confusing results with the rate appearing to decrease and then increase as a function of nucleotide concentration. Fitting by computer simulation handles the data by taking into account the concentrations of all reactants as the reaction proceeds to completion. Finally, by fitting the data globally, we can avoid fitting the data to multiple exponential functions, which is often problematic (23). For example, the slow release of pyrophosphate seen in the H932Y/A mutant misincorporation studies shows complicated kinetics where enzyme-bound product equilibrates with nucleotide binding at the active site. This causes the observed rate to be nearly constant or decrease as the amplitude increases as a function of nucleotide concentration. This effect can be easily resolved by fitting the data globally but is difficult to resolve with conventional fitting methods, as illustrated by our earlier work on the incorporation of 8-oxo-dGTP (24).
We used confidence contour analysis to establish that the fitted parameters were well constrained by the data when fitting to either Scheme 1 or 2. The confidence contour analysis does not prove the model but, rather, can only be used to evaluate whether the model is adequately constrained by the data. It does not eliminate a more complex model. In particular, it should be noted that the full time course of all sequential misincorporation events seen with the H932Y mutant (Figs. 3, C and D, and 4) could also be fit to a model in which pyrophosphate release was readily reversible. The model also required explicitly including DNA translocation so that pyrophosphate could immediately rebind after releasing from the E-DNA complex, but after translocation only dNTP could bind. Although this model could account for the data, the parameters were not well constrained, requiring additional experiments to define the kinetics of pyrophosphate release and rebinding.
It should also be noted that the kinetic constants for misincorporation reported here were determined using the exonuclease-deficient enzymes. Quantification of the full effect of mutation on the fidelity requires analysis of the rates of excision by the exonuclease versus extension by the addition of the next correct nucleotide. These experiments are currently under way.
Previous kinetic analyses have shown that substrate-induced conformational change plays a crucial role in the discrimination between the correct and incorrect base pairs by controlling whether a nucleotide will be incorporated or rapidly released (9, 27). Perhaps His-932 plays a critical role in the conformational change presumed to occur after incorporation and preceding product release and translocation. Generally the steps after nucleotide incorporation, PPi release and translocation, are assumed to be fast and to be limited by the rate of a conformational change from closed to open state after nucleotide incorporation (9). By making pyrophosphate release slow or readily reversible, the enzyme can retain the product of the reaction, ultimately favoring the reverse reaction and substrate release. In the case of 8-oxo-dGTP and AZT incorporation it was suggested that this phenomenon provided a mechanism used by the enzyme to reduce the incorporation of these nucleotides (24, 25). Because the His-932 H-bonds to the β phosphate, the positive charge stabilizes the leaving group (pyrophosphate), and one might expect that mutation of His-932 would lead to increased rates of pyrophosphate release. However, in the absence of this interaction, the rate of pyrophosphate release appears slower. It is possible that there is a conformational change after chemistry that misaligns residues to slow catalysis and promotes product release (27). According to this model, one might predict that His-932 is involved in this conformational change step. However, it is not easy to explain why this effect is observed for the misincorporation events only and not in the incorporation of the correct dNTP or the ddNTP. It is also important to note that we observed multiple misincorporation events after formation of the T:T mismatch by H932Y (Fig. 3C). This observation implies a model in which pyrophosphate release is faster but readily reversible, which requires additional testing.
The physiological consequences of the H932Y mutation may be attributable to the reduction in kcat/Km, which would slow the rate of mitochondrial DNA replication. However, kpol is unaffected by the mutation, whereas the Km is increased to a value of 100 μm, which is comparable with the physiological nucleotide concentration; therefore, the mutation should slow the rate of replication by only 2-fold. Although a 150-fold reduction in replication rate may be lethal, a 2-fold reduction in rate may be tolerated well enough for survival but could lead to slow onset effects. Thus, for a mitochondrial mutation to appear in the human population, it must not be so severe as to prevent growth and development. On the other hand, more subtle effects of the mutation must lead to cumulative damage that over time cause reduced mitochondrial function. Accordingly, quantification of the effects of disease related mutations in Pol γ require careful measurement. Physiological effects are probably not attributable to changes in fidelity of incorporation. However, it is significant that the H932Y mutant rapidly extended mismatches, which would decrease the fidelity contribution of the proofreading exonuclease, and this could have significant physiological effects over time, leading to PEO.
Correlation of a POLG mutation with its induced clinical phenotype and the biochemical defects is complex. In general, most mutations following a dominant mode of inheritance tend to show dominant negative biochemical behavior. Because human cells express both copies of POLG, expression of a single WT copy of POLG is sufficient to avoid disease (33). Recessive mutations in POLG produce defective polymerases that minimally interfere with the mitochondrial DNA replication by the WT enzyme as opposed to the enzyme produced by a PEO allele. This is supported by the fact that a number of mitochondrial disorders result when a recessive mutation is heterozygous with other POLG mutations and the clinical severity and phenotype is determined by the nature of the mutation in the other POLG allele. For example, the recessive mutation A467T is involved in a large number of mitochondrial disorders and is heterozygous with other mutations, which could determine the clinical severity and the phenotype (34, 35). H932Y is found as a compound with T251I/P587L and G1051R, and thus, the clinical phenotype and severity of H932Y mutation would be largely modulated by these mutations on the other allele. Although the defective polymerase from recessive mutation may not directly compete with the WT enzyme, it may compete indirectly through interaction with other protein in the replication fork. Knowledge of the complex interactions of all the proteins in the mitochondrial replisome may be needed to refine the model to address this more difficult question.
Acknowledgment
The rapid chemical quench-flow instruments and KinTek Explorer software were donated by KinTek Corporation. Financial conflict of interest: K. A. Johnson is President of KinTek Corp.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM 044613 (to K. A. J.). This work was also supported by the Welch Foundation (F-1604) (to K. A. J.).
- pol
- polymerase
- PEO
- opthalmoplegia
- dd
- dideoxy.
REFERENCES
- 1.Roitberg A. E., Okur A., Simmerling C. (2007) J. Phys. Chem. B. 111, 2415–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suomalainen A., Majander A., Wallin M., Setälä K., Kontula K., Leinonen H., Salmi T., Paetau A., Haltia M., Valanne L., Lonnqvist J., Peltonen L., Somer H. (1997) Neurology 48, 1244–1253 [DOI] [PubMed] [Google Scholar]
- 3.Lee Y. S., Kennedy W. D., Yin Y. W. (2009) Cell 139, 312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steitz T. A., Smerdon S., Jäger J., Wang J., Kohlstaedt L. A., Friedman J. M., Beese L. S., Rice P. A. (1993) Cold Spring Harb. Symp. Quant. Biol. 58, 495–504 [DOI] [PubMed] [Google Scholar]
- 5.Doublié S., Tabor S., Long A. M., Richardson C. C., Ellenberger T. (1998) Nature 391, 251–258 [DOI] [PubMed] [Google Scholar]
- 6.Johnson A. A., Tsai Y., Graves S. W., Johnson K. A. (2000) Biochemistry 39, 1702–1708 [DOI] [PubMed] [Google Scholar]
- 7.Li Y., Korolev S., Waksman G. (1998) EMBO J. 17, 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doublié S., Sawaya M. R., Ellenberger T. (1999) Structure Fold. Des. 7, R31-R35 [DOI] [PubMed] [Google Scholar]
- 9.Tsai Y. C., Johnson K. A. (2006) Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellinger M. W., Johnson K. A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 7734–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graziewicz M. A., Longley M. J., Bienstock R. J., Zeviani M., Copeland W. C. (2004) Nat. Struct. Mol. Biol. 11, 770–776 [DOI] [PubMed] [Google Scholar]
- 12.Di Fonzo A., Bordoni A., Crimi M., Sara G., Del Bo R., Bresolin N., Comi G. P. (2003) Hum. Mutat. 22, 498–499 [DOI] [PubMed] [Google Scholar]
- 13.Mancuso M., Filosto M., Bellan M., Liguori R., Montagna P., Baruzzi A., DiMauro S., Carelli V. (2004) Neurology 62, 316–318 [DOI] [PubMed] [Google Scholar]
- 14.Stumpf J. D., Bailey C. M., Spell D., Stillwagon M., Anderson K. S., Copeland W. C. (2010) Hum. Mol. Genet. 19, 2123–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carrodeguas J. A., Theis K., Bogenhagen D. F., Kisker C. (2001) Mol. Cell 7, 43–54 [DOI] [PubMed] [Google Scholar]
- 16.Yakubovskaya E., Chen Z., Carrodeguas J. A., Kisker C., Bogenhagen D. F. (2006) J. Biol. Chem. 281, 374–382 [DOI] [PubMed] [Google Scholar]
- 17.Patel S. S., Wong I., Johnson K. A. (1991) Biochemistry 30, 511–525 [DOI] [PubMed] [Google Scholar]
- 18.Johnson A. A., Johnson K. A. (2001) J. Biol. Chem. 276, 38090–38096 [DOI] [PubMed] [Google Scholar]
- 19.Lee Y. S., Lee S., Demeler B., Molineux I. J., Johnson K. A., Yin Y. W. (2010) J. Biol. Chem. 285, 1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong I., Patel S. S., Johnson K. A. (1991) Biochemistry 30, 526–537 [DOI] [PubMed] [Google Scholar]
- 21.Johnson K. A., Simpson Z. B., Blom T. (2009) Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 22.Johnson K. A. (1993) Annu. Rev. Biochem. 62, 685–713 [DOI] [PubMed] [Google Scholar]
- 23.Johnson K. A., Simpson Z. B., Blom T. (2009) Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 24.Hanes J. W., Thal D. M., Johnson K. A. (2006) J. Biol. Chem. 281, 36241–36248 [DOI] [PubMed] [Google Scholar]
- 25.Hanes J. W., Johnson K. A. (2007) Nucleic Acids Res. 35, 6973–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Truniger V., Lázaro J. M., Esteban F. J., Blanco L., Salas M. (2002) Nucleic Acids Res. 30, 1483–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson K. A. (2008) J. Biol. Chem. 283, 26297–26301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson K. A. (2010) Biochim. Biophys. Acta 1804, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabor S., Richardson C. C. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 6339–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson A. A., Ray A. S., Hanes J., Suo Z., Colacino J. M., Anderson K. S., Johnson K. A. (2001) J. Biol. Chem. 276, 40847–40857 [DOI] [PubMed] [Google Scholar]
- 31.Lee H., Hanes J., Johnson K. A. (2003) Biochemistry 42, 14711–14719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H. R., Wang M., Konigsberg W. (2009) Biochemistry 48, 2087–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan S. S., Longley M. J., Naviaux R. K., Copeland W. C. (2005) DNA Repair 4, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 34.Nguyen K. V., Sharief F. S., Chan S. S., Copeland W. C., Naviaux R. K. (2006) J. Hepatol. 45, 108–116 [DOI] [PubMed] [Google Scholar]
- 35.Luoma P. T., Luo N., Löscher W. N., Farr C. L., Horvath R., Wanschitz J., Kiechl S., Kaguni L. S., Suomalainen A. (2005) Hum. Mol. Genet. 14, 1907–1920 [DOI] [PubMed] [Google Scholar]