


Abstract
A novel multiple affinity purification (MAFT) or tandem affinity purification (TAP) tag has been constructed. It consists of the calmodulin binding peptide, six histidine residues, and three copies of the hemagglutinin epitope. This ‘CHH’ MAFT tag allows two or three consecutive purification steps, giving high purity. Active Clb2–Cdc28 kinase complex was purified from yeast cells after inserting the CHH tag into Clb2. Associated proteins were identified using mass spectrometry. These included the known associated proteins Cdc28, Sic1 and Cks1. Several other proteins were found including the 70 kDa chaperone, Ssa1.
INTRODUCTION
Improvements in mass spectrometry combined with large-scale sequencing projects now allow the identification of protein from sub-picomolar samples. This in turn means that the members of protein complexes can be identified by mass spectrometry even when only very tiny amounts of the complex are available. The rate-limiting step is generally purification of the complex. Often, one or more member proteins of a complex are known, and the problem is to identify the associated proteins. In such cases it is sometimes possible to attach an affinity tag to the known protein and use this tag to purify the complex.
Use of affinity tags has several advantages over classical purification procedures (1–6). A well-chosen tag can allow a very high degree of purification in a single step, often using mild conditions that preserve the complex and its biological activity. Perhaps even more importantly, the conditions for purification are generally more dependent on the affinity tag than on the protein to which it is attached. Thus, purification methods can be standardized, allowing many different proteins or complexes to be purified by essentially identical methods. In this way, high throughput can be achieved.
However, no single tag allows purification to homogeneity. To circumvent this problem, Rigaut et al. (7) recently developed the ‘tandem affinity purification’ tag, or TAP tag, containing the calmodulin binding peptide (CBP) and the IgG binding domains of protein A. The inclusion of two affinity tags allows two consecutive affinity purification steps, thus achieving much higher purification than could be achieved by a single tag. We have independently developed the same idea. In our case, the tag contains (i) the calmodulin binding peptide for purification on calmodulin resin (8,9); (ii) six consecutive histidine residues (6× His) for purification on nickel resin (10,11); and (iii) three copies of hemagglutinin (3× HA) tag for immunoprecipitation with commercially available monoclonal antibody 12CA5 (12,13). Since the basis of each purification is different, the final degree of purification may be very high. We call this general strategy ‘multiple affinity purification’, or MAFT, and this particular version of a MAFT tag we have named the CHH tag (calmodulin, histidine, hemagglutinin).
We have developed two versions of CHH cassette, pSH6 and pSH26. pSH6 contains CHH tag on a NotI fragment that can be inserted at the C- or N-terminus of any protein. Another CHH cassette, pSH26, contains the URA3 gene flanked by direct repeats of CHH tags. This allows a CHH tag to be inserted into any yeast gene of interest with the help of PCR and in vivo recombination (14).
Here we demonstrate the use of the CHH tag to purify the Clb2–Cdc28 protein kinase complex from Saccharomyces cerevisiae. Two consecutive steps of affinity purification produced an extremely pure preparation of Clb2–Cdc28 protein kinase complex with associated proteins. The kinase was highly active. The associated proteins could be identified by mass spectrometry.
MATERIALS AND METHODS
Construction of the CHH (CBP-HIS6-HA3) tag
Oligonucleotides and plasmids used are listed in Tables 1 and 2. The CHH MAFT tag was generated by using crossover PCR to combine two independent PCR products: CBP-HIS6 and HIS6-HA3. Oligonucleotides PrS2 and PrS3 were designed to amplify the CBP-HIS6 domain of the pSH2 plasmid generating a 152 bp fragment. To generate HIS6-HA3, a triple HA epitope in plasmid pSH4 was amplified using oligonucleotides PrS1 and PrS4, generating a 209 bp fragment. The PCR fragments generated above had 119 bp of overlapping sequence, so crossover PCR was used to generate the CHH MAFT tag by mixing the gel purified 152 and 209 bp PCR fragments with oligonucleotides PrS1 and PrS4, followed by amplification. The resulting 242 bp band was gel-purified, quantified, cloned (see below) and sequenced in its entirety. The structure and sequence of the CHH tag is shown in Figure 1.
Table 1. Oligonucleotides.
| Oligonucleotide |
Sequence |
| PrS1 (HA-1) | GCT ATC TGC CTC GAG ACT AGT GCG GCC GCA CTG AGC AGC |
| PrS2 (CAL-1) | ACG ATC GTC GAA TTC GAG CTC GGC GGC CGC AAG CGA CGA TGG AAA AAG AAT TTC ATA GCC |
| PrS3 (CAL-HIS-2) | GCT ATC TGC CTC GAG ACT AGT GCG GCC GCA ATG ATG ATG ATG ATG ATG TGC CCC GGA GGA TGA GAT TTT CTT AAA GC |
| PrS4 (HA-HIS6) | AAG ATC TCA TCC TCC GGG GCA CAT CAT CAT CAT CAT CAT ATC TTT TAC CCA TAC GAT GTT CC |
| PrS5 (URA3-1) | AAG CTT GCA TGC CTG CAG GTC G |
| PrS6 (URA3-2) | GCA TAT TTG AGA AGA TGC GGC |
| PrS7 (Triple-dwn) | GCT ATC TGC CTC GAG ACT AGT TTC TCA GCG GCC GCA CTG AGC |
| PrS8 (CLB-1) | AGC TTC TAT AAT TTC CGT CC |
| PrS9 (CDC28) | CCA CCC AGT AAT ACC TCC GG |
| PrS10 (3′-CDC28) | CCC ATC ATA ATG TTG AAA CTG |
| PrS11 (5′-BAMCDC28) | TTC TAA GTT TGG ATC CAA AAA ATG AGC GGT GAA TTA GCA AAT TAC |
| PrS12 (3′-BAMCDC28) | TTT CCA AGT TGG ATC CCA TAC AAT GCG TTA TTT CGT TTT |
Text in parentheses represents other names assigned to oligonucleotides.
Table 2. Plasmids.
| Name |
Vector |
Insert |
| pSH2 | YCplac22 | GAL1-CLB2-CAL-HIS6 |
| pSH4 | YCplac22 | GAL1-CLB2-HA3 |
| pSH5 | YCplac22 | GAL1-CLB2 |
| pSH6 | YCplac22 | GAL1-CLB2-CAL-HIS6-HA3 |
| pSH13 | PGBDU-2 | GAL4 DNA binding domain + CDC28K40R |
| pSH14 | YEP213 | CDC28 |
| pMR438 | pBR322 | GAL1,10 |
| pSH16 | pMR438 | GAL1-CDC28K40R |
| pSH22 | pKS(–) | URA3 |
| pSH23 | pSH22 | CAL-HIS6-HA3 |
| pSH26 | pSH23 | Two copies of CAL-HIS6-HA3 |
Figure 1.

The structure (A) and sequence (B) of TAP tag (CHH) consisting of calmodulin binding peptide (CBP), six histidine residues (HIS6), and three copies of the hemagglutinin epitope (HA3), flanked by NotI sites.
Construction of GAL-CLB2-CHH and GAL-CDC28K40R (pSH16)
Plasmid pSH6 (GAL-CLB2-CBP-HIS6-HA3) was constructed starting with pSH4 (pGAL-CLB2) which contains a NotI site in the appropriate reading frame just before the stop codon of CLB2. pSH4 was digested with NotI, and mixed with the purified 242 bp PCR product containing the CHH tag (see above), which had likewise been digested with NotI. Ligation generated plasmid pSH6 (GAL-CLB2-CBP-HIS6-HA3). Because a BglII site had been engineered into oligonucleotide PrS4, the orientation of the insert was determined by digestion with BglII. Subsequently, the entire CHH tag was sequenced.
Plasmid pSH16 (GAL-CDC28 K40R) was constructed by using three plasmids; pSH13 (pGBDU2CDC28K40R), pSH14 (YEP213-CDC28) and pMR438. Oligonucleotide PrS11, consisting of a BamHI site and 24 bases of CDC28 starting from the ATG, was designed to amplify the 5′-region of CDC28K40R on plasmid pSH13 (previously cut with EcoRI and PstI) using one-sided PCR. The 3′-region of CDC28, including its transcription terminator, was amplified by performing one-sided PCR on HindIII, XbaI-cut pSH14 using oligonucleotide PrS10. One-sided PCR products generated above had overlapping sequences, and crossover PCR was done by mixing both PCR products and oligonucleotides PrS10 and PrS11, to generate a CDC28 K40R construct flanked by BamHI sites. The resulting 1.3 kb PCR product was gel-purified, quantified, digested with BamHI and cloned into the BamHI site of pMR438 to generate pSH16 (GAL-CDC28-K40R). The orientation of CDC28-K40R was determined by sequencing the entire CDC28-K40R fragment using PrS9 and PrS10 oligonucleotides.
Construction of pSH26
To generate a vector for PCR epitope tagging of genes in yeast (14), we cloned the CHH tag on either side of the URA3 marker in a yeast integrating vector. Oligonucleotides PrS2 and PrS7 were used to amplify a 261 bp fragment containing the CHH tag on plasmid pSH6. This PCR fragment was cut with SacI and SpeI and ligated to a similarly-cut pSH22 plasmid. Subsequently, the plasmid generated from this ligation, pSH23 was cut with EcoRI and XhoI and ligated to a similarly-cut PCR fragment generated above to create plasmid pSH26. Finally, both of the cloned CHH tags were sequenced using oligonucleotides PrS5 and PrS6.
Strains and culture conditions
Yeast strains YSH16 (untagged GAL-CLB2; WT CDC28), YSH19 (GAL-CLB2-CHH; WT CDC28) and YSH24 (GAL-CLB2-CHH CDC28 GAL-cdc28-K40R) used in this study were prepared by transforming plasmids (pSH5, pSH6 and pSH6+pSH16, respectively) into a S.cerevisiae diploid strain W303 (MATa/MATα), which is homozygous for ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3-1 can1-100 and is [psi+] (15). CLB2 (tagged or untagged) and kinase-dead cdc28-K40R were expressed from the GAL promoter. Standard conditions were used for culturing and manipulating yeast (16).
Affinity chromatography and immunoprecipitation
Yeast were grown in YEP (yeast extract peptone) media containing 2% raffinose w/v as carbon source, at 30°C to 2.0 × 107 cells/ml. Galactose (1% final) was added to each culture and cells were grown for an additional 90 min. Cells were harvested by centrifugation at 4°C, washed in ice-cold water, and resuspended in a volume of lysis buffer (Table 3) equal to the volume of the cell pellet. Cells were lysed at 4°C with acid-washed glass beads and a Mini Bead-beater (Biospec Products). Resulting cell extracts were centrifuged at 14 000 r.p.m. in a microcentrifuge at 4°C. Protein concentrations, as determined by the Bradford assay (17), ranged from 3.0 to 6.5 mg/ml. Prior to use, calmodulin affinity resin (Stratagene) or Ni-NTA resin (Qiagen) was washed four times with six bed-volumes of lysis buffer (Table 3). Between washes, resin was collected using a 2 min centrifugation at 1000 r.p.m. at 4°C. Protein extract with protein concentrations ranging from 3.0 to 6.0 mg/ml was mixed with one-twentieth volume of pre-equilibrated affinity resin (see above). (For example, from 6 l of culture, we typically obtained 20 ml of extract with a protein concentration of ∼4.5 mg/ml, which was then mixed with 1 ml of resin.) The suspension was rocked at 4°C for 2 h. Unbound material was then removed by washing the affinity resin four times with a volume of lysis buffer (Table 3) equal to six bed-volumes of the resin, with pulse-spins between each wash followed by the removal of the supernatant. This unbound material is referred to as ‘flow-through’. Proteins were eluted from the affinity resin by eluting six times, each time with two bed-volumes of elution buffer (Table 3), containing 2 mM EGTA (for calmodulin resin) or 250 mM imidazole (for Ni-NTA resin). (For example, for a 6 l culture, 1 ml of resin was eluted six times, with 2 ml each time, yielding a total of 12 ml.) Eluted fractions containing partially purified Clb2–Cdc28 complex were pooled, and passed through an empty Bio-Spin chromatography column (Bio-Rad) to filter out residual resin beads. This filtered eluate was subjected to immunoprecipitation, for which we used monoclonal antibody 12CA5 covalently coupled to Immunopure protein A–Sepharose beads (Pierce) (see below). Typically, immunoprecipitation was carried out by mixing 10 ml of partially purified Clb2–Cdc28 complex from the affinity resin with 100 µl of protein A–Sepharose beads crosslinked to 12CA5. The suspension was rocked at 4°C for 2 h. Precipitated immune complex was washed four times with cold immunoprecipitation buffer (Table 3). Resulting immunocomplexes containing purified Clb2–Cdc28 complex on protein A beads were stored at –70°C.
Table 3. Buffers.
| Cell lysis buffers | 1a | 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% Triton X-100, 10 mM β-mercaptoethanol, 1 mM magnesium acetate, 1 mM imidazole, 2 mM calcium chloride, 1 mM DTT, 0.05% deoxycholic acid, 10 mM sodium fluoride and PIsb |
| 2a | 50 mM NaH2PO4 pH 8.0, 150 mM NaCl, 10 mM imidazole, 50 mM sodium fluoride and PIsb | |
| Elution buffers | 1a | 50 mM Tris–HCl pH 8.0, 250 mM NaCl, 50 mM sodium fluoride, 2 mM EGTA, 10 mM β-mercaptoethanol and PIsb |
| 2a | 50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 50 mM sodium fluoride, 250 mM imidazole and PIsb | |
| Immunoprecipitation buffer | 50 mM Tris–HCl pH 7.5, 250 mM NaCl, 0.5% Triton X-100, 50 mM sodium fluoride, 5 mM EDTA and PIsb | |
| 2× kinase reaction buffer | 100 mM Tris–HCl pH 7.5, 20 mM MgCl2, 2 mM DTT |
a1: used for calmodulin–peptide binding affinity column; 2: used for Ni affinity column.
bPIs, protease inhibitors: 1.5 mM PMSF, 10 µg/ml leupeptin, 5 µg/ml pepstatin A, 10 µg/ml TPCK, 2 mM benzamidine, 0.2 mg/ml bacitracin.
Crosslinking of 12CA5 monoclonal antibody to protein A–Sepharose beads
Crosslinking was adapted from the method described by Harlow and Lane (18). One milliliter of immunopure protein A–Sepharose beads (50% slurry) was washed three times in PBS (136 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.4), then resuspended in 10 ml PBS. To this, 1 ml (6–10 mg protein) of ascites fluid from the 12CA5 cell line was added, and the suspension was incubated at room temperature for 1 h with gentle rocking. A percentage of the suspension (0.1%, 11 µl) was saved for later analysis. Beads were harvested by gentle centrifugation (3000 g, 5 min), and 11 µl of the supernatant was saved for later analysis. Beads were washed twice with 5 ml (10 vol) of 0.2 M sodium borate pH 9. Washed beads were resuspended in 5 ml (10 vol) of 0.2 M sodium borate pH 9, and 0.1% (5.5 µl) of the suspension was saved for later analysis. The monoclonal antibody bound to the protein A beads was then covalently coupled to the beads by the addition of the crosslinking agent dimethyl suberimidate (DMS) to a final concentration of 20 mM. The suspension was incubated for 30 min at room temperature with gentle rocking. A percentage of the suspension (0.1%, 5.5 µl) was saved for later analysis. The crosslinking reaction was quenched by washing the beads once with 5 ml of 0.2 M ethanolamine pH 8, and then incubating for 2 h at room temperature in 5 ml of 0.2 M ethanolamine pH 8, with gentle mixing. Beads were washed in PBS, and finally resuspended in 0.5 ml of PBS with 0.01% merthiolate to preserve sterility.
To check the efficiency of antibody binding and coupling, the volume of each fraction was adjusted to 12 ml with PBS, an equal volume of 2× sample buffer was added, and then samples were boiled for 2 min and loaded on an SDS–10% polyacrylamide gel. Samples before crosslinking showed abundant free IgG, while the sample after crosslinking showed little or no free IgG.
Protein kinase assay using synthetic peptides
Protein kinase activity was assayed by incorporation of [γ-32P]ATP in a synthetic peptide (19). The experimental 14 amino acid synthetic peptide (RRRAQNATPNKAAG) used in this study had one consensus Cdc28 phosphorylation site (TPNK), while the control peptide (RRRAQNAAPNKAAG) was identical except for the substitution of alanine for the phosphorylatable threonine at position 8. A typical kinase reaction mixture, in a total volume of 20 µl, contained 2× kinase reaction buffer (Table 3), 1 µCi [γ-32P]ATP, 5 µM ATP, 0.5 mg/ml BSA, 1.5 mM experimental or control peptide and the protein kinase. The reaction was carried out by incubating the contents at 30°C for 15 min. Aliquots (15 µl) of supernatant from each kinase reaction sample was pipetted onto 2 cm squares of P81 Whatman paper. The squares were dried at room temperature for 10 min, then washed three times with 75 mM orthophosphoric acid to remove any unbound [γ-32P]ATP. Squares were dried at room temperature and counted for radioactivity in scintillation vials containing 10 ml organic scintillation fluid using an LKB 1209 Rackbeta Liquid Scintillation Counter.
Immunoblot analysis
Immunoblot analysis was carried out as previously described (20). Samples containing Clb2–Cdc28 complex were mixed with an equal volume of 2× protein sample buffer for SDS–PAGE. Samples were heated to boiling for 2 min then loaded on 10% SDS–polyacrylamide gels. Proteins were transferred to HybondTM ECLTM (Amersham) membrane using a semi-dry transfer apparatus. Proteins were visualized by staining with Ponceau S (18). The membrane blot was blocked using 5% non-fat dry milk in a buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl and 0.1% Tween-20. For detection of proteins tagged with 3× HA, blots were incubated with 12CA5 monoclonal antibody ascites fluid at a dilution of 1/5000 followed by secondary HRP-conjugated anti-mouse antibody (Amersham) at a dilution of 1/10 000. For detection of Cdc28, a rabbit polyclonal generated against the N-terminal 12 amino acids of Cdc28 was used (B.Futcher, unpublished results). For detection of Sic1, a rabbit polyclonal antibody generated against full-length recombinant Sic1 was used (M.Tyers, personal communication). HRP-linked donkey anti-rabbit antibody (Amersham) was used as a secondary antibody at a dilution of 1/10 000 for the detection of Cdc28 and Sic1. An enhanced chemiluminescence (ECL) detection system (Amersham) was used for protein visualization according to the manufacturer’s instructions.
Tandem mass spectrometry
For the identification of proteins associated with the tandem affinity purified Clb2–Cdc28 complex, samples were prepared from a 12 l culture of yeast, and Clb2–Cdc28 was purified using a calmodulin resin followed by immunoprecipitation with 12CA5 as described above. Proteins were separated by 10% SDS–PAGE and visualized by silver staining (21). Silver-stained bands were excised and in-gel digested with trypsin following the procedure of Shevchenko et al. (22). The digested proteins were identified by using the nano-LC/MS/MS method described by Gatlin et al. (23).
RESULTS
Choice of affinity tags
We chose the calmodulin binding peptide (CBP) and the hexahistidine peptide (6× His) as affinity tags for three reasons. First, they each allow reasonable purification in good yield of tagged proteins. Second, elution of tagged proteins from the affinity resin occurs under mild conditions, which would allow many protein complexes to remain intact. Third, they are both relatively small, which makes them easier to work with at the DNA level, and may lessen their impact on the function of the tagged protein. In addition, we chose the 3× HA peptide because it is an excellent epitope tag: the 12CA5 monoclonal antibody recognizes it with high affinity and avidity; it works well in both western blotting and immunoprecipitation; and 12CA5 and other antibodies recognizing 3× HA are commercially available.
Tagging of Clb2
We chose to test the CHH tag using the non-abundant yeast mitotic cyclin Clb2 (24,25). Clb2 forms a complex with and activates the cyclin-dependent kinase Cdc28 (26,27). The Clb2–Cdc28 protein kinase activity can be assayed in vitro using a variety of substrates, which typically bear the consensus phosphorylation site S/T-P-X-K/R, where X is any amino acid. The Clb2–Cdc28 complex is known to associate with several other proteins such as Sic1 and Cks1 (28–30). Thus, this test system allows us to ask whether enzymatic activity and protein complexes survive the purification procedure. For these experiments, the CLB2 gene was over-expressed from the GAL promoter, which in this case gives ∼10-fold over-expression. Even with over-expression, however, Clb2 remains an extremely non-abundant protein.
The relevant in vivo substrates of Clb2–Cdc28 are largely unknown. These substrates are amongst the most interesting proteins that might co-purify with the Clb2–Cdc28 complex. Unfortunately, the association of substrates with this protein kinase complex may well be weak and transient, and so unlikely to survive purification. In an effort to stabilize the interaction between the kinase complex and its substrates, we created a mutant form of the Cdc28 kinase, Cdc28-K40R, which lacks a lysine crucial for enzymatic activity and so cannot phosphorylate proteins (31). The interactions of Cdc28-K40R with the normal substrates of Cdc28 might be somewhat stabilized, since enzyme–substrate interactions are typically stronger than enzyme–product interactions. In a two-hybrid assay, the Cdc28-K40R protein interacts more strongly than wild-type Cdc28 with certain putative substrates (G.Sherlock and B.Futcher, unpublished results). Some (but not all) of our purifications of Clb2-CHH used a strain that also over-expressed Cdc28-K40R, with the idea that substrates might remain associated with the Clb2-CHH/Cdc28-K40R complex.
Purification of Clb2–Cdc28 complexes
Cells containing tagged or untagged Clb2 were grown, induced with galactose and lyzed. Protein extract was purified using either a nickel resin (for purification of the 6× His tag) (Fig. 2A), or using a calmodulin resin (for purification of the CBP tag) (Fig. 2B, lanes 1 and 2). Proteins were then separated by PAGE, and visualized by silver staining. Both the nickel and the calmodulin resin gave a large purification, in the sense that only a small fraction of the protein applied was retained on the resin, but nevertheless many different proteins were retained, and Clb2 constituted only a tiny fraction of the eluate (Fig. 2). Similarly, immunoprecipitation with 12CA5 as the only purification step greatly enriches for Clb2, but leaves many contaminating proteins (data not shown).
Figure 2.
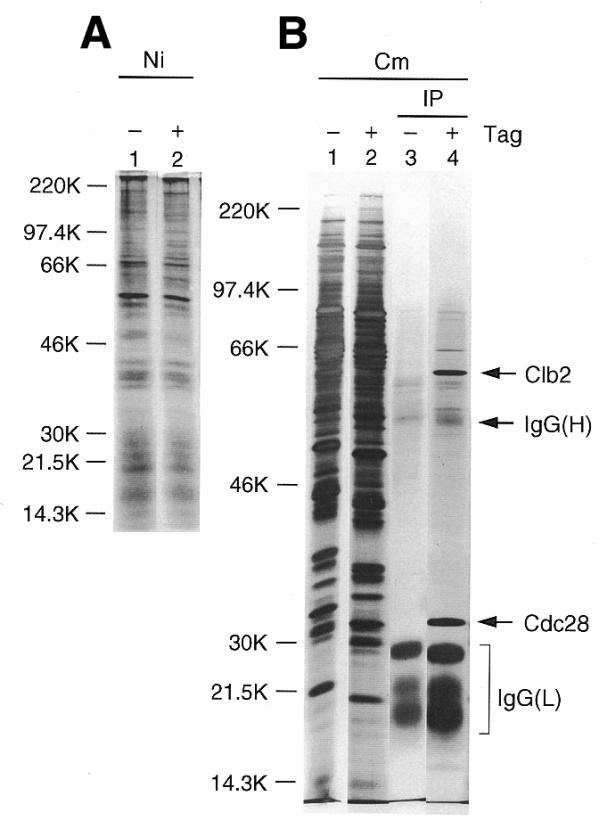
TAP of Clb2–Cdc28 complex. Untagged Clb2 (odd-numbered lanes) and Clb2-CHH (even-numbered lanes) were expressed under the control of the GAL promoter in strains YSH16 and YSH19. (A) Untagged (lane 1) or tagged (lane 2) cell extracts were purified on a nickel (Ni) affinity resin, and visualized after 10% SDS–PAGE and silver staining. (B) Untagged (lane 1) or tagged (lane 2) cell extracts were purified on a calmodulin (Cm) affinity resin. Pooled eluted fractions from the calmodulin affinity resin were further purified by immunoprecipitation (IP) with 12CA5 monoclonal antibody on protein A beads (lane 3, untagged control; lane 4, Clb2-CHH). Proteins were separated using 10% SDS–PAGE and visualized by silver staining. Most of the material visible in lane 3 is heavy (H) or light (L) chain IgG.
However, when protein purified on the calmodulin resin was then subjected to immunoprecipitation as a second purification step, a striking purification was obtained (Fig. 2B, lanes 3 and 4). Nearly all of the contaminating yeast proteins were removed, and a 56 kDa band corresponding to Clb2 was one of the strongest bands on the gel. Another strong band migrating at ∼34 kDa proved to be Cdc28 (see below), which is expected to co-purify with Clb2 in stoichiometric quantities (32).
Similar results were obtained using the nickel resin followed by immunoprecipitation (data not shown), although our impression is that the nickel/12CA5 combination is somewhat less effective than the calmodulin/12CA5 combination.
Sequential purifications with the nickel resin and the calmodulin resin were also attempted. Unfortunately, there is an incompatibility between the buffer requirements for the two resins: the elution buffer for the calmodulin resin is incompatible with binding to the nickel resin, and vice versa. Our attempts at buffer exchange were disappointing, in that there was a significant loss of yield at the buffer exchange step, at least for small-scale purifications. Buffer exchange was not attempted for any large-scale purification.
Western analysis of protein purified by calmodulin/12CA5 showed that the prominent 56 kDa band of Figure 2B is Clb2, and that the prominent 34 kDa band is Cdc28 (Fig. 3). Furthermore, western analysis showed that Sic1, another protein expected to co-purify with Clb2 (28), was also present (Fig. 3). Sic1 is sub-stoichiometric, so it is not visible by silver staining in Figure 2, but it is visible by silver staining in similarly purified material when a larger amount of protein is loaded on the gel (see below). Note that the relative intensities of the bands seen by western analysis do not indicate the relative abundance of each protein, because the antibodies used to detect the three proteins are quite different.
Figure 3.
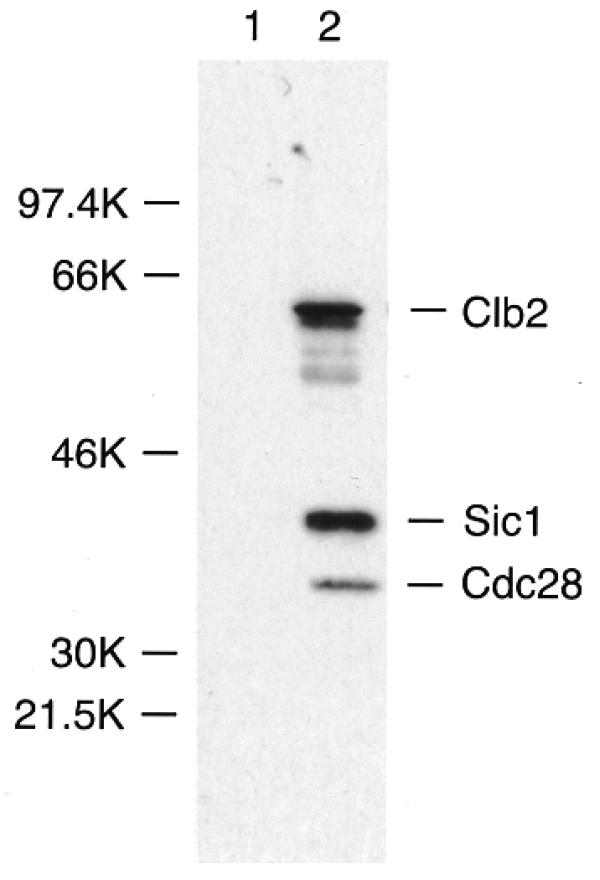
Western analysis of proteins associated with Clb2–Cdc28 complex. Cell extracts prepared from untagged Clb2 (lane 1) and Clb2-CHH (lane 2) were subjected to sequential purifications on calmodulin affinity resin followed by immunoprecipitation with 12CA5 monoclonal antibody on protein A beads. Immunoprecipitates were applied to 10% SDS–PAGE gels and immunoblotted with antibodies against HA (monoclonal 12CA5), Sic1 (rabbit polyclonal) and Cdc28 (rabbit polyclonal).
Protein kinase assays with a synthetic peptide substrate showed that the purified Clb2–Cdc28 complexes retained kinase activity (Fig. 4). The protein kinase assay suggests that the immunoprecipitation step is quite efficient, since the activity remaining after precipitation is ∼60% of the activity before precipitation.
Figure 4.

Kinase activity of various fractions. Untagged (fractions 1–3) or CHH-tagged Clb2 (fractions 4–9) was purified by binding to calmodulin or nickel resin, eluted, then immunoprecipitated with 12CA5. Equal proportions of various fractions were assayed for protein kinase activity using [γ-32P]ATP, peptide substrates and a filter binding assay (Materials and Methods). Fraction 1, untagged Clb2, flow-through from calmodulin resin. Fraction 2, untagged Clb2, eluate from calmodulin resin. Fraction 3, untagged Clb2, immunoprecipitate (IP) of eluate. Fraction 4, Clb2-CHH, flow-through from calmodulin resin. Fraction 5, Clb2-CHH, eluate from calmodulin resin. Fraction 6, Clb2-CHH, IP of eluate from calmodulin resin. Fraction 7, Clb2-CHH, flow-through from nickel resin. Fraction 8, Clb2-CHH, eluate from nickel resin. Fraction 9, IP of eluate from nickel resin. The data are means of triplicate determinations. Standard deviation in each case was <15% of the mean.
Identification of co-purifying proteins by tandem mass spectrometry
The usefulness of the CHH tag for identification of members of a protein complex was tested. Twelve liter cultures of cells carrying untagged Clb2 (control) or Clb2-CHH or Clb2-CHH in a CDC28 GAL-cdc28K40R strain were grown, induced with galactose, and lyzed. Tagged Clb2 was purified first on calmodulin resin, and then immunoprecipitated using 12CA5. Proteins in the immunoprecipitates were resolved using SDS–10% PAGE and visualized by silver staining (Fig. 5). Very little yeast protein was present in the untagged control (lane 1), while several bands, including the previously identified Clb2 and Cdc28 bands, were prominent in the Clb2-CHH lane (lane 2). Several bands were more prominent when Clb2-CHH was purified from the cdc28K40R strain than when purified from the CDC28 strain (compare lane 3 with lane 2), suggesting that the corresponding proteins might enjoy a tighter association with the kinase-dead version of Cdc28 than with the wild-type protein.
Figure 5.
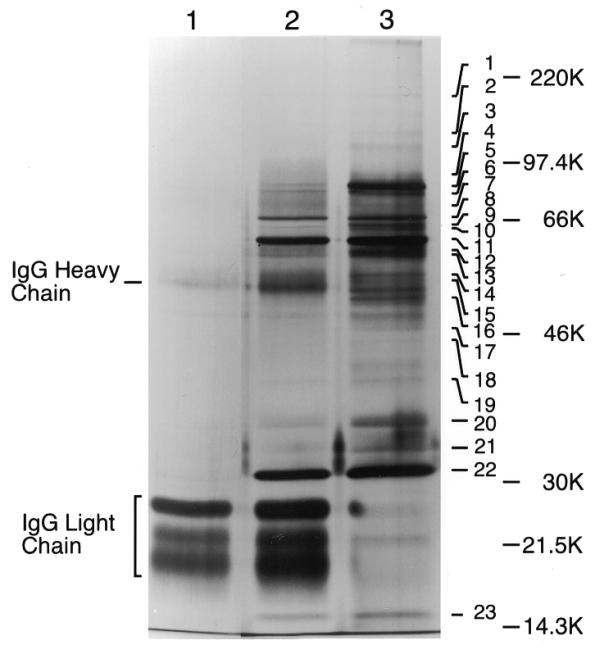
Resolution of TAP Clb2–Cdc28 complex on silver-stained gel. Extracts from 12 l cultures of yeast strains YSH16, YSH19 and YSH24 carrying untagged Clb2 and Cdc28 (lane 1), Clb2-CHH and Cdc28 (lane 2), or Clb2-CHH, Cdc28 and Cdc28K40R (lane 3), respectively, were purified on calmodulin affinity resin and then immunoprecipitated with 12CA5 monoclonal antibody. Purified proteins were resolved using 10% SDS–PAGE and proteins were visualized by silver staining. Silver-stained protein band nos 1–23 (lane 3) were excised, digested with trypsin and fragments were used for the identification of proteins by mass spectrometry.
A total of 23 protein bands were excised (from lane 3, Fig. 5). The gel slices were treated with trypsin to digest proteins, and the fragments were analyzed by tandem mass spectrometry. Using the SEQUEST algorithm, acquired fragmentation spectra of peptides were correlated with predicted amino acid sequences from the standard S.cerevisiae open reading frames (ORFs). In this way, the proteins present in each of bands 1–23 (Fig. 5) were identified (Table 4).
Table 4. Proteins co-purifying with Clb2-CHH.
| Banda |
No. peptides detected |
ORF/proteinb |
|
Mol. wt (kDa) |
| 1 | 7 | YDR172W/SUP35 | (3) | 76 |
| YOR204W/DED1 | (4) | 65 | ||
| 2 | * | |||
| 3 | 2 | YCR030C | (1) | 96 |
| YER151C/UBP3 | (1) | 101 | ||
| 4 | * | |||
| 5 | 25 | YDR172W/SUP35 | (17) | 76 |
| YIL105C | (7) | 78 | ||
| YLL054C | (1) | 89 | ||
| 6 | * | |||
| 7 | * | |||
| 8 | 18 | YAL005C/SSA1 | (18) | 69 |
| YLL024C/SSA2c | (14) | 69 | ||
| YBL075C/SSA3c | (5) | 70 | ||
| YER103W/SSA4c | (3) | 69 | ||
| 9 | 6 | YDL229W/SSB1 | (6) | 66 |
| YNL209W/SSB2c | (5) | 66 | ||
| 10 | 20 | YOR204W/DED1 | (16) | 65 |
| YPL119C/DBP1 | (4) | 68 | ||
| 11 | 17 | YPR119W/CLB2 | (17) | 56 |
| YGR108W/CLB1c | (3) | 55 | ||
| 12 | 15 | YPR119W/CLB2 | (15) | 56 |
| YGR108W/CLB1c | (1) | 55 | ||
| 13 | * | |||
| 14 | 6 | YPR119W/CLB2 | (6) | 56 |
| 15 | 4 | YPR119W/CLB2 | (4) | 56 |
| 16 | 3 | YLL054C | (1) | 89 |
| YPR119W/CLB2 | (2) | 56 | ||
| 17 | 2 | YPR080W/TEF1 | (2) | 50 |
| YBR118W/TEF2c | (2) | 50 | ||
| 18 | 1 | YDL055C/PSA1 | (1) | 39 |
| 19 | 3 | YPR119W/CLB2 | (3) | 56 |
| 20 | 10 | YLR079W/SIC1 | (10) | 40 |
| 21 | * | |||
| 22 | 11 | YBR160W/CDC28 | (11) | 34 |
| YPL031C/PHO85c | (2) | 35 | ||
| 23 | 5 | YBR135W/CKS1 | (5) | 17 |
aIndicates the position of protein band on silver-stained gel shown in Figure 4.
bValues in parentheses represent the number of peptides identified from a particular protein.
cIndicates that the peptides of the marked ORF are identical to peptides of another ORF assigned to the same protein band. For instance, for band 11, 17 peptides characteristic of Clb2 were found. Three of these same peptides are also present in Clb1. Thus, there is no evidence for the presence of Clb1 in band 11, though its presence cannot be excluded.
*Only trypsin, a contaminant from the digestion procedure, was detected.
These proteins included six different forms of Clb2 itself. Some of these might be proteolytic fragments, while others might be differently phosphorylated forms. In addition, there were three proteins known or strongly suspected to be associated with Clb2. These were Cdc28, the protein kinase catalytic subunit (27); Sic1, a protein known to bind and inhibit Clb–Cdc28 complexes (28,33); and Cks1, a protein of somewhat uncertain function, but widely conserved and known to associate with cyclin–CDK complexes (30,34). These are the only three proteins known to bind tightly to Clb2 (or the Clb2–Cdc28 complex) in fairly large amounts.
Neglecting trypsin (a contaminant from the digestion procedure), and neglecting Psa1, Ubp3 and Ycr030c, which were identified by only a single peptide and whose presence is therefore somewhat uncertain, there were eight other proteins found. These were Sup35, Yil105c, Yll054c, Ded1, Dbp1, Tef1, Ssa1 and Ssb1. The significance of these proteins is unknown (but see below). Five of the eight (Sup35, Ded1, Dbp1, Tef1 and Ssb1) are abundant proteins involved in protein synthesis. There is no apparent connection between these proteins and Clb2, and yet it is not clear why they would be present as non-specific contaminants, since they are not present in the untagged control, and many other equally or more-abundant proteins involved in protein synthesis are not present.
DISCUSSION
The MAFT strategy worked very well. A non-abundant complex was purified to near homogeneity. Purification on a scale sufficient for mass spectrometry required only 12 l of cell culture, and the purification was completed within 1 day. The kinase activity of the complex was preserved, and at least three genuine Clb2-associated proteins co-purified with Clb2-CHH. It is likely that exactly the same purification method will be similarly successful with many other proteins and protein complexes.
The broad strategy of MAFT can be implemented in many different ways. Seraphin and co-workers (7) have developed a multiple affinity tag called the TAP tag, while here, we have developed the CHH tag. In our studies, we did an initial purification with either a calmodulin or a nickel resin, and then a second purification by immunoprecipitation. A disadvantage of these particular protocols is that the final purified material is associated with antibody and protein A beads, rather than being in solution (though incubation with a quadruple HA peptide can elute up to 50% of the bound material, data not shown). We had hoped to purify proteins using the calmodulin resin and the nickel resin in sequence, yielding a soluble product. Unfortunately, the buffers used to elute material from the calmodulin resin are incompatible with binding to the nickel resin, and vice versa. Consecutive purifications using these two resins should be possible if buffer is exchanged between steps, but this introduces extra complexity and loss of yield. It is likely that additional MAFT cassettes can be designed incorporating other tags that can be used consecutively and immediately, and that will yield a soluble product.
We did not measure the yield of Clb2, though it seemed good compared to other methods. Our experience with immunoprecipitation of a wide variety of tagged proteins suggests that an important factor in determining yield will be the accessibility of the tag on the protein of interest. In cases where yield is unacceptably low, it may be beneficial to move the tag to the other end of the protein.
There is an abundance of evidence showing that Cdc28, Sic1 and Cks1 are strongly associated with Clb2 (27,28,30), and indeed we found all three of these proteins. There is some evidence for association of Swi4 (35) and Nap1 (36), but we found neither of these. Swi4 is itself a non-abundant protein, and perhaps would be found in a larger-scale purification. Nap1 has a mass of 48 kDa, and migrates in a region of the gel containing contaminants from the IgG heavy chain. We did not examine any fragments from this region of the gel by mass spectrometry, and so would not have seen Nap1 even if it were present. Finally, there are four proteins suggested to associate with Clb2 by two-hybrid analysis (but by no other evidence) (37), but we found none of these. Thus, out of a total of nine proteins where there is at least a trace of evidence for direct association with Clb2, we found three, but these are the three for which evidence of association is most compelling.
We also found eight proteins for which there was no previous evidence of association with Clb2; at present, we do not know whether any of these are truly associated, or whether they are simply co-purifying. These proteins were found in a strain over-expressing a mutant form of Cdc28 which is defective in ATP binding. Conceivably the lack of ATP leads to a folding defect, and this somehow promotes association with chaperones. With regard to Sup35 and Yil105c, the host yeast strain used for production of Clb2 was W303. This strain is [psi+] (15), meaning that it contains the prion (aggregated) form of Sup35 (38). Remarkably, YIL105c (37) contains a region near its N-terminus resembling the glutamine-rich prion region of Sup35. Possibly, the presence of one prion in this strain has induced aggregation in Yil105c, and the aggregation of these two proteins has somehow allowed them to co-purify with Clb2.
A large hydrophobic face of the cyclin makes contact with the cyclin-dependent kinase (39). What is the state of this hydrophobic face after the cyclin has folded, but before it has paired with its CDK partner? It would not be surprising if chaperones were temporarily associated with the hydrophobic face of the cyclin. Indeed, the Ssa ‘chaperones’ are required for the folding of proteins (40) and, furthermore, the chaperone-like protein Cdc37 is already known to be involved in forming the cyclin–CDK complex (41), and two other proteins involved in protein folding are somehow needed for cyclin–CDK function (42). Possibly, the chaperone Ssa1 is also involved; it would be an appropriate molecule for protecting the hydrophobic face of Clb2 until a complex with Cdc28 could be formed. This could be the reason Ssa1 (and possibly also Ssb1) was found associated with Clb2. In this context, it is interesting that there is an allele of SSA1, ssa1-134 (43), which has a mutant phenotype reminiscent of the phenotypes of multiple clb mutants.
We purified Clb2 from a strain expressing a K40R mutation of Cdc28, in the hope that substrates might more stably associate with this inactive kinase complex. Although several proteins did seem more abundant in the complex isolated from the cdc28K40R strain, only one of these, Yll054c, has any resemblance to a candidate substrate. Yll054c is an uncharacterized protein (44), but it resembles a zinc-finger transcription factor, and it has several possible SP and TP sites which could be phosphorylated by Cdc28 (though none of the sites has the full CDK consensus). Nevertheless, this strategy might still be successful if purification were carried out on a larger scale.
Acknowledgments
ACKNOWLEDGEMENT
This study was supported by National Institutes of Health grant GM39978 to B.F.
References
- 1.Phizicky E.M. and Fields,S. (1995) Protein–protein interactions: methods for detection and analysis. Microbiol. Rev., 59, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Field J., Nikawa,J., Broek,D., MacDonald,B., Rodgers,L., Wilson,I.A., Lerner,R.A. and Wigler,M. (1988) Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol ., 8, 2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolodziej P.A., Woychik,N., Liao,S.M. and Young,R.A. (1990) RNA polymerase II subunit composition, stoichiometry, and phosphorylation. Mol. Cell. Biol., 10, 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugano S., Kim,D.W., Yu,Y.S., Mizushima-Sugano,J., Yoshitomo,K., Watanabe,S., Suzuki,F. and Yamaguchi,N. (1992) Use of an epitope-tagged cDNA library to isolate cDNAs encoding proteins with nuclear localization potential. Gene, 120, 227–233. [DOI] [PubMed] [Google Scholar]
- 5.Reisdorf P., Maarse,A.C. and Daignan-Fornier,B. (1993) Epitope-tagging vectors designed for yeast. Curr. Genet., 23, 181–183. [DOI] [PubMed] [Google Scholar]
- 6.Surdej P. and Jacobs-Lorena,M. (1994) Strategy for epitope tagging the protein-coding region of any gene. Biotechniques, 17, 560–565. [PubMed] [Google Scholar]
- 7.Rigaut G., Shevchenko,A., Rutz,B., Wilm,M., Mann,M. and Seraphin,B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- 8.Carr D.W., Stofko-Hahn,R.E., Fraser,I.D., Bishop,S.M., Acott,T.S., Brennan,R.G. and Scott,J.D. (1991) Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem., 266, 14188–14192. [PubMed] [Google Scholar]
- 9.Stofko-Hahn R.E., Carr,D.W. and Scott,J.D. (1992) A single step purification for recombinant proteins. Characterization of a microtubule associated protein (MAP 2) fragment which associates with the type II cAMP-dependent protein kinase. FEBS Lett., 302, 274–278. [DOI] [PubMed] [Google Scholar]
- 10.Janknecht R., de Martynoff,G., Lou,J., Hipskind,R.A., Nordheim,A. and Stunnenberg,H.G. (1991) Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. Proc. Natl Acad. Sci. USA, 88, 8972–8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann A. and Roeder,R.G. (1991) Purification of his-tagged proteins in non-denaturing conditions suggests a convenient method for protein interaction studies. Nucleic Acids Res., 19, 6337–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson I.A., Niman,H.L., Houghten,R.A., Cherenson,A.R., Connolly,M.L. and Lerner,R.A. (1984) The structure of an antigenic determinant in a protein. Cell, 37, 767–778. [DOI] [PubMed] [Google Scholar]
- 13.Tyers M., Tokiwa,G., Nash,R. and Futcher,B. (1992) The Cln3-Cdc28 kinase complex of S.cerevisiae is regulated by proteolysis and phosphorylation. EMBO J., 11, 1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider B.L., Seufert,W., Steiner,B., Yang,Q.H. and Futcher,A.B. (1995) Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast, 11, 1265–1274. [DOI] [PubMed] [Google Scholar]
- 15.Thomas B.J. and Rothstein,R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- 16.Guthrie C. and Fink,G.R. (1991) Guide to Yeast Genetics and Molecular Biology. In Methods in Enzymology. Vol. 194, Academic Press, New York. [PubMed]
- 17.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 18.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Glass D.B., Masaracchia,R.A., Feramisco,J.R. and Kemp,B.E. (1978) Isolation of phosphorylated peptides and proteins on ion exchange papers. Anal. Biochem., 87, 566–575. [DOI] [PubMed] [Google Scholar]
- 20.Honey S., O’Keefe,P., Drahushuk,A.T., Olson,J.R., Kumar,S. and Sikka,H.C. (2000) Metabolism of benzo(a)pyrene by duck liver microsomes. Comp. Biochem. Physiol. C, 126, 285–292. [DOI] [PubMed] [Google Scholar]
- 21.Rabilloud T., Carpentier,G. and Tarroux,P. (1988) Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. Electrophoresis, 9, 288–291. [DOI] [PubMed] [Google Scholar]
- 22.Shevchenko A., Wilm,M., Vorm,O. and Mann,M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem., 68, 850–858. [DOI] [PubMed] [Google Scholar]
- 23.Gatlin C.L., Kleemann,G.R., Hays,L.G., Link,A.J. and Yates,J.R.,III (1998) Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal. Biochem., 263, 93–101. [DOI] [PubMed] [Google Scholar]
- 24.Fitch I., Dahmann,C., Surana,U., Amon,A., Nasmyth,K., Goetsch,L., Byers,B. and Futcher,B. (1992) Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell, 3, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahmann C. and Futcher,B. (1995) Specialization of B-type cyclins for mitosis or meiosis in S.cerevisiae. Genetics, 140, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yaglom J., Linskens,M.H., Sadis,S., Rubin,D.M., Futcher,B. and Finley,D. (1995) p34Cdc28-mediated control of Cln3 cyclin degradation. Mol. Cell. Biol., 15, 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nasmyth K. (1993) Control of the yeast cell cycle by the Cdc28 protein kinase. Curr. Opin. Cell Biol., 5, 166–179. [DOI] [PubMed] [Google Scholar]
- 28.Schwob E., Bohm,T., Mendenhall,M.D. and Nasmyth,K. (1994) The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S.cerevisiae. Cell, 79, 233–244. [DOI] [PubMed] [Google Scholar]
- 29.Hadwiger J.A., Wittenberg,C., Mendenhall,M.D. and Reed,S.I. (1989) The Saccharomyces cerevisiae CKS1 gene, a homolog of the Schizosaccharomyces pombe suc1+ gene, encodes a subunit of the Cdc28 protein kinase complex. Mol. Cell. Biol., 9, 2034–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang Y. and Reed,S.I. (1993) The Cdk-associated protein Cks1 functions both in G1 and G2 in Saccharomyces cerevisiae. Genes Dev., 7, 822–832. [DOI] [PubMed] [Google Scholar]
- 31.Thuret J.Y., Valay,J.G., Faye,G. and Mann,C. (1996) Civ1 (CAK in vivo), a novel Cdk-activating kinase. Cell, 86, 565–576. [DOI] [PubMed] [Google Scholar]
- 32.Surana U., Robitsch,H., Price,C., Schuster,T., Fitch,I., Futcher,A.B. and Nasmyth,K. (1991) The role of CDC28 and cyclins during mitosis in the budding yeast S.cerevisiae. Cell, 65, 145–161. [DOI] [PubMed] [Google Scholar]
- 33.Schneider B.L., Yang,Q.H. and Futcher,A.B. (1996) Linkage of replication to start by the Cdk inhibitor Sic1. Science, 272, 560–562. [DOI] [PubMed] [Google Scholar]
- 34.Kaiser P., Moncollin,V., Clarke,D.J., Watson,M.H., Bertolaet,B.L., Reed,S.I. and Bailly,E. (1999) Cyclin-dependent kinase and Cks/Suc1 interact with the proteasome in yeast to control proteolysis of M-phase targets. Genes Dev., 13, 1190–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amon A., Tyers,M., Futcher,B. and Nasmyth,K. (1993) Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell, 74, 993–1007. [DOI] [PubMed] [Google Scholar]
- 36.Kellogg D.R., Kikuchi,A., Fujii-Nakata,T., Turck,C.W. and Murray,A.W. (1995) Members of the NAP/SET family of proteins interact specifically with B-type cyclins. J. Cell Biol., 130, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uetz P., Giot,L., Cagney,G., Mansfield,T.A., Judson,R.S., Knight,J.R., Lockshon,D., Narayan,V., Srinivasan,M., Pochart,P. et al. (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 38.Glover J.R., Kowal,A.S., Schirmer,E.C., Patino,M.M., Liu,J.J. and Lindquist,S. (1997) Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S.cerevisiae. Cell, 89, 811–819. [DOI] [PubMed] [Google Scholar]
- 39.Jeffrey P.D., Russo,A.A., Polyak,K., Gibbs,E., Hurwitz,J., Massague,J. and Pavletich,N.P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature, 376, 313–320. [DOI] [PubMed] [Google Scholar]
- 40.Unno K., Kishido,T., Hosaka,M. and Okada,S. (1997) Role of Hsp70 subfamily, Ssa, in protein folding in yeast cells, seen in luciferase-transformed ssa mutants. Biol. Pharm. Bull., 20, 1240–1244. [DOI] [PubMed] [Google Scholar]
- 41.Gerber M.R., Farrell,A., Deshaies,R.J., Herskowitz,I. and Morgan,D.O. (1995) Cdc37 is required for association of the protein kinase Cdc28 with G1 and mitotic cyclins. Proc. Natl Acad. Sci. USA, 92, 4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crenshaw D.G., Yang,J., Means,A.R. and Kornbluth,S. (1998) The mitotic peptidyl-prolyl isomerase, Pin1, interacts with Cdc25 and Plx1. EMBO J., 17, 1315–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oka M., Nakai,M., Endo,T., Lim,C.R., Kimata,Y. and Kohno,K. (1998) Loss of Hsp70-Hsp40 chaperone activity causes abnormal nuclear distribution and aberrant microtubule formation in M-phase of Saccharomyces cerevisiae. J. Biol. Chem., 273, 29727–29737. [DOI] [PubMed] [Google Scholar]
- 44.Winzeler E.A., Shoemaker,D.D., Astromoff,A., Liang,H., Anderson,K., Andre,B., Bangham,R., Benito,R., Boeke,J.D., Bussey,H. et al. (1999) Functional characterization of the S.cerevisiae genome by gene deletion and parallel analysis. Science, 285, 901–906. [DOI] [PubMed] [Google Scholar]