

Abstract
Although metastasis tumor antigen 1 (MTA1) contributes to the responsiveness of macrophages to LPS, the underlying mechanism remains unknown. Here, we investigated the role of MTA1 in the regulation of expression and function of MyD88, a proximal component of NF-κB signaling. We discovered that MTA1 targets MyD88 and that MyD88 is a NF-κB-responsive gene in LPS-stimulated macrophages. We found that MTA1 is required for MyD88-dependent stimulation of NF-κB signaling and expression of proinflammatory cytokines such as IL-1β, MIP2, and TNF-α as MTA1 depletion leads to a substantial reduction in the expression of NF-κB target genes. In addition, LPS-mediated stimulation of MyD88 transcription was accompanied by an enhanced recruitment of MTA1, RNA polymerase II, and p65RelA complex to the NF-κB consensus sites in the MyD88 promoter. Interestingly, the recruitment of both MTA1 and MyD88 expression is effectively blocked by NF-κB inhibitor parthenolide. Selective knockdown of MyD88 by a dominant negative mutant of MyD88 or selective siRNA also impairs the ability of LPS to stimulate the NF-κB target genes. These findings reveal an inherent coregulatory role of MTA1 upon the expression of MyD88 and suggest that MTA1 regulation of MyD88 may constitute at least one of the mechanisms by which MTA1 stimulates LPS-induced NF-κB signaling in stimulated macrophages.
Keywords: Chromatin Immunoprecipitation (ChIP), Cytokine Induction, Gene Regulation, Lipopolysaccharide (LPS), Macrophage, MyD88, Promoters, Transcription Repressor, MTA1, Coregulator
Introduction
Toll-like receptors (TLRs)2 play a key role in the innate immune system, particularly in the inflammatory response against various microorganisms by recognizing pathogen-associated molecular patterns (1–4). Upon the recognition of pathogen-associated molecular patterns, TLR signaling promptly induces immune responses that signal through adaptor molecules, the myeloid differentiation primary response gene 88 (MyD88), Toll/interleukin (IL)-1 receptor (TIR) domain containing adaptor protein (TIRAP), TIR domain containing adaptor-inducing interferon (IFN)-β (TRIF), and TRIF-related adaptor molecules (TRAM), eventually to activate transcriptional factors such as nuclear factor (NF)-κB, activator protein 1 (AP-1), and IFN regulatory factors to induce antibacterial and antiviral responses (5, 6). Among the TLRs, TLR4 is the major receptor involved in the detection of Gram-negative bacteria and their associated endotoxins such as lipopolysaccharide (LPS) (7) and signal through MyD88, leading to the subsequent downstream activation of NF-κB and mitogen-associated protein kinase (MAPK) signaling pathways (8). These signaling cascades are responsible for induction of proinflammatory cytokines and chemokines (9).
LPS activates two signal transduction pathways, i.e. MyD88-dependent and -independent pathways (10–13). The MyD88-dependent pathway activates NF-κB, which results in the production of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α and ultimately leads to the activation and accumulation of monocytes (14). In contrast, the MyD88-independent pathway activates interferon regulatory factor-3, resulting in the expression of IFN-β and IFN-inducible genes. MyD88-dependent TLR signals brought the spontaneous T cell and myeloid cell activation, cachexia, and premature lethality in the A20-deficient mice (15). Constitutive MyD88-dependent TLR signals provide beneficial, noninflammatory signals (16), triggering spontaneous inflammation. For example, MyD88-mediated innate immune signaling and inflammatory cell recruitment to the lung are required for the protection from lethal SARS-CoV infection caused by a human corona virus (CoV) (17). Also, TLR/MyD88 signaling is critical for induction of innate immune responses after partial hepatectomy as liver regeneration after partial hepatectomy requires innate immune responses (18). Several reports have suggested that inflammation plays a critical role in tumorigenesis and invasion, and the underlying mechanisms involved in these processes have been elucidated (19, 20). Now it is evident that an inflammatory microenvironment is an essential component of all tumors (21, 22).
Metastasis tumor antigen 1 (MTA1) chromatin modifier plays a critical role in modifying DNA accessibility for cofactors, largely as a part of the nucleosome remodeling and histone deacetylation complex (23–25). In addition, MTA1 is one of the most up-regulated genes in human cancer and has been shown to play a role in tumorigenesis (23, 26, 27). Recently, this laboratory has shown that MTA1 also plays a critical homeostatic role in inflammatory responses both as a target and as a component of the NF-κB signaling by regulating a subset of LPS-induced proinflammatory cytokines such as IL-1β, MIP2, and TNF-α (28). Because LPS-induced proinflammatory cytokine production is widely thought to be mediated through the stimulation of the MyD88-dependent NF-κB pathway, here we attempted to delineate the role of MTA1 in regulation of MyD88-dependent signaling. We discovered that MTA1 is required for MyD88-dependent activation of NF-κB signaling and production of proinflammatory cytokines in LPS-stimulated cells. Further, we found that MyD88 is a direct target of MTA1 and that LPS-induced MyD88 expression requires MTA1 in macrophages.
EXPERIMENTAL PROCEDURES
Antibodies and Cell Culture
Antibodies against MTA1 (catalogue number A300-280A) and RNA polymerase II (catalogue number A300-653A) were purchased from Bethyl Laboratories (Montgomery, TX). HDAC2 (sc-9959), NF-κB p65 (sc-372), and NF-κB p65 (286-H) X (sc-7151 X), ERK (sc-154), and JNK (sc-571) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-ERK (catalogue number 9106) and phospho-JNK (catalogue number 9255) were purchased from Cell Signaling Technology. Antibody against MyD88 (catalogue number AB 2068) was purchased from Abcam (Cambridge, MA). Normal mouse IgG, rabbit IgG, and antibodies against vinculin were purchased from Sigma. All cells were cultured in Dulbecco's modified Eagle's medium/F12 medium supplemented with 10% fetal bovine serum.
Cloning of Murine MyD88 Promoter
Full-length murine MyD88 promoter was generated by PCR using mouse genomic DNA and cloned into PGL3 luciferase reporter vector (Promega, Madison, WI) with the Infusion 2.0 Dry-Down PCR cloning kit (Clontech). The PCR amplification was carried out using the forward primer 5′-CGCCCACTCACCCCTTGCCCCA-3′ and reverse primer 5′-GGCCCCCGGTGCCGTCCAGTACGC-3′.
Quantitative PCR (qPCR)
For qPCR, total RNA was extracted using TRIzol reagent (Invitrogen), and first-strand cDNA synthesis was carried out with SuperScript II reverse transcriptase (Invitrogen) using 2 μg of total RNA and oligo(dT) primer. cDNA from macrophages was synthesized using the FastLane Cell cDNA synthesis kit (Qiagen, Valencia, CA). qPCR was performed with the gene-specific primers listed in supplemental Table 1 using a 7900HT sequence detection system (Applied Biosystems, Foster City, CA). The levels of mRNA of all the genes were normalized to that of β-actin mRNA.
Chromatin Immunoprecipitation (ChIP) Assay, Reporter Assays, and Western Blot Analysis
ChIP assays were done by using NF-κB, MTA1, RNA polymerase II, or HDAC2 antibodies as described elsewhere (29). The primers used for ChIP are listed in supplemental Table 2. MTA1-luc, IL-1β-luc, MIP2-luc, and MyD88-luc assays were performed according to the manufacturer's instructions (Promega), and the results were standardized against the β-galactosidase activity, an internal control. Some assays were performed in the presence of control siRNA or MTA1 siRNA or MyD88 siRNA. Western blot analysis was performed as described previously (29).
Isolation of Peritoneal Macrophages
After LPS treatment, peritoneal lavage was done with 10 ml of sterile ice-cold PBS, and the peritoneal lavage fluid was collected. The cells were washed and resuspended in Dulbecco's modified Eagle's medium/F12 medium supplemented with 10% fetal bovine serum, cultured overnight, and then washed to remove nonadherent cells.
siRNA Transfection
siRNA against MTA1 and negative control siRNA was purchased from Dharmacon (Lafayette, CO), and MyD88 siRNA was purchased from Santa Cruz Biotechnology. Raw cells were seeded at 40% density the day before transfection in 6-well plates, and siRNA transfections were performed with Oligofectamine reagent (Invitrogen) according to the manufacturer's instructions. Cells were harvested 48 h after transfection, and cell lysates were prepared.
EMSA
Nuclear extracts were prepared using a Nonidet P-40 lysis method (30). EMSA for NF-κB DNA binding was performed using the annealed and [γ-32P]ATP end-labeled oligonucleotides in a 20-μl reaction mixture for 15 min at 20 °C. Samples were run on a nondenaturing 5% polyacrylamide gel and imaged by autoradiography. Specific competitions were performed by adding a 100 molar excess of competitor to the incubation mixture, and supershift EMSAs were performed by adding 1.5 μl of the NF-κB p65 (Santa Cruz 286-H) antibody. Oligonucleotides used are shown in the supplemental Table 3.
Statistical Analysis and Reproducibility
The results are given as the means ± S.E. Statistical analysis of the data was performed by using Student's t test.
RESULTS AND DISCUSSION
MTA1 Regulates MyD88 Expression in LPS-stimulated Macrophages
Recent findings from this laboratory have established that MTA1 status plays a critical coregulatory role in controlling the expression of a subset of NF-κB target genes in LPS-stimulated macrophages (28). During the course of these studies, we also found that MTA1 influences the status of NF-κB signaling, at least in part by regulating the expression of a proximal signaling component in stimulated macrophages (28). Because MyD88 is a LPS response gene (31) as well as an adaptor/integrator of TLR4-initiated signals (14), here we investigated the contribution of MTA1 on the levels of MyD88.
To this end, we first determined the effect of MTA1 knockdown on the levels of MyD88 expression in the Raw 264.7 (Raw), a widely used macrophage cell line in studying TLR signaling and inflammatory mechanisms. We found that mediated reduction in the levels of endogenous MTA1 by specific siRNA in the Raw cells compromised the ability of LPS to induce the expression of MyD88 protein and mRNA (Fig. 1, A and B). Because the outcome of siRNA-mediated knockdown studies is dependent on the extent of target knockdown, we next examined the effect of genetic MTA1 depletion in MEFs (mouse embryonic fibroblasts) and cultured peritoneal macrophages from wild-type and MTA1−/− mice (32) on the ability of Escherichia coli-derived LPS on MyD88 expression. We found that MTA1 depletion substantially compromised the ability of LPS to induce MyD88 protein and mRNA expression in MEFs (Fig. 1, C and D) as well as in MTA−/− macrophages (Fig. 1E), suggesting that MTA1 may be a new mediator of MyD88 expression by LPS. Consistent with this possibility, during the course of these studies, we consistently observed that MTA1 knockdown reduced the expression of MyD88 expression in LPS-treated cells, suggesting the potential role of MTA1 in supporting the MyD88 pathway in LPS-stimulated macrophages.
FIGURE 1.
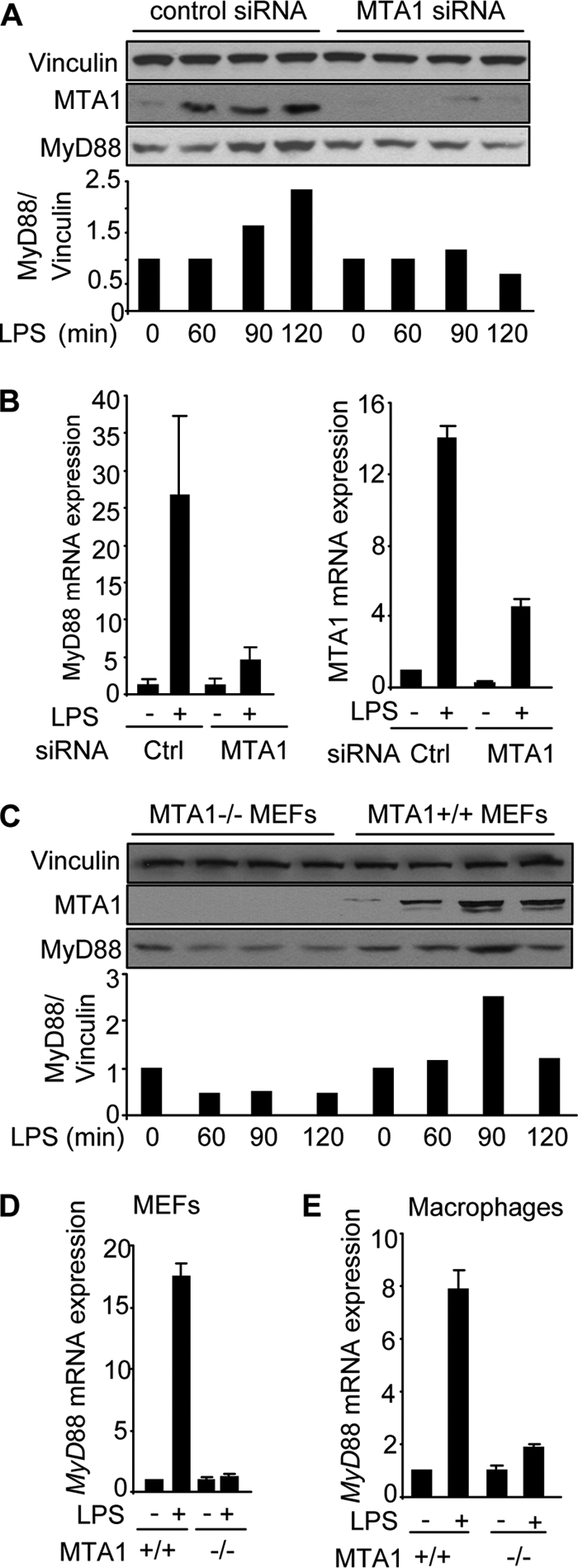
MTA1 regulates MyD88 expression. A, upper, Western blot analysis of MyD88 in LPS-stimulated Raw cells with or without MTA1 knockdown. Lower, densitometric analysis of MyD88 protein expression. B, qPCR analysis of MyD88 mRNA in LPS-stimulated Raw cells with or without MTA1 knockdown. C, upper, expression of MyD88 protein in LPS stimulation of MTA1+/+ and MTA1−/− MEFs. Lower, densitometric analysis of MyD88 expression. D, qPCR analysis of MyD88 mRNA in LPS-stimulated wild type and MTA1−/− MEFs. E, qPCR analysis of MyD88 mRNA in LPS-stimulated murine macrophages isolated from wild type and MTA1−/− mice.
MTA1 Stimulates MyD88 Transcription
To evaluate the role of MTA1 in the regulation of MyD88 expression, we next cloned the MyD88 promoter fragment (+67 to −1233) into a TATA-less pGL3-luc reporter. We found that LPS is a potent inducer of MyD88 transcription and that depletion of the endogenous MTA1 by specific siRNA in the Raw cells (Fig. 2A) and in MEFs (Fig. 2B) compromises the ability of LPS to induce MyD88 promoter activity. Consistent with these observations, we found that MTA1 overexpression in the Raw cells induces MyD88 promoter activity as well as MyD88 mRNA (Fig. 2, C and D). These results suggest that MTA1 may be a mechanistic mediator of MyD88 stimulation in LPS-stimulated cells.
FIGURE 2.

MTA1 regulates MyD88 transcription. A, MyD88 promoter-luciferase activity in LPS (1 μg/ml for 4 h) stimulated Raw cells with or without MTA1 knockdown. B, MyD88 promoter-luciferase activity in LPS (1 μg/ml for 4 h)-stimulated wild type and MTA1−/− MEFs. C, MyD88 promoter-luciferase activity in LPS-stimulated Raw cells after cotransfecting with control vector pcDNA or MTA1. D, qPCR analysis of MyD88 mRNA in LPS (4 h) stimulated Raw cells after cotransfecting with control vector pcDNA or MTA1.
To gain a deeper insight into the mechanism of LPS stimulation of the MyD88 transcription, we next conducted a ChIP-based promoter-walk in the Raw cells stimulated with or without LPS and mapped the recruitment of MTA1 onto two regions of the MyD88 promoter at (+81 to −93) and (−280 to −569) (Fig. 3, A and B). We also found that MTA1·Pol II coactivator complexes are corecruited to these regions of MyD88 promoter in LPS-stimulated macrophages (Fig. 3C). Interestingly, we also observed an in-parallel, de-recruitment of the MTA1·HDAC2 corepressor complex from the above regions of the MyD88 promoter in LPS-stimulated Raw cells (Fig. 3D). Together, these findings suggest that MTA1 targets MyD88 chromatin in LPS-stimulated macrophages.
FIGURE 3.

MyD88 is a MTA1 target gene. A, binding region of MTA1 on murine MyD88 promoter. B, recruitment of MTA1 to MyD88 chromatin in Raw cells treated with LPS for 1 h. C, ChIP analysis of MTA1 followed by RNA polymerase II recruitment to MyD88 chromatin in Raw cells treated with LPS for 1 h. D, ChIP analysis of MTA1 followed by HDAC2 recruitment to MyD88 chromatin in Raw cells treated with LPS for 1 h.
MyD88 Is a NF-κB-regulated Gene
Because MTA1 status affects both NF-κB signaling (28) and MyD88 expression in LPS-stimulated Raw cells (this study) and because LPS is a potent inducer of NF-κB (33), we next determined the mechanism of LPS regulated MyD88 expression. As illustrated in Fig. 4A, inhibition of the NF-κB pathway by the pharmacologic inhibitor parthenolide suppressed LPS-mediated stimulation of MyD88 promoter activity and MyD88 protein expression in the Raw cells, suggesting that LPS may regulate MyD88 via NF-κB pathway. In support of these findings, we found that transiently expressed p65RelA increases MyD88 promoter activity under both basal and LPS-inducible conditions in the Raw cells (Fig. 4B).
FIGURE 4.

MyD88 is target gene of NF-κB. A, MyD88 promoter-luciferase activity followed by Western blot analysis for MyD88 expression in Raw cells treated with LPS in the presence of parthenolide. B, MyD88 promoter-luciferase activity in Raw cells treated with LPS after cotransfecting with p65. C, four NF-κB motifs in mouse MyD88 promoter. The NF-κB consensus sequences were identified by using the program alibaba 2.1. The functionally active NF-κB sites are in bold. D, ChIP analysis of p65, p65 followed by RNA polymerase II, and MTA1 followed by p65 recruitment to MyD88 chromatin in Raw cells treated with LPS for 1 h. E, effect of parthenolide on the recruitment of p65 onto MyD88 promoter (+81 to −93 and −280 to −569) in LPS-stimulated Raw cells. F, EMSA analysis of p65 binding to the mouse MyD88 promoter using the oligonucleotide encompassing NF-κB consensus sequences (Site 1, Site 2, and Site 3) in Raw cells treated LPS for 2 h.
To elucidate the molecular details of MTA1 regulation of MyD88 transcription, we scanned the MyD88 promoter for the putative NF-κB consensus motifs. This exercise led to the identification of three perfect NF-κB motifs in the MyD88 promoter for the first time (Fig. 4C). Results from anti-p65RelA antibody-based ChIP analysis of the MyD88 promoter indicated that LPS stimulation enhances the recruitment of p65RelA to the regions 1 (+81 to −93) and (−280 to −569) (Fig. 4D) and not to region 3 (−877 to −1053) of the MyD88 promoter. We also found that p65RelA·Pol II and MTA1·p65RelA complex also recruited to these regions in the MyD88 promoter (Fig. 4D). Importantly, p65RelA recruitment to the MyD88 chromatin was effectively prevented by the NF-κB inhibitor parthenolide (Fig. 4E).
Region 1 of the MyD88 promoter contains a single NF-κB site (site 1), whereas region 2 contains two NF-κB sites (sites 2 and 3) (Fig. 4C). To demonstrate a potential direct binding of p65RelA to the mouse MyD88 promoter DNA, we next performed EMSA using a 26-bp oligonucleotide encompassing sites 1, 2, and 3 and the nuclear extracts from LPS-stimulated Raw cells. We found evidence of distinct p65RelA·DNA complex formation using probes encompassing sites 1 and 3 but not site 2 of the MyD88 promoter DNA (Fig. 4F). The specificity of the detected protein·DNA complex was further verified by supershift experiments using anti-p65RelA but not control IgG or MTA1-antibody (Fig. 4F, compare lanes 4 and 14 with lanes 2 and 12, respectively). These results suggest that LPS stimulates MyD88 transcription via a direct binding of p65RelA binding to the MyD88 promoter and that MyD88 is a NF-κB-responsive gene.
MyD88 Regulates NF-κB Target Genes
To determine whether MyD88 plays a mechanistic role in LPS-stimulation of the NF-κB target genes, we used a well characterized dominant negative mutant of MyD88, known as TIR-MyD88 (12). We found that transient expression of TIR-MyD88 impaired the ability of LPS to stimulate the NF-κB-luc activity (Fig. 5A) as well as TNF-α promoter-luc activity (Fig. 5B). Consistent with these findings, TIR-MyD88 also impaired the ability of LPS to stimulate MTA1 promoter-luc activity and MTA1 expression (Fig. 5C), which itself is a NF-κB-regulated gene (28). These findings suggested a potential role of MyD88 in the expression of LPS-inducible genes.
FIGURE 5.

Selective knockdown of MyD88 compromised NF-κB, TNF-α, and MTA1 expression. A, NF-κB promoter-luciferase activity in LPS-stimulated Raw cells with or without TIR-MyD88. B, TNF-α promoter-luciferase activity in LPS-stimulated Raw cells with or without TIR-MyD88. C, MTA1-promoter-luciferase activity and qPCR analysis of MTA1 mRNA expression in LPS-stimulated Raw cells with or without TIR-MyD88, a dominant negative mutant of MyD88.
To validate the role of MyD88 in the expression of LPS-inducible genes independently, we next showed that selective siRNA-mediated knockdown of MyD88 in the Raw cells is accompanied by a significant reduction in the ability of LPS to induce promoter activities driven from TNF-α, MIP2, IL-1β, and MTA1 (Fig. 6A) and the mRNAs expression of TNF-α, IL-1β, MIP2, and MTA1 (Fig. 6B) in Raw cells. To understand further the possible involvement of other TLRs in mediating the observed LPS-induced cytokine production, we next examined the mRNA levels of TLR4, TLR3, and TLR2 along with INF-β, one of the targets of TLR3 in LPS-stimulated MEFs and macrophages isolated from MTA1+/+ and MTA1−/− mice. MTA1 depletion substantially compromised the ability of LPS to induce TLR4 mRNA expression, whereas the mRNA levels of TLR2, TLR3, and INF-β were not compromised in MEFs and macrophages (Fig. 7, A and B), suggesting the involvement of TLR4 MyD88-dependent signaling as modified by the status of MTA1. In addition to these findings, we also observed the complete abolition of the MAPK activation in response to LPS treatment of MTA1−/− MEFs (Fig. 7C). However, an early activation of MAPKs was observed in MTA1+/+ MEFs in response to LPS as expected (Fig. 7C). These results also validated the role of MAPKs in TLR4 MyD88-dependent signaling. Together, these findings revealed that MTA1 regulation of MyD88 expression may constitute, at least in part, one of the mechanisms by which MTA1 modulates LPS-induced NF-κB signaling in stimulated macrophages (Fig. 7D).
FIGURE 6.

MyD88 regulates NF-κB target genes. A, TNF-α, MIP2, IL-1β, MTA1 promoter-luciferase activities in LPS (1 μg/ml for 4 h) stimulated Raw cells after knocking down MyD88 using MyD88-siRNA. B, qPCR analysis of TNF-α, MIP 2, IL-1β, MTA1, and MyD88 mRNA expression in LPS-stimulated Raw cells after knocking down MyD88 using MyD88-siRNA is shown.
FIGURE 7.

MTA1 regulates LPS response via TLR4 MyD88-dependent signaling. A and B, qPCR analysis of TLR4, TLR2, TLR3, and INF-β mRNA expressions in LPS-stimulated MEFs and macrophages isolated from MTA1+/+ and MTA1−/− mice, respectively. C, Western blot analysis of phospho-ERK (P-ERK), ERK, phospho-JNK (P-JNK), and JNK in MTA1+/+ and MTA1−/− MEFs in the presence and absence of LPS. D, schematic representation of MTA1 regulating LPS response via MyD88-dependent signaling: LPS stimulation of macrophages is accompanied by transcriptional stimulation of MTA1 through NF-κB pathway. Once MTA1 is induced, it acts as a coactivator and gets recruited to the MyD88 promoter, leading to increased MyD88 expression, which in turn, further potentiates NF-κB signaling as well as MyD88 transcription.
In brief, our finding of MTA1 regulation of MyD88 has introduced a new regulatory player to the cascade of event leading to the induction of immune and proinflammatory genes by TLRs. In general, TLR signaling is regulated at multiple levels, and some, but not all, of the pathways are MyD88-dependent (34–36). For example, MyD88 regulates proinflammatory cytokines in a class-specific manner (37) and also needed for the recruitment of NF-κB, RNA polymerase II, and TATA-binding proteins to secondary response gene promoters (Cxcl2, Cxcl1, and Tnf genes which encode MIP2, GRO1, and TNF-α, respectively) (37). Accordingly, expression levels of all of these (primary and secondary responsive genes) are significantly reduced in MyD88−/− macrophages (37). Chromatin modifications of TLR-dependent genes have been also implicated in the negative regulation of the inflammatory response (38). In addition to inflammation, recent studies have also linked a role of MyD88 in NF-κB-mediated proliferation and invasion of ovarian, colon, and pancreatic cancer cells (20, 39). However, the precise molecular mechanisms responsible for the regulation of MyD88 at the transcriptional level in cancer and immune cells remain poorly understood. In this context, our present data demonstrate that MTA1 contributes to the regulation of the LPS response via modulating the MyD88-dependent signaling and expression of proinflammatory cytokines. Emerging data suggest that MTA1 regulates its target genes either by acting as a corepressor (40, 41) or as transcriptional coactivator via interacting with RNA polymerase II (42, 43). Our results showed that MyD88 is target of MTA1, and it regulates the MyD88 expression at the transcription level by directly binding to its promoter as a part of the p65 and RNA polymerase II complex (Figs. 3 and 4). These findings raises the possibility that MTA1 may protect the animal by regulating the innate immune response either by directly modulating the NF-κB signaling (28) or by regulating the MyD88-dependent signaling. Because MTA1 is widely up-regulated in human cancer, these findings further add to our understanding of the complex regulatory mechanisms involved between the immune system and the cancer.
Supplementary Material
Acknowledgment
We thank Katherine A. Fitzgerald for providing the dominant negative MyD88 construct.
This work was supported, in whole or in part, by National Institutes of Health Grants CA98823 and CA98823-S1 (to R. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1–3.
- TLR
- Toll-like receptor
- ChIP
- chromatin immunoprecipitation
- MEF
- mouse embryonic fibroblast
- MTA1
- metastatic tumor antigen 1
- MyD88
- myeloid differentiation primary response gene 88
- qPCR
- quantitative PCR
- TIR
- Toll/interleukin-1 response.
REFERENCES
- 1.Takeda K., Akira S. (2004) Semin. Immunol. 16, 3–9 [DOI] [PubMed] [Google Scholar]
- 2.Zhang D., Zhang G., Hayden M. S., Greenblatt M. B., Bussey C., Flavell R. A., Ghosh S. (2004) Science 303, 1522–1526 [DOI] [PubMed] [Google Scholar]
- 3.Chen R., Alvero A. B., Silasi D. A., Steffensen K. D., Mor G. (2008) Oncogene 27, 225–233 [DOI] [PubMed] [Google Scholar]
- 4.Gorden K. B., Gorski K. S., Gibson S. J., Kedl R. M., Kieper W. C., Qiu X., Tomai M. A., Alkan S. S., Vasilakos J. P. (2005) J. Immunol. 174, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 5.Akira S., Uematsu S., Takeuchi O. (2006) Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 6.Beutler B., Jiang Z., Georgel P., Crozat K., Croker B., Rutschmann S., Du X., Hoebe K. (2006) Annu. Rev. Immunol. 24, 353–389 [DOI] [PubMed] [Google Scholar]
- 7.Beutler B. (2004) Immunity 21, 134–135 [DOI] [PubMed] [Google Scholar]
- 8.Muzio M., Ni J., Feng P., Dixit V. M. (1997) Science 278, 1612–1615 [DOI] [PubMed] [Google Scholar]
- 9.McDermott E. P., O'Neill L. A. (2002) J. Biol. Chem. 277, 7808–7815 [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. (2003) Nat. Immunol. 4, 1144–1150 [DOI] [PubMed] [Google Scholar]
- 11.Oshiumi H., Sasai M., Shida K., Fujita T., Matsumoto M., Seya T. (2003) J. Biol. Chem. 278, 49751–49762 [DOI] [PubMed] [Google Scholar]
- 12.Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., McMurray D., Smith D. E., Sims J. E., Bird T. A., O'Neill L. A. (2001) Nature 413, 78–83 [DOI] [PubMed] [Google Scholar]
- 13.Kawai T., Adachi O., Ogawa T., Takeda K., Akira S. (1999) Immunity 11, 115–122 [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi O., Akira S. (2010) Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 15.Turer E. E., Tavares R. M., Mortier E., Hitotsumatsu O., Advincula R., Lee B., Shifrin N., Malynn B. A., Ma A. (2008) J. Exp. Med. 205, 451–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakoff-Nahoum S., Medzhitov R. (2009) Nat. Rev. Cancer 9, 57–63 [DOI] [PubMed] [Google Scholar]
- 17.Sheahan T., Morrison T. E., Funkhouser W., Uematsu S., Akira S., Baric R. S., Heise M. T. (2008) PLoS Pathog. 4, e1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iimuro Y., Seki E., Son G., Tsutsui H., Nakanishi K., Fujimoto J. (2007) J. Gastroenterol. Hepatol. 22, Suppl. 1, S57–S58 [DOI] [PubMed] [Google Scholar]
- 19.Karin M. (2006) Nature 441, 431–436 [DOI] [PubMed] [Google Scholar]
- 20.Ikebe M., Kitaura Y., Nakamura M., Tanaka H., Yamasaki A., Nagai S., Wada J., Yanai K., Koga K., Sato N., Kubo M., Tanaka M., Onishi H., Katano M. (2009) J. Surg. Oncol. 100, 725–731 [DOI] [PubMed] [Google Scholar]
- 21.Grivennikov S. I., Greten F. R., Karin M. (2010) Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mantovani A., Allavena P., Sica A., Balkwill F. (2008) Nature 454, 436–444 [DOI] [PubMed] [Google Scholar]
- 23.Toh Y., Nicolson G. L. (2009) Clin. Exp. Metastasis 26, 215–227 [DOI] [PubMed] [Google Scholar]
- 24.Manavathi B., Kumar R. (2007) J. Biol. Chem. 282, 1529–1533 [DOI] [PubMed] [Google Scholar]
- 25.Denslow S. A., Wade P. A. (2007) Oncogene 26, 5433–5438 [DOI] [PubMed] [Google Scholar]
- 26.Toh Y., Pencil S. D., Nicolson G. L. (1994) J. Biol. Chem. 269, 22958–22963 [PubMed] [Google Scholar]
- 27.Kumar R., Wang R. A., Bagheri-Yarmand R. (2003) Semin. Oncol. 30, 30–37 [DOI] [PubMed] [Google Scholar]
- 28.Pakala S. B., Bui-Nguyen T. M., Reddy S. D., Li D. Q., Peng S., Rayala S. K., Behringer R. R., Kumar R. (2010) J. Biol. Chem. 285, 23590–23597 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Kumar R., Wang R. A., Mazumdar A., Talukder A. H., Mandal M., Yang Z., Bagheri-Yarmand R., Sahin A., Hortobagyi G., Adam L., Barnes C. J., Vadlamudi R. K. (2002) Nature 418, 654–657 [DOI] [PubMed] [Google Scholar]
- 30.Bui-Nguyen T. M., Pakala S. B., Sirigiri R. D., Xia W., Hung M. C., Sarin S. K., Kumar V., Slagle B. L., Kumar R. (2010) Oncogene 29, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noman A. S., Koide N., Hassan F., I-E-Khuda I., Dagvadorj J., Tumurkhuu G., Islam S., Naiki Y., Yoshida T., Yokochi T. (2009) Innate Immun. 15, 33–41 [DOI] [PubMed] [Google Scholar]
- 32.Manavathi B., Peng S., Rayala S. K., Talukder A. H., Wang M. H., Wang R. A., Balasenthil S., Agarwal N., Frishman L. J., Kumar R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13128–13133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q., Verma I. M. (2002) Nat. Rev. Immunol. 2, 725–734 [DOI] [PubMed] [Google Scholar]
- 34.Chang J., Kunkel S. L., Chang C. H. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 18327–18332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liew F. Y., Xu D., Brint E. K., O'Neill L. A. (2005) Nat. Rev. Immunol. 5, 446–458 [DOI] [PubMed] [Google Scholar]
- 36.Lang T., Mansell A. (2007) Immunol. Cell Biol. 85, 425–434 [DOI] [PubMed] [Google Scholar]
- 37.Kayama H., Ramirez-Carrozzi V. R., Yamamoto M., Mizutani T., Kuwata H., Iba H., Matsumoto M., Honda K., Smale S. T., Takeda K. (2008) J. Biol. Chem. 283, 12468–12477 [DOI] [PubMed] [Google Scholar]
- 38.Foster S. L., Hargreaves D. C., Medzhitov R. (2007) Nature 447, 972–978 [DOI] [PubMed] [Google Scholar]
- 39.Kelly M. G., Alvero A. B., Chen R., Silasi D. A., Abrahams V. M., Chan S., Visintin I., Rutherford T., Mor G. (2006) Cancer Res. 66, 3859–3868 [DOI] [PubMed] [Google Scholar]
- 40.Molli P. R., Singh R. R., Lee S. W., Kumar R. (2008) Oncogene 27, 1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazumdar A., Wang R. A., Mishra S. K., Adam L., Bagheri-Yarmand R., Mandal M., Vadlamudi R. K., Kumar R. (2001) Nat. Cell Biol. 3, 30–37 [DOI] [PubMed] [Google Scholar]
- 42.Balasenthil S., Gururaj A. E., Talukder A. H., Bagheri-Yarmand R., Arrington T., Haas B. J., Braisted J. C., Kim I., Lee N. H., Kumar R. (2007) Cancer Res. 67, 7132–7138 [DOI] [PubMed] [Google Scholar]
- 43.Gururaj A. E., Singh R. R., Rayala S. K., Holm C., den Hollander P., Zhang H., Balasenthil S., Talukder A. H., Landberg G., Kumar R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6670–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.