

Abstract
AIM: To investigate the transforming growth factor-β (TGF-β) isoforms in the peripheral and hepatic venous blood of primary biliary cirrhosis (PBC) patients.
METHODS: We examined TGF-β1, TGF-β2 and TGF-β3 (enzyme-linked immunosorbent assay), in 27 stage IV PBC patients (27 peripheral and 15 hepatic vein sera), 35 early (I-II) PBC and 60 healthy controls. As disease controls 28 hepatitis C virus (HCV) cirrhosis (28 peripheral and 17 hepatic vein serum), 44 chronic HCV hepatitis and 38 HCV-related hepatocellular carcinomas were included. We also tested liver tissue by immunohistochemistry to identify localization of TGF isoforms.
RESULTS: TGF-β1 was significantly decreased in all cirrhotics (PBC III-IV: median 13.4 ng/mL; range, 7.4-26.2, HCV cirrhosis: 11.6 ng/mL; range, 5.0-33.8), compared to controls (30.9 ng/mL; range, 20.9-37.8). TGF-β2 was increased in viral cirrhosis but not in PBC and chronic hepatitis. TGF-β3 (47.2 pg/mL; range, 27.0-79.7 in healthy controls) was increased in early and late PBC (I-II: 94.3 pg/mL; range, 41.5-358.6; III-IV: 152.8 pg/mL; range, 60.4-361.2; P < 0.001) and decreased in viral cirrhosis (37.4 pg/mL; range, 13.3-84.0; P < 0.05). Hepatic vein TGF-β levels were analogous to those in peripheral blood. Immunohistochemistry identified all isoforms in portal tract lymphocytes, sinusoidal cells and cholangiocytes. TGF-β3 was additionally overexpressed in hepatocytes in PBC patients.
CONCLUSION: The serum profile of TGF-β isoforms is different in cirrhotics. Increased TGF-β3 is characteristic of PBC. These findings may be related to the immunological abnormalities of PBC.
Keywords: Transforming growth factor-β, Primary biliary cirrhosis, Liver fibrosis, Cirrhosis
INTRODUCTION
Primary biliary cirrhosis (PBC) is an autoimmune disease characterized by lymphocytic infiltration of the portal tracts and selective destruction of intrahepatic small bile ducts with progressive fibrosis and finally cirrhosis[1]. T-regulatory cells (T-reg) may play an important role[2]. Transforming growth factor-β (TGF-β) is a pleomorphic cytokine regulating many cellular functions including proliferation, differentiation, migration and survival[3,4]. Three members of the TGF-β family have been identified with TGF-β1 being predominant in the immune system[5]. Many aspects of TGF-β functions may be related to the pathogenesis of PBC.
T-reg downregulate immune responses and there is a reciprocal relationship between T-reg and the recently described Th17 cells[6]. TGF-β induces both the differentiation[7,8] and maintenance[9] of T-reg Fox P3-positive cells, while in conjunction with the pro-inflammatory cytokine interleukin-6 (IL-6) induces the generation of the proinflammatory Th17 cells[10-12].
Evidence from transgenic mice indicate that impaired TGF-β signaling is involved in the pathogenesis of PBC, through deregulation of T-reg cells[13]. TGF-β is also involved in the fibrotic process. TGF-β stimulates the synthesis of laminin, collagen IV and entactin by liver endothelial cells[14] and stellate cells, but myofibroblasts are not responsive to TGF-β[15]. Liver fibrosis is reduced by 50%-70% after TGF-β blockade[16].
Despite experimental evidence for the involvement of TGF-β in the pathogenesis of PBC, data in humans is scarce.
We therefore studied serum and liver tissue TGF-β isoforms in patients with PBC in comparison with normal controls and patients with other chronic liver diseases.
MATERIALS AND METHODS
Patients
Informed consent was obtained from each patient included in the study. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of the University Hospital. Diagnosis and fibrosis staging in all patients was verified by liver biopsy. Patients with early PBC (35 stage I-II) and late PBC (27 stage III-IV, according to Ludwig) were diagnosed on the basis of the histology plus the presence of antimitochondrial antibodies (AMA) and M2 antibodies by enzyme-linked immunosorbent assay (ELISA) and increased alkaline phosphatase. Patient demographics are presented in Table 1. Hepatitis C virus (HCV) patients were used as the disease control group (28 HCV cirrhosis, 44 chronic HCV hepatitis, genotype 1, and 38 HCV cirrhosis-associated hepatocellular carcinoma (HCC). All had positive anti-HCV tests (Abbott) and a positive qualitative and quantitative determination of HCV RNA. Sixty age-matched normal controls were included (from blood donors, staff and visitors of the unit). Blood samples were collected from the hepatic vein of 15 cirrhotic PBC patients and 17 HCV cirrhotics during measurement of intrahepatic wedge pressure. All sera were stored at -80°C until assayed.
Table 1.
Basic characteristics of patients
| PBC | Chronic hepatitis | HCC | Viral cirrhosis | |
| No. of patients | 62 | 44 | 38 | 28 |
| Mean age | 69.1 ± 8.8 | 53 ± 11 | 62 ± 8.3 | 64 ± 10.3 |
| Mayo score (mean) | 5.19 ± 1.44 | |||
| Gender (F/M) | 56/6 | 36/8 | 13/25 | 16/12 |
| Bilirubin (mg/dL) | 1.5 ± 0.6 | 1.2 ± 0.4 | 1.4 ± 0.5 | 1.8 ± 0.5 |
| Albumin (g/dL) | 4.2 ± 1.0 | 4.5 ± 0.8 | 3.6 ± 0.4 | 3.0 ± 0.9 |
| Alk phos (UI/L) (normal < 125) | 185 ± 36 | 110 ± 15 | 152 ± 28 | 138 ± 18 |
| γGt (UI/L) (normal < 50) | 98 ± 25 | 39 ± 18 | 65 ± 23 | 57 ± 13 |
| ALT (UI/L) | 62 ± 13 | 92 ± 36 | 71 ± 11 | 66 ± 17 |
| AST (UI/L) | 55 ± 17 | 86 ± 32 | 65 ± 21 | 59 ± 12 |
| IgM (mg/dL) | 428 ± 52 | 165 ± 32 | 138 ± 28 | 139 ± 47 |
| Histological stage | ||||
| Ludwig I-II | 35 | |||
| Ludwig III-IV | 27 | |||
| Ishak 1-3 | 30 | |||
| Ishak 4-5 | 14 |
PBC: Primary biliary cirrhosis; HCC: Hepatocellular carcinoma; Alk phos: Alkaline phosphatase; γGt: γ-glutamyl transpeptidase; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; Ig: Immunoglobulin.
Immunohistochemistry was performed on liver tissue from 15 PBC patients (7 stage I-II and 8 stage IV), 10 patients with chronic hepatitis C and 10 patients with HCV-related cirrhosis.
Methods
ELISAs: TGF-β1, TGF-β2 and TGF-β3 were measured with commercially available ELISAs (DuoSet® ELISA Development System, human TGF-β1, human TGF-β2, human TGF-β3, R&D Systems Abingdon, UK), according to the manufacturer’s instructions.
Immunocytochemistry: TGF-β isoform expressions were studied in paraffin sections by the alkaline phosphatase method or by immunofluorescence using monoclonal antibodies.
For the alkaline phosphatase method the DAKO APAAP Mouse Real™ Detection System, was used according to the manufacturer’s instructions.
Incubation with the primary antibody (Mouse anti-human TGF-β1, TGF-β2 and TGF-β3 R&D Systems diluted 1:10 with the DAKO Rear™Antibody diluent) was done overnight at room temperature, followed by incubation with primary antibody enhancer for 25 min and with the secondary alkaline phosphatase conjugated antibody for 30 min. A-naphthol phosphate and Fast Red were used to demonstrate the presence of TGF-β isoforms.
For immunofluorescence, antigen retrieval was achieved by incubation with citrate buffer (1.8 mmol/L citric acid and 8.2 mmol/L sodium citrate) for 2 h at 37°C.
After blocking with phosphate-buffered saline containing 0.2% TritonX-100, 2 mmol/L MgCl2 and 1% gelatin from cold water fish skin (Sigma-Aldrich, Germany) for 10 min, sections were incubated overnight at 4°C with mouse anti-human TGF-β1 (R&D Systems, UK, dilution 1/10) in blocking buffer. Sections were washed with blocking buffer and incubated with Alexa fluor 488 F(ab’)2 fragment of goat anti-mouse IgG (H+L) (Invitrogen, UK, dilution 1/1000) for 1 h at room temperature.
For detection of TGF-β2 and TGF-β3 (R&D Systems, UK, dilution 1/20) we followed the same procedure, except for an incubation time of 2 h.
Negative controls for both alkaline phosphatase and immunofluorescence were run by omitting the primary antibody.
Statistical analysis
The results of serum TGF-β1, TGF-β2 and TGF-β3 were not normally distributed according to Bartlett’s test. The Kruskal-Wallis non-parametric analysis of variance was used for comparisons among all groups, with Dunn’s procedure for multiple comparisons. The Kolmogorov and Smirnov test was used to assess Gaussian distributions All tests were performed with the SPSS version 15 statistical package. P < 0.05 was considered significant. All values are expressed as median and range.
RESULTS
TGF-β1
Overall there was a significant difference in TGF-β1 among different groups (P < 0.0001). Patients with cirrhosis, whether viral or PBC, had lower values compared to normal controls (HCV cirrhosis 11.6 ng/mL; range, 5.0-33, PBC stages III-IV 13.4 ng/mL; range, 7.4-26.2 ng/mL vs 30.9 ng/mL; range, 20.9-37.8 for healthy controls). Early PBC patients also had lower levels than controls (22.5 ng/mL; range, 7.2-38.3). TGF-β1 levels were also low in the blood from hepatic veins (Figure 1A).
Figure 1.

Transforming growth factor-β1 (A), transforming growth factor-β2 (B) and transforming growth factor-β3 (C). A: Box plots showing the interquartile range (box), median (horizontal line) and range (vertical lines) of the serum levels of transforming growth factor-β1 (TGF-β1) in normal controls, patients with hepatocellular carcinoma (HCC), viral cirrhosis, chronic active hepatitis C (CAH), primary biliary cirrhosis (PBC) stages I-II, PBC stages III-IV, in hepatic vein blood from PBC and viral cirrhosis patients (central). B, C: Box plots showing the interquartile range (box), median (horizontal line) and range (vertical lines) in the peripheral and hepatic vein serum levels of TGF-β2 and TGF-β3 of the same group of patients. aP < 0.05, bP < 0.01, dP < 0.001.
TGF-β2
There was also a significant difference in TGF-β2 among groups (P < 0.0001). However, patients with viral cirrhosis (442.4 pg/mL; range, 282.8-967.0) had higher levels than controls (370.5 pg/mL; range, 235.0-495.7) while HCC cirrhotics had the highest values. By contrast, patients with either early PBC (349.2 pg/mL; range, 208.4-616.2) or late PBC (384.7 pg/mL; range, 298.9-543.8) had levels comparable with controls. This was also reflected in values from the hepatic veins (Figure 1B).
TGF-β3
Although the overall difference in TGF-β3 among groups was also highly significant (P < 0.0001) the profile was completely different. Patients with early (94.3 pg/mL; range, 41.5-358.6) and late PBC (152.8 pg/mL; range, 60.4-361.2; P < 0.001) had high values compared to controls (47.2 pg/mL; range, 27.0-79.7 in healthy controls), but viral cirrhotics (37.4 pg/mL; range, 13.3-84.0; P < 0.05) had statistically lower values compared to either controls (P < 0.005) or to both groups of PBC patients (P < 0.05 and P < 0.01, respectively). Levels were high in the hepatic vein from PBC patients and low in HCV cirrhotics (Figure 1C).
Immunohistochemistry
TGF-β1: There was TGF-β1 expression in bile duct epithelium and in numerous mononuclear cells in portal tracts in all disease groups. Endothelial cells of either the hepatic artery or the portal vein were positive (Figure 2A). Sinusoidal cells were also positive. Periportal hepatocytes showed a variable expression of TGF-β1 with either a diffuse cytoplasmic or a more globular pattern.
Figure 2.
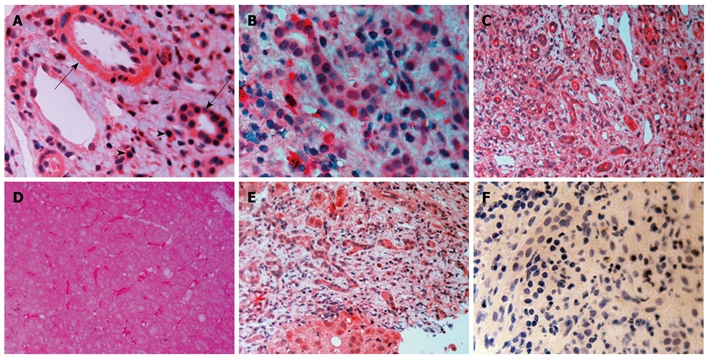
Immunocytochemistry for transforming growth factor-β1, transforming growth factor-β2 and transforming growth factor-β3. A: Transforming growth factor-β1 (TGF-β1) in viral cirrhosis. Positive cholangiocytes (arrows), cells in the hepatic arterial wall and many positive lymphocytes (arrowheads); B: Primary biliary cirrhosis (PBC) stage II, TGF-β2. Numerous positive cholangioles and positive mononuclear cells, probably lymphocytes; C: TGF-β3 in viral cirrhosis. Many positive hyperplastic cholangioles and positive lymphocytes; D: PBC stage I, TGF-β3. Hepatocytes vaguely stained. Strong staining of sinusoidal cells; E: PBC stage IV, TGF-β3. Positive hepatocytes, cholangioles and mononuclear cells; F: PBC stage IV, negative control. Magnifications are A, B and F × 400; C-E, × 200.
TGF-β2: A similar picture was also present. (Figure 2B). Globular deposition of TGF-β2 was evident in many hepatocytes. Some cholangioles were negative in PBC patients.
TGF-β3: Although the pattern was similar in all disease groups (Figure 2C-E), hepatocytes from patients with PBC showed a strong staining compared with the either chronic HCV hepatitis or HCV cirrhosis patients (Figure 3B). In addition, on immunofluorescence, positive lymphocytes infiltrating portal bile ductules demonstrated weaker rim fluorescence compared with other positive portal lymphocytes (Figure 3A).
Figure 3.
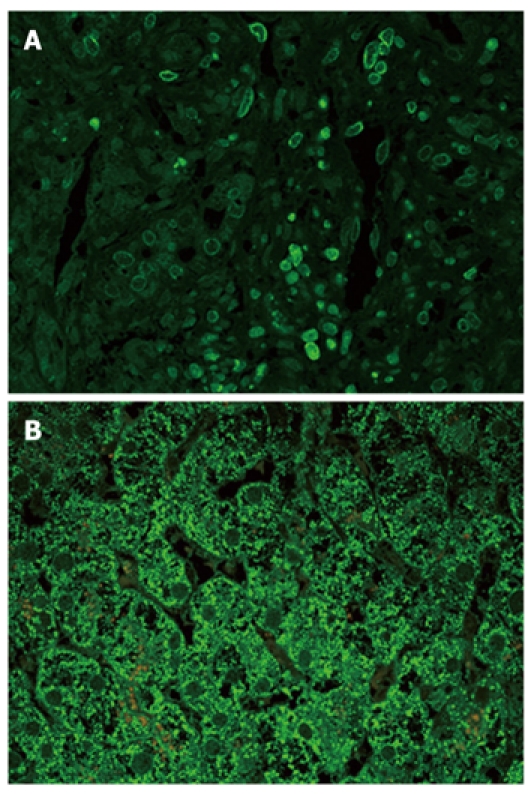
Immunofluorescence for transforming growth factor-β in PBC 3. A: Many positive mononuclear cells. Rim staining of varying intensity is evident. Some cells with low rim staining seem to infiltrate a bile ductule; B: Hepatocytes strongly stained in stage IV.
DISCUSSION
We have demonstrated an increase in TGF-β3 in both peripheral blood and blood from the hepatic vein in patients with PBC. Interestingly, the increase in TGF-β3 was observed in early stage PBC, indicating that this is a disease-specific finding and not merely the result of the cirrhotic process. TGF-β1 was decreased in all cirrhotic patients, either HCV-related or PBC and this was also found in the blood of hepatic veins. TGF-β2 was the isoform that was increased in viral cirrhosis but not in PBC, a finding also verified in hepatic veins. In addition, we identified cholangiocytes as a source of liver TGF-β isoforms, in addition to portal lymphocytes and sinusoidal cells. Hepatocytes were also positive for TGF isoforms but it was TGF-β3 that was strongly expressed in hepatocytes from PBC patients.
There is strong experimental evidence that TGF-β is implicated in the pathogenesis of PBC, probably through deregulation of T-reg.
Transgenic mice with the dominant-negative TGF type II receptor (dn TGF-βRII), spontaneously develop biliary disease resembling human PBC[13]. The normal liver contains only a few quiescent CD4 FoxP3 T-reg but it can rapidly recruit these cells when CD8 T blasts appear[17].
In PBC liver, destruction of biliary cells is mediated by liver infiltrating T-cells, especially CD8 cytotoxic cells which are highly enriched in PBC livers[18]. However in the CD25-negative mouse model, lack of CD8 cells attenuates but does not abolish bile ductular destruction, indicating that an additional mechanism of destruction is also present in PBC[19].
TGF-βRII is also directly implicated in liver fibrosis[20].
Mice deficient in the IL-2 receptor (CD25) demonstrate a PBC-like lesion in the liver[21]. A child with a complete deficiency of the α subunit of the IL-2 receptor in peripheral CD25 lymphocytes developed a PBC-like disease[22], indicating that animal models may be relevant in human disease. Similarly, experimental evidence that T-reg are reduced in a model of PBC[23] have been verified in human disease where decreased numbers of peripheral and liver T-reg were found in PBC patients and most importantly in the peripheral blood of their relatives[24].
A substantial number of FoxP3 T-reg are negative for CD25[25]. It seems therefore that the lymphocytes positive for TGF-β histochemically identified in our study are either CD25-FoxP3 T-reg or other conventional T-cells capable of producing TGF-β isoforms[4,26].
Peripheral blood levels of TGF-β have not been adequately studied in PBC. Circulating TGF-β has been reported to reflect the severity of PBC and is reduced after 2 years of treatment with ursodeoxycholic acid[27].
Although the 3 isoforms of TGF-β are considered to have similar functions, there is evidence that this might not be so. Loss of TGF-β1 is compatible with postnatal survival, but the liver and other tissues are infiltrated with mononuclear cells, while loss of TGF-β2 and TGF-β3 leads to perinatal death[28,29].
Hepatic stellate and Kupffer cells are reported to be the main sources of TGF-β[30]. However the origin of TGF-β isoforms is not well established. Studies from experimental animals indicate that in the normal state Kupffer cells express all 3 isoforms[31]. Endothelial cells express mostly TGF-β1 and lower levels of TGF-β2 and TGF-β3 while stellate cells express very little TGF-β, and hepatocytes have no constitutional expression[32]. After a fibrogenic injury all forms of TGF-β increase in stellate cells probably through stimulation by retinoic acid produced by hydrolysis of retinol esters during activation of stellate cells[33]. Induced T-reg secrete TGF-β[34]. TGF-β from other sources such as dendritic cells may also be important[35]. Therefore analysis of peripheral levels is difficult due to the many potential sources of TGF-β that may reach the circulation. In particular, the cellular source of TGF-β required for T-reg maintenance in PBC or other liver disease is not clear.
Our histochemical findings indicate that portal lymphocytes are sources of TGF-β isoforms in both PBC and chronic HCV and so are sinusoidal cells. As already mentioned, lymphocytes are probably T-reg although detailed studies are required. We identified a novel source of TGF-β isoforms as intrahepatic bile ductules were strongly positive for TGF-β. It is conceivable that the increase in TGF-β3 observed in PBC may result from an overproduction by intrahepatic cholangiocytes.
Clearance of TGF-β is complex. It may be sequestered by α2 macroglobulin[36], undergo renal clearance or be taken up by hepatocytes[37]. This might explain the positive globules observed within hepatocytes in our study, at least for TGF-β1 and TGF-β2. However, hepatocytes from patients with PBC particularly from stage IV were strongly positive for TGF-β3 compared with the other 2 isoforms (Figure 3), a finding which is unlikely to be due to increased uptake. Hepatocytes therefore are an additional source for the increased levels of TGF-β3 in patients with PBC.
Levels in the hepatic vein blood followed the same pattern as in peripheral blood both for TGF-β3 and TGF-β2, confirming the liver as the source of TGF-β isoform differences found in our study.
In our study, TGF-β2 and TGF-β3 circulate in pg quantities as compared to ng for TGF-β1. In fibroblast cultures, TGF-β3 was proved to be 3-5 times more potent than TGF-β1[38]. TGF-β3 is also significantly more potent than the other 2 isoforms in stimulation of neovascularization[29]. The in vitro inhibitory activity of TGF-β3 on an epithelial cell line is in the range of 10-50 pg/mL[39] while transformation and proliferation of rat fibroblasts is achieved at a concentration of 100 pg/mL. Therefore, the levels we found are biologically relevant.
Based on previous reports and on the findings of the present study, we suggest a hypothetical model for the pathogenesis of PBC based on a dual mechanism for bile duct destruction. Our findings may provide a link to explain the reported immunological abnormalities in PBC.
Differences in TGF-β isoforms may lead to a deranged balance between T-reg and Th17 cells. It is TGF-β1 that modulates FoxP3 expression and the regulatory activity of CD4 cells[40]. The presence of increased local levels of TGF-β3 (and the concomitant relative lack of TGF-β1) in conjunction with increased levels of IL-6 may shift the balance towards an increased activity of the proinflammatory Th17 cells adding to the CD8 cytotoxic lymphocytes destructive mechanism, while at the same time it leading to functionally defective T-reg.
Although plasma levels of IL-6 are decreased in PBC[41], monocytes from PBC patients secrete more IL-1 and IL-6 after in vitro challenge with Toll-like receptor ligands[42], and increased expression of IL-6 has been described in the liver of PBC patients[43]. The increased presence of TGF-β3 instead of TGF-β1 could be a further mechanism that favors Th17 activity since one can postulate that TGF-β3 could either be associated with functionally defective T-reg or could directly favor the increase of Th17. Further evidence for such a hypothesis is provided by a recent report that mice with mutation of the gene encoding the FoxP3 transcriptional factor developed AMA positivity and features resembling PBC with increased levels of IL-17 and IL-23, cytokines associated with Th17 cells[44]. There is also recent evidence that Th17 cells are implicated in the pathogenesis of PBC as they are increased in the portal tracts of PBC patients[45].
As we found cholangiocytes to be a major source of TGF-β isoforms in the liver, one can further postulate that these cells may be the origin of the aberrant production of TGF-β isoforms in PBC. This requires further clarification.
Recent experimental evidence adds further support on the proposal for a crucial role of TGF-β3 in the pathogenesis of PBC. TGF-β3 was reported to decrease collagen synthesis and tissue inhibitor of metalloproteinases-1 expression, and increase matrix metalloproteinase-9 in CCl4-induced liver fibrosis[46]. This finding could offer a potential explanation for the delayed development of fibrosis and cirrhosis in PBC compared with other chronic liver diseases.
In our study we also found significantly increased levels of TGF-β2 in HCV-associated cirrhosis with the highest values found in cases with HCC in accordance with previous reports of increased TGF-β expression in human HCC[47,48]. Interestingly, T-reg are increased in numbers and correlate with disease progression and survival in patients with HCC[49].
In summary, we have demonstrated that the serum profile of the 3 TGF-β isoforms is different in the disease groups studied. An increase in TGF-β3 is a characteristic of PBC irrespective of stage and therefore it may be pathogenetically important. Results from the hepatic vein samples indicate that the liver seems to be the source of the TGF-β abnormalities. We also propose that these findings may be the causative link leading to T-reg and/or Th17 deregulations reported in PBC.
COMMENTS
Background
Primary biliary cirrhosis (PBC) is an autoimmune disease characterized by mononuclear infiltration of the portal tracts and destruction of intrahepatic small bile ducts, fibrosis and finally cirrhosis. transforming growth factor-β (TGF-β) is a cytokine regulating many cellular functions including proliferation, differentiation, migration and survival of hepatic stellate cells. Three members of the TGF-β family have been identified with TGF-β1 being the most studied. Many aspects of TGF-β functions may be related to the pathogenesis of PBC as it regulates both immune responses and fibrosis in the liver.
Research frontiers
The role of the 3 different isoforms of TGF-β in chronic liver disease has not been investigated. Different isoforms may be related to the pathogenesis of different chronic liver diseases. With this purpose in mind, the authors aimed to investigate the levels these isoforms in different chronic liver diseases both in peripheral and in hepatic vein blood samples in order to identify the pattern of their increase in these liver diseases.
Innovations and breakthroughs
Although TGF-β1 was significantly decreased in all cirrhotics compared to controls. TGF-β2 was increased in viral cirrhosis but not in PBC and chronic hepatitis. Interestingly TGF-β3 was increased in both early and late PBC and decreased in viral cirrhosis with the levels of this marker in the hepatic vein being analogous to the peripheral blood. TGF-β3 was also overexpressed in the hepatocytes of PBC patients. This is the first report of differences in the expression of TGF-β isoforms in a disease characterized by progressive fibrosis.
Applications
Increased TGF-β3 levels found in PBC might be genetically controlled and can induce immunological responses and profibrotic processes during disease progression. The findings of this study may provide support of further research in the field that will further elucidate the role of the TGF-β isoforms during liver disease progression and could lead to novel therapeutic approaches in PBC. Furthermore, the findings of this study suggest that TGF-β isoforms may also be regulated differently in other diseases characterized by autoimmune and fibrotic phenomena.
Terminology
TGF-β is a protein that regulates immune responses and fibrotic progression during liver diseases. Although the 3 isoforms of TFG-β have been known for many years the question whether they have distinct roles in different liver diseases has not been studied.
Peer review
This paper finds differences in TGF-β isotypes production in PBC. Until now we do not have a good answer for the role of TGF-β isoforms, then we can not approach the pathological reason why TGF-β3 is increased in PBC directly. The findings of this paper will indicate the new mechanism of PBC.
Footnotes
Supported by Research funds of the University of Crete
Peer reviewer: Shinji Shimoda, MD, PhD, Medicine and Biosystemic Science, Kyushu University Graduate School of Medical Sciences, 3-1-1 Maidashi, Higashi-Ku, Fukuoka 812-8582, Japan
S- Editor Wang YR L- Editor Cant MR E- Editor Lin YP
References
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 2.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 3.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 4.Wan YY, Flavell RA. 'Yin-Yang' functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev. 2007;220:199–213. doi: 10.1111/j.1600-065X.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 6.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 7.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 8.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 9.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 11.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 13.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, Ansari AA, Coppel RL, Li MO, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 14.Neubauer K, Krüger M, Quondamatteo F, Knittel T, Saile B, Ramadori G. Transforming growth factor-beta1 stimulates the synthesis of basement membrane proteins laminin, collagen type IV and entactin in rat liver sinusoidal endothelial cells. J Hepatol. 1999;31:692–702. doi: 10.1016/s0168-8278(99)80350-x. [DOI] [PubMed] [Google Scholar]
- 15.Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology. 2000;31:1094–1106. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]
- 16.George J, Roulot D, Koteliansky VE, Bissell DM. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bochtler P, Riedl P, Gomez I, Schirmbeck R, Reimann J. Local accumulation and activation of regulatory Foxp3+ CD4 T(R) cells accompanies the appearance of activated CD8 T cells in the liver. Hepatology. 2008;48:1954–1963. doi: 10.1002/hep.22559. [DOI] [PubMed] [Google Scholar]
- 18.Kita H, Lian ZX, Van de Water J, He XS, Matsumura S, Kaplan M, Luketic V, Coppel RL, Ansari AA, Gershwin ME. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–123. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu W, Zhang W, Tsuneyama K, Moritoki Y, Ridgway WM, Ansari AA, Coppel RL, Lian ZX, Mackay I, Gershwin ME. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(-/-) mice. Hepatology. 2009;49:133–140. doi: 10.1002/hep.22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 21.Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, Yang GX, Ridgway W, Ueno Y, Ansari AA, et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–1249. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 22.Aoki CA, Roifman CM, Lian ZX, Bowlus CL, Norman GL, Shoenfeld Y, Mackay IR, Gershwin ME. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50–53. doi: 10.1016/j.jaut.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Salas JT, Banales JM, Sarvide S, Recalde S, Ferrer A, Uriarte I, Oude Elferink RP, Prieto J, Medina JF. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134:1482–1493. doi: 10.1053/j.gastro.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, Chuang YH, Nakamura T, Saito S, Shimoda S, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006;43:729–737. doi: 10.1002/hep.21123. [DOI] [PubMed] [Google Scholar]
- 25.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 27.Neuman M, Angulo P, Malkiewicz I, Jorgensen R, Shear N, Dickson ER, Haber J, Katz G, Lindor K. Tumor necrosis factor-alpha and transforming growth factor-beta reflect severity of liver damage in primary biliary cirrhosis. J Gastroenterol Hepatol. 2002;17:196–202. doi: 10.1046/j.1440-1746.2002.02672.x. [DOI] [PubMed] [Google Scholar]
- 28.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 29.Cox DA. Transforming growth factor-beta 3. Cell Biol Int. 1995;19:357–371. doi: 10.1006/cbir.1995.1082. [DOI] [PubMed] [Google Scholar]
- 30.Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. 2010;34:1–6. doi: 10.1016/j.jaut.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Roth S, Gong W, Gressner AM. Expression of different isoforms of TGF-beta and the latent TGF-beta binding protein (LTBP) by rat Kupffer cells. J Hepatol. 1998;29:915–922. doi: 10.1016/s0168-8278(98)80119-0. [DOI] [PubMed] [Google Scholar]
- 32.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96:447–455. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okuno M, Moriwaki H, Imai S, Muto Y, Kawada N, Suzuki Y, Kojima S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology. 1997;26:913–921. doi: 10.1053/jhep.1997.v26.pm0009328313. [DOI] [PubMed] [Google Scholar]
- 34.Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–214. doi: 10.1034/j.1600-065x.2001.1820117.x. [DOI] [PubMed] [Google Scholar]
- 35.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schüftan GG, Bachem MG. Alpha2-macroglobulin reduces paracrine- and autocrine-stimulated matrix synthesis of cultured rat hepatic stellate cells. Eur J Clin Invest. 1999;29:519–528. doi: 10.1046/j.1365-2362.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- 37.Coffey RJ Jr, Kost LJ, Lyons RM, Moses HL, LaRusso NF. Hepatic processing of transforming growth factor beta in the rat. Uptake, metabolism, and biliary excretion. J Clin Invest. 1987;80:750–757. doi: 10.1172/JCI113130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ten Dijke P, Iwata KK, Goddard C, Pieler C, Canalis E, McCarthy TL, Centrella M. Recombinant transforming growth factor type beta 3: biological activities and receptor-binding properties in isolated bone cells. Mol Cell Biol. 1990;10:4473–4479. doi: 10.1128/mcb.10.9.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ten Dijke P, Iwata KK, Thorikay M, Schwedes J, Stewart A, Pieler C. Molecular characterization of transforming growth factor type beta 3. Ann N Y Acad Sci. 1990;593:26–42. doi: 10.1111/j.1749-6632.1990.tb16097.x. [DOI] [PubMed] [Google Scholar]
- 40.Pyzik M, Piccirillo CA. TGF-beta1 modulates Foxp3 expression and regulatory activity in distinct CD4+ T cell subsets. J Leukoc Biol. 2007;82:335–346. doi: 10.1189/jlb.1006644. [DOI] [PubMed] [Google Scholar]
- 41.Chuang YH, Lian ZX, Tsuneyama K, Chiang BL, Ansari AA, Coppel RL, Gershwin ME. Increased killing activity and decreased cytokine production in NK cells in patients with primary biliary cirrhosis. J Autoimmun. 2006;26:232–240. doi: 10.1016/j.jaut.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 42.Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, Coppel RL, Shimoda S, Ishibashi H, Gershwin ME. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–808. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 43.Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, Matsumura S, Yabushita K, Okano N, Tsuji T. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–427. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Sharma R, Ju ST, He XS, Tao Y, Tsuneyama K, Tian Z, Lian ZX, Fu SM, Gershwin ME. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology. 2009;49:545–552. doi: 10.1002/hep.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lan RY, Salunga TL, Tsuneyama K, Lian ZX, Yang GX, Hsu W, Moritoki Y, Ansari AA, Kemper C, Price J, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Liu P, Gao X, Qian W, Xu K. rAAV2-TGF-β(3) Decreases Collagen Synthesis and Deposition in the Liver of Experimental Hepatic Fibrosis Rat. Dig Dis Sci. 2010;55:2821–2830. doi: 10.1007/s10620-009-1119-3. [DOI] [PubMed] [Google Scholar]
- 47.Abou-Shady M, Baer HU, Friess H, Berberat P, Zimmermann A, Graber H, Gold LI, Korc M, Büchler MW. Transforming growth factor betas and their signaling receptors in human hepatocellular carcinoma. Am J Surg. 1999;177:209–215. doi: 10.1016/s0002-9610(99)00012-4. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzaki K, Date M, Furukawa F, Tahashi Y, Matsushita M, Sakitani K, Yamashiki N, Seki T, Saito H, Nishizawa M, et al. Autocrine stimulatory mechanism by transforming growth factor beta in human hepatocellular carcinoma. Cancer Res. 2000;60:1394–1402. [PubMed] [Google Scholar]
- 49.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]