

Abstract
Gene therapy using viral vectors for liver diseases, particularly congenital disorders, is besought with difficulties, particularly immunologic reactions to viral antigens. As a result, nonviral methods for gene transfer in hepatocytes have also been explored. Gene repair by small synthetic single-stranded oligodeoxynucleotides (ODNs) produces targeted alterations in the genome of mammalian cells and represents a great potential for nonviral gene therapy. To test the feasibility of ODN-mediated gene repair within chromosomal DNA in human hepatocytes, two new cell lines with stably integrated mutant reporter genes, namely neomycin and enhanced green fluorescent protein were established. Targeting theses cells with ODNs specifically designed for repair resulted in site-directed and permanent gene conversion of the single-point mutation of the reporter genes. Moreover, the frequency of gene alteration was highly dependent on the mitotic activity of the cells, indicating that the proliferative status is an important factor for successful targeting in human hepatocytes. cDNA array expression profiling of DNA repair genes under different cell culture conditions combined with RNA interference assay showed that mismatch repair (MMR) in actively growing hepatocytes imposes a strong barrier to efficient gene repair mediated by ODNs. Suppression of MSH2 activity in hepatocytes transduced with short hairpin RNAs (shRNAs) targeted to MSH2 mRNA resulted in 25- to 30-fold increase in gene repair rate, suggesting a negative effect of MMR on ODN-mediated gene repair. Taken together, these data suggest that under appropriate conditions nonviral chromosomal targeting may represent a feasible approach to gene therapy in liver disease.
Introduction
The liver plays a central role in many inherited and acquired genetic disorders. Hepatocytes are considered a favorable candidate for gene therapy but pose formidable obstacles. During the last decade, considerable effort has been devoted to the development of gene therapy using virus-based approaches to deliver therapeutic genes into the hepatocytes (Ferry and Heard, 1998; HERZOG, 2005). However, virus-mediated gene delivery has been rather challenging (HERZOG, 2005). Because most hepatocytes in the adult liver divide infrequently, they have proved to be poor targets for retrovirus-mediated gene transfer that establishes integrated proviruses only in dividing cells (Roe et al., 1993). Although adenovirus vectors efficiently deliver genes to cultured hepatocytes and to mouse liver cells in vivo (Herz and Gerard, 1993), adenoviral vectors cause immune and inflammatory responses, which usually lead to the rapid elimination of transduced cells (KAY, 1998). Newer generations of adenovirus vectors are being developed to reduce the expression of viral genes and thereby alleviate this problem (Engelhardt et al., 1994), but it is unclear whether it can be totally eliminated. Some success in liver targeting with adeno-associated viruses has been reported, although even large doses of virions transduced fewer than 5% of hepatocytes (Xiao et al., 1998). Despite these attempts, successful viral-based gene therapy in liver cells still remains elusive.
To overcome the problems associated with viral-based systems, several nonviral gene therapy approaches, including oligonucleotide-based strategies for targeted gene alteration, have emerged (Igoucheva et al., 2004a). RNA–DNA chimeric oligonucleotides (RDO) have been used to direct nucleotide changes in cultured human hepatoma cell lines and animals, including the Gunn rat model for Cringle–Najjar syndrome (Kren et al., 1997, 1998; Bandyopadhyay et al., 1999). Although repair frequencies as high as 40% have been reported in liver, the reproducibility of gene repair by RDO has been questioned because many researchers have failed to achieve a significant level of gene alteration (Yoon et al., 2002).
Site-specific gene modification at the chromosomal locus by modified single-stranded oligonucleotides (ODNs) has been developed as an alternative strategy to correct or generate point mutations in genomic DNA of mammalian cells (Igoucheva et al., 2001). Several features of the ODNs make them an attractive tool for in vivo applications. The primary advantage is the ability to make a permanent specific alteration of a gene of interest in a single step, without disturbing the genomic organization required for endogenous expression and regulation of the targeted gene. Moreover, this process might be used to correct dominant or gain-of-function mutations that are not amenable to gene replacement strategy. In the past few years, we have made significant progress in our understanding of the mechanism responsible for gene alteration by ODNs and provided clear evidence that this approach can be used to introduce permanent site-specific changes into episomal and chromosomal DNA of somatic cells and embryonic stem (ES) cells (Alexeev et al., 2002; Igoucheva et al., 2003; Pierce et al., 2003; Igoucheva et al., 2004a, 2004b; Igoucheva and Yoon, 2005; Igoucheva et al., 2006a).
Previously, site-specific chromosomal targeting by ODN in cultured human hepatoma HuH-7 cells was reported (Kren et al., 2002). However, several concerns have been raised about potential artifacts in this study. First, gene correction was detected by RCR amplification of genomic DNA, thereby producing “corrected amplicons” generated by priming of oligonucleotides to the DNA during PCR amplification. Second, potential cross contamination between cells could not be excluded because neither clonal analysis nor direct confirmation at the protein level was performed. Third, human hepatoma cells used in this study lack enzymatic activities of freshly isolated hepatocytes and cannot be employed for enzymatic assays.
In the present study, a new experimental system allowing for selection of corrected cells, clonal expansion, genotype characterization, and determination of factors influencing gene repair by ODN in hepatocytes was developed. Correction of single-point mutations in reporter genes by modified ODNs was assessed in VA13 cells, a human hepatoma cell line derived from HepG2 cells stably transduced with murine alcohol dehydrogenase, as a model hepatoma system closely resembling hepatocytes in alcohol metabolism (Clemens et al., 1995). We showed that (1) ODNs can produce site-specific, single-base alterations in genomic DNA of mutant neomycin and enhanced green fluorescent protein (EGFP) genes with an efficiency of up to 10−4. The restoration of reporter activity and the genomic sequence in corrected hepatocytes was stable, indicating permanent gene repair. Moreover, (2) efficiency of gene repair strongly depended on the cell proliferative state, whereby ODNs were more effective in dense hepatocyte cultures, in which the proliferative rate is considerably less than that in sparse cultures. Importantly, (3) the efficiency of gene targeting correlated with the activity of mismatch repair (MMR) system, as determined by profiling of DNA repair genes. (4) Downregulation of MSH2 expression resulted in increased ODN-mediated gene repair by more than an order of magnitude. These findings suggest that the interplay between MMR and gene targeting in human hepatocytes operates in an antagonistic manner.
Material and Methods
Synthesis and purification of oligonucleotides, plasmids
The 45mer oligonucleotides were synthesized and purchased from Operon Biotechnologies, Inc. (Huntsville, AL). Fluorescent oligodeoxynucleotides (FITC-ODN) were synthesized by incorporating fluorescein phosphoramidite at the 5′-end, as described previously (Igoucheva et al., 2001). All oligonucleotides were synthesized using 10 μmol scale and purified by HPLC. To protect oligonucleotides from nuclease degradation, four 2′-O-methyl RNA residues were incorporated at each end of the oligonucleotide, as described previously (Igoucheva et al., 2001).
The pcDNA3.1-EGFP− plasmid encoding mutant EGFP was constructed by cloning the mutant EGFP gene into multiple cloning site of a pcDNA3.1 vector as described previously (Pierce et al., 2003). Site-directed mutagenesis was used to mutate the 67th codon of the EGFP gene from GGC to CGC. A plasmid containing puromycin-resistant and neomycin-sensitive genes (pNeoS1Pur) was constructed by cloning the puromycin cassette from a pPur vector (Clontech, Mountain View, CA) into multiple cloning site of a pcDNA3.1+ vector (Invitrogen, Carlsbad, CA) as described previously (Murphy et al., 2007). Site-directed mutagenesis (Stratagene, La Jolla, CA) was used to mutate the 22nd codon of the neomycin-resistant gene from TAT to TAG. Both pNeoS1Pur and pcDNA3.1-EGFP− plasmids were kindly provided by Dr. E. Pierce (University of Pennsylvania, PA). Predesigned short hairpin RNA (shRNA) plasmids for the human MSH2 gene were purchased from SuperArray (Frederick, MD). Each vector expressed the specific shRNA, under control of the U1 promoter, and the neomycin-resistant gene that permitted the selection of stably transfected cells. An experimentally verified shRNA design algorithm assured gene specificity and efficacy. An advanced specificity search in addition to BLAST built into the algorithm helped to reduce the potential off-target effects. All plasmid DNAs were prepared using a large-scale isolation protocol (Qiagen, Santa Clara, CA).
Cell cultures, ODN, and plasmid DNA delivery
Human hepatic VA13 cells were kindly provided by Dr. D. Clemens (University of Nebraska Medical Center and Veterans Affairs Medical Center, Omaha, NB). These cells were derived from the human hepatocellular carcinoma cell line, HepG2, by inserting the coding region of the murine alcohol dehydrogenase gene Adh-1 using a eukaryotic expression plasmid, pIVL-2. VA13 recombinant cells stably expressing Adh-1 were selected in the presence of 400 μg/ml zeocin (Clemens et al., 1995). VA13 cells were cultured in high glucose DMEM medium supplemented with 25 mM HEPES, 0.1 mM nonessential amino acids, and 10% heat-inactivated FBS, at 37°C and 5% CO2. All tissue culture reagents were purchased from Invitrogen (Carlsbad, CA).
For uptake studies, three different delivery methods were used: Amaxa nucleofection, SuperFect liposomes, and HepG2 transfection reagent. Complex formation of fluorescent-conjugated ODN (FITC-ODN, 7.5 μg per transfection) or pmaxEGFP plasmid DNA (2.0 μg per transfection) and liposomes was performed according to the manufacturer's protocols. The Amaxa Nucleofection Kit V (program, T-28) was used according to the manufacturer's protocol. Delivery of pmaxEGFP plasmid was assessed 24 hours after transfection. In case of FITC-ODN, cells were kept with ODN–liposome complexes for 6 hours, washed with PBS and fixed with 10% formalin at 37°C for 20 minutes. In all cases, cellular delivery was monitored by fluorescence microscopy. For quantitative analysis, cells treated with FITC-ODN were harvested 6 hours after transfection, washed with PBS, and subjected to flow cytometry. Untreated controls were included in every experiment. Data were collected on a BD FACSCalibur flow cytometer (BD Bioscience) and analyzed with FlowJo software (Tree Star, Inc.).
Establishment of VA13 cells with integrated Puror/Neos and EGFP− mutant genes
Stable VA13 cell lines containing the integrated cassette expressing puromycin-resistant/neomycin-sensitive genes and mutant EGFP gene were generated by nucleofection of the pNeoS1Pur and pcDNA3.1-EGFP− constructs (2 μg per nucleofection), respectively. The Amaxa Nucleofection Kit V (program, T-28) was used according to the manufacturer's protocol. Forty-eight hours after nucleofection, the cells were split into new 10-cm plates and selected for 3 weeks with 2 μg/ml of puromycin selective agent. Selected cells were characterized at the DNA sequence level by RT–PCR analysis, followed by DNA sequencing using an automatic DNA sequencer (ABI 373A; Applied Biosystems, Foster City, CA) and direct primers. Total RNA was isolated using the Qiagen RNeasy Mini Kit (Valencia, CA). RT-PCR was conducted with a Reverse-iT One Step Kit (ABgene, Inc., Rochester, NY) according to the manufacturer's protocol. The mutant neomycin gene was amplified using the primer pair: Neo-1 5′-ATGATTGAACAAGATGGATTGC-3′ and Neo-4 5′TCAGAAGAACTCGTCAAGAAGG-3′. The mutant EGFP was amplified using the primer pair: EGFP-1005 5′-ATGGTGAGCAAGGGCGAGGA-3′ and EGFP-1002 5′-TGCCGTCCTCGATGTTGTGG-3′. Amplified products were purified using the Qiagen purification kit (Valencia, CA) and subsequently subjected to a direct DNA sequencing.
Several stable VA13-EGFP− cell lines containing the integrated predesigned shRNA plasmids for the MSH2 gene were produced by nucleofection of the C1, C2, C3, and C4 constructs, respectively. Forty-eight hours after nucleofection, the cells were selected with 500 μg/ml G418 for 3 weeks. The level of MSH2 expression was determined by RT-PCR analysis using hMSH2–1 5′-CTCAAGGACAAAGACTTGTTAACC-3′ and hMSH2–2 5′-AGTCACAAAAACTGCCAACAAT-3′ primers. Human β-actin was used as the loading control and was amplified with a primer pair purchased from R&D Systems (Minneapolis, MN).
Western blot analysis
Nuclear proteins were isolated using a NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce, Rockford, IL). Proteins were separated by a 4%–12% gradient SDS–PAGE and transferred to PVDF membrane followed by western blot analysis using monoclonal antihuman MSH2 antibodies (EMD Bioscience, San Diego, CA) and monoclonal antihuman β-actin antibodies (Cell Signaling Technology, Danvers, MA). Immunocomplexes were detected with HRP labeled antimouse secondary antibodies (Promega, Madison, WI) and were visualized using SuperSignal WestFemto substrates (Pierce, Rockford, IL).
Chromosomal targeting
For chromosomal targeting, 1 × 105 cells (sparse culture) and 5 × 105 cells (dense culture) were seeded in a 6-well plate and kept in culture until they reached 20%–30% confluence for sparse and 60%–70% confluence for dense cultures, respectively. Targeting oligonucleotides (7.5 μg) were diluted with OptiMEM and were preincubated with SuperFect according to the manufacturer's protocol. Preformed complexes were added directly to cells with 2 ml of complete growth medium. Six hours later, cells were fed with 2 ml of fresh medium and allowed to grow. When VA13-Puror/Neos cells were used, selection with G418 (500 μg/ml) was started 48 hours after transfection and continued for 10 days. The total number of G418-resistant clones was counted. When VA13-EGFP− cells were used, the expression of wild-type EGFP was detected using fluorescence microscopy and EGFP+ cells were counted. For further analysis, several clones were picked using a cloning cylinder, propagated, and subjected to verification at the DNA sequence level by RT-PCR analysis followed by direct DNA sequencing.
cDNA microarrays
VA13 cells were seeded in 6-well plates at different densities. In all cases, total RNA was extracted using the Qiagen RNAeasy extraction kit (Valencia, CA) according to the manufacturer's protocol. GEArray Q Series Human (HS-029) DNA Damage Signaling Pathways Gene Arrays were purchased from SuperArray (Frederick, MD). All steps including prehybridization and hybridization were performed according to the manufacturer's protocol, as described previously (Igoucheva et al., 2006b). For cDNA probe synthesis, 5 μg of total RNA was reverse transcribed and labeled with [α-32P]-dCTP using the Super Array AmpoLabeling-LPR kit (Frederick, MD). Hybridization was carried out for 16–18 hours at 60°C, followed by washing and autoradiography. Exposure time was adjusted so that the extent of hybridization was in a linear range.
Acquisition of cDNA hybridization signals was performed as described previously. The net expression of each gene was calculated by the mean intensity of dot minus the mean of the negative (background) intensity. To introduce a normalization step, the average ratio of expression levels of two constitutive genes (GAPDH and β-actin) was determined and introduced as a correction factor (CF). After normalization, a 2-fold difference in expression was considered as significant.
Results
Establishment of transgenic VA13 cell with reporter genes
Plasmid delivery optimization
To achieve optimum delivery of targeted plasmid DNAs into VA13 hepatocytes, three delivery methods, including Amaxa nucleofection, SuperFect, and HepG2 transfection reagents, were tested. The efficiency of delivery was judged 24 hours after transfection by the expression of pmaxEGFP plasmid encoding full-length wild-type EGFP gene, using fluorescence microscopy. As shown in Fig. 1A, Amaxa nucleofection resulted in the highest efficiency approaching 80%, whereas SuperFect transfection produced modest delivery, about 40%–50% (Fig. 1B). In contrast, HepG2 transfection reagent showed no appreciable delivery as shown by few EGFP-expressing cells (Fig. 1C). Therefore, nucleofection was used in all plasmid delivery experiments in VA13 cells.
FIG. 1.
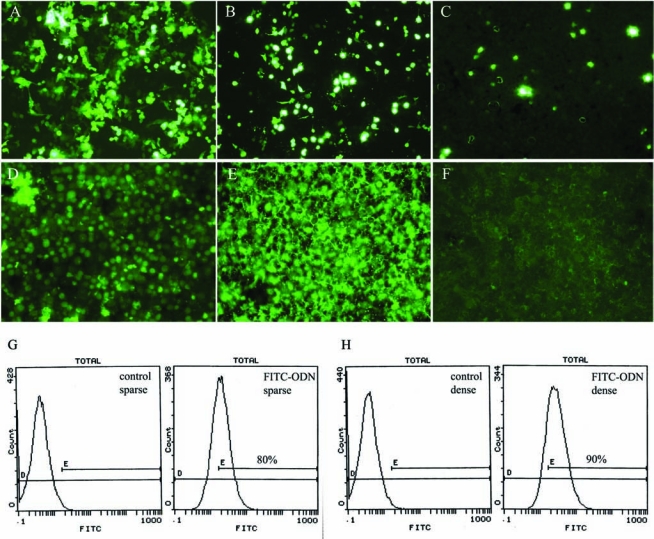
(A–C) Cellular uptake of pmaxEGFP plasmid and (D-F) FITC-ODN in actively growing VA13 cells using fluorescence microscopy. Nucleic acids were delivered using three different transfection methods: (A, D) Amaxa nucleofection, (B, E) SuperFect liposomes, and (C, F) HepG2 transfection reagent. Assessment of cellular delivery of pmaxEGFP plasmid was performed 24 hours after transfection, whereas FITC-ODN uptake was completed 6 hours after transfection. (G) Analysis of FITC-ODN uptake in sparse and (H) dense VA13 cells by flow cytometry. Flow cytometric analysis was performed 6 hours after delivery of FITC-ODN. Nontreated sparse and dense VA13 cells were used as a control, respectively.
Reporter genes cell lines
To investigate the feasibility of ODN-mediated gene targeting at the chromosomal level in human hepatocytes, we generated two VA13 cell lines with stably integrated mutant reporter genes.
The VA13-neos cell line is based on the neomycin phosphotransferase II (neo) gene of transposon Tn5. The neo gene encodes a protein of 264 amino acids and inactivates antibiotics by catalyzing the transfer of the terminal phosphate of ATP to the drug. An inactive mutant version of the neo gene (neos) was created by introduction of a mutation into codon 22 (Y22X), which creates a stop codon and completely inactivates the phosphotransferase activity of the neo gene (Fig. 2A).
FIG. 2.

Sequences of ODNs and targeted sequences of Neo and EGFP genes. (A) The pNeoS1Pur plasmid contained a TAG stop at codon 22 of the neomycin resistance gene. (B) The pcDNA3.1-EGFP− plasmid contained a CGC at codon 67. The All ODNs consist of 45 nucleotide complementary sequence to the Neo and EGFP coding strand (antisense orientation), respectively, except the mismatch G/A (Neo) or C/C (EGFP). Targeted sites are underlined. Corresponding control ODNs do not have a mismatch and are fully homologous to the mutant sequence. For protection of ODN from nuclease degradation, four residues of 2′O-methyl uridine are incorporated at both 5′ and 3′ ends.
The VA-EGFP cell line is based on a mutant of the EGFP. Since EGFP autofluoresces, it renders itself as an easily detectable marker. The mutation G67R in the EGFP chromophore (Fig. 2B) produced no detectable fluorescence, as determined by both microscopy and fluorescence-activated cell sorting (FACS) analysis.
VA13 lines with stably integrated puromycin-resistant–neomycin-susceptible cassette and mutant EGFP gene were established by nucleofection of VA13 cells with pNeoS1Pur and pcDNA3.1-EGFP plasmids, respectively, followed by puromycin selection. Resistant colonies were individually picked and expanded into distinct clonal lines of neomycin-susceptible and EGFP-mutant cells, VA13-Puror/Neos and VA13-EGFP−, respectively. Integration of the reporter cassettes into chromosomal DNA of the established cell lines was confirmed by Southern blot analysis using the full-length neo and EGFP genes cDNA as a probe, respectively (data not shown). Mutated phenotype was confirmed by RT-PCR analysis with gene specific primers followed by sequencing (data not shown). In addition, full inactivation of the Neos gene in the generated VA13-Puror/Neos clonal line was demonstrated by culturing the cells in the presence of a G418 selective agent (200 μg/ml). No viable cells were detected after 14 days, confirming the neomycin-susceptible phenotype of established line. For systemic studies, the clonal line with a one copy of Neos and EGFP genes, respectively, was chosen for further experimentation.
Delivery of ODN and chromosomal gene repair in transgenic human VA13 cell lines
As a first step toward ODN-mediated gene targeting, the delivery of ODN into actively growing VA13 cells was optimized by monitoring the accumulation of nuclear fluorescence from FITC-ODN. As shown in Fig. 1D–F, FITCODN accumulation and distribution were highly dependent on delivery agent. A massive nuclear internalization (∼90% nuclei positive) was observed when FITC-ODN was delivered by SuperFect (Fig. 1E). Moreover, the nuclear distribution was homogeneous as shown by the intensity of fluorescence. By contrast, a substantial majority of FITC-ODN complexed with HepG2 transfection reagent was detected only in the cell cytoplasm as punctuate fluorescence and was largely absent from the nucleolus compartment (Fig. 1F). Once conditions for ODN delivery were optimized, the efficiency of ODN/SuperFect lipofection was assayed in VA13 cells at different plating density using FACS analysis. Data showed that there are no significant differences in the transfection efficiency of sparse (80%) and dense cultures (90%; Fig. 1G and H).
A series of ODNs was synthesized to test their activity in gene correction in established cell lines. In all cases, chromosomal targeting was performed using ODNs with antisense sequence and a 45 nt homology to the targeted chromosomal sequence in accordance with original structure (Fig. 2A and B). As a control, ODNs fully complementary to the mutant sequences were used. For chromosomal targeting, VA13-Puror/Neos and VA13-EGFP− cells at 20%–30% and 60%–70% confluency were transfected with corresponding ODNs. Gene correction of each individual gene was accessed according to its selective nature. To determine Neor resistant cells in targeted VA13-Puror/Neos cells, selection with 200 μg/ml G418 was initiated 48 hours after transfection with Neo-1 ODN, and continued until neomycin-resistant colonies appeared and grew large enough to be picked (usually 10–14 days) and then propagated for further analysis. The VA13-EGFP− cells transfected with EGFP-1 ODN designed to restore EGFP function were examined for expression of wild-type EGFP by direct counting of EGFP+ cells using fluorescence microscopy, or subjected to live sorting to enrich the EGFP+ fraction for further analysis. For instance, sorted EGFP+ cells were seeded into a 60-mm dish with complete medium, and persistence of EGFP+ expression was monitored daily. During next 2 weeks, five clones of each targeted line were picked and propagated for further analysis. In each case, the efficiency of gene correction was determined from at least five independent experiments as the proportion of cells with the corrected phenotype divided by the total number of cells used for analysis.
A summary of results from these experiments is presented in Table 1. As expected, control transfections using the Y22X-Neo and G67R-EGFP ODNs, which share complete homology with mutant sequences, demonstrated no detectable gene repair. In contrast, both Neo-1 and EGFP-1 ODNs designed for correction were able to produce gene repair in both cell lines. Nevertheless, the number of colonies that emerged after live-sorting of genetically corrected VA13-EGFP+ cells corresponded to only two-thirds of the initial number of sorted cells. For instance, only 8 out of 15 EGFP+ cells were able to produce viable clones expressing wild-type EGFP protein in sparse cultures, whereas 35 out of 60 EGFP+ cells gave rise to colonies in dense cultures. Moreover, the gene correction rates were highly dependent on the proliferative status of the targeted cells. For instance, cells transfected at low plating density, where there is active cell division, showed much lower correction activity (∼1.8–2.5 × 10−5) than slowly dividing cells (∼0.9–1.0 × 10−4), suggesting that the proliferative status is an important factor for successful targeting by ODN in human hepatocytes.
Table 1.
The Frequency of Chromosomal Gene Correction by ODNs in Hepatic VA13 Cells
| Cell line | ODN | Targeting frequency in sparse culturesa | Targeting frequency in dense culturesa |
|---|---|---|---|
| VA13-Puror/Neos | Neo-control Neo-1 | <10−5 (1.8 ± 0.9) × 10−5 | <10−5 (0.9 ± 1.0) × 10−4 |
| VA13-EGFP− | EGFP-control EGFP-1 | <10−5 (2.5 ± 1.6) × 10−5 | <10−5 (1.0 ± 1.5) × 10−4 |
The frequency of gene correction was determined among the results obtained from five independent transfection experiments by dividing the number of cells with corrected phenotype by the number of initially targeted cells.
To confirm the corrected phenotype in subcloned VA13-Puror/Neor and VA13-EGFP+ cells, their DNA was isolated and the regions surrounding the point mutation were amplified and subsequently sequenced using direct gene-specific primers. Three clones for each cell line exhibited a correction of TAG codon to TAT at position 22 for the Neo gene, and CGC codon to GGC at position 67 for the EGFP gene, respectively (Fig. 3A and B). Moreover, no other DNA sequence changes were observed in the amplified region that flanked the corrected base confirming the site-specific manner of gene correction. The data presented here show that modified ODNs can create site-specific alterations in the genomic DNA of human hepatocytes, and that gene repair rates are highly dependent on the proliferative status of cells.
FIG. 3.
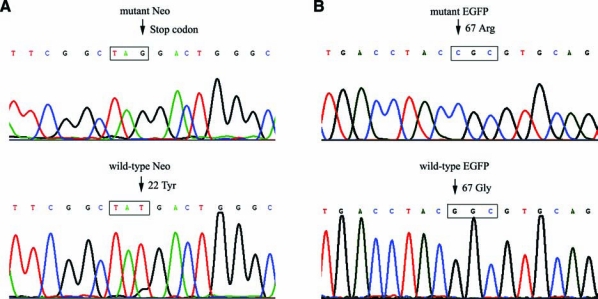
Representative DNA sequencing analysis of PCR products generated from (A) VA13-Puror/Neor and (B) VA13-EGFP+ corrected clones, respectively. Arrows indicate the position of the mutation/correction.
Correlation between hepatic cell density and gene expression profile
In order to determine the factors influencing ODN-mediated gene repair in human hepatocytes, transcription profiles for genes involved in DNA damage signaling and DNA repair were investigated, in VA13 cells with varied densities. For this purpose, two populations of VA13-EGFP−cells, ranging from sparse (25%–30% confluency) to dense (70%–80% confluency), were analyzed using a focused cDNA microarray, as described in Material and Methods. Each array contained 96 cDNA fragments from the genes associated with specific biological pathways: (1) ATM/ATR signaling network, (2) cell cycle arrest pathway, (3) apoptosis pathway, and (4) genome stability/repair pathway. The hybridization intensity of each spot was measured using an image analysis system to quantify each specific cDNA for its hybridization signature. An increase or a decrease greater than 2-fold was considered to be significant for data analysis.
Results of cDNA arrays are presented in Fig. 4 and Table 2. Many genes were downregulated in the growth arrested sparse VA13-EGFP− cells relative to proliferating cell cultures possibly owing to a generalized decrease in metabolic activity, protein content, or cell size (Fig. 4B). Noticeably, many genes involved in DNA damage response were upregulated in sparse cultures (Fig. 4A). The altered expression was found in all categories of genes, including ATM/ATR signaling network (6 out of 15 genes), cell cycle arrest pathway (3 out of 24), apoptosis pathway (4 out of 15), and genome stability/DNA repair pathway (6 out of 46 genes). Interestingly, expression levels of specific genes participating in MMR showed significant increase in hybridization signals, suggesting that this repair pathway may be directly implicated in ODN-directed gene alteration.
FIG. 4.
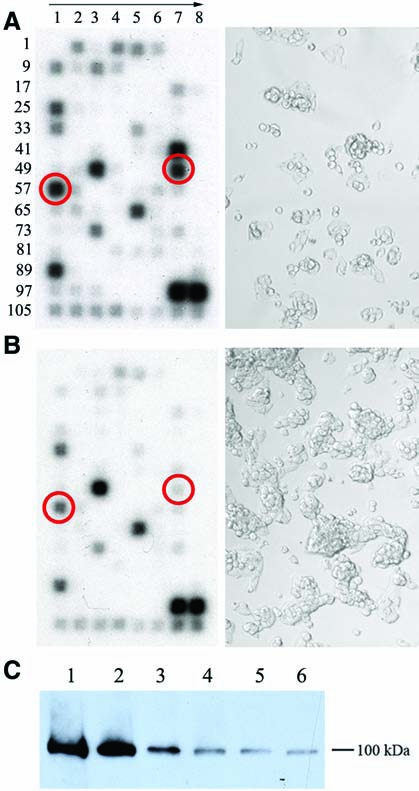
cDNA gene expression profiles in VA13 cells at different densities. Autoradiograms of microarrays hybridized to RNA isolated from (A) sparse and (B) dense cells, respectively. The direction of orientation is indicated by arrow and the position numbers of genes on the array coincides with the numbers listed in Table 2. Phase contrast images corresponding to sparse and dense populations of VA13 are shown on left panels, respectively, next to autoradiograms. Location of hybridization signals for mismatch repair genes, MSH2 and MSH6, is shown in red circles. (C) Immunodetection of MSH2 in VA13 cells at different plating densities: 10% (Lane 1), 20% (Lane 2), 40% (Lane 3), 60% (Lane 4), 80% (Lane 5), and 100% (Lane 6). For each sample, 5 μg of nuclear proteins were resolved on SDS–PAGE gel, and MSH2 protein was detected with monoclonal antihuman MSH2 antibodies as described in Material and Methods.
Table 2.
Gene Expression Profile of VA13 Cells at Various Plating Density
| Position | Genebank | Description | Gene name | LDa | HDa |
|---|---|---|---|---|---|
| ATR/ATM, signaling network | |||||
| 4 | NM_000051 | Ataxia telangiectasia mutated | ATM | 4 | 2 |
| 5 | NM_001184 | Homo sapiens ataxia telangiectasia and and Rad3 related | ATR | 4 | 2 |
| 23 | NM_001263 | CDP-diacylglycerol synthase | CDS1 | 3 | 1 |
| 25 | AF016582 | CHK1 (checkpoint, S.pombe) homolog | Chk1 | 6 | 1 |
| 58 | AF058696 | Nijmegen breakage syndrome 1 (NBS1) | Nibrin | 2 | 1 |
| 69 | NM_002853 | RAD1 homolog (S.pombe) | RAD1 | 6 | 5 |
| DNA damage response pathways Cell cycle arrest | |||||
| 47 | X67155 | Kinesin-like 5 (mitotic kinesin-like protein 1, KNSL5) | MKLP-1 | 12 | 1 |
| 51 | Z12020 | Mouse double minute 2, human homolog of; p53-binding protein | mdm2 | 9 | 9 |
| 89 | NM_003390 | Wee1+ (S.pombe) homolog | WEE1 | 6 | 4.5 |
| Apoptosis | |||||
| 2 | NM_001160 | Apoptotic protease activating factor | Apaf-1 | 3 | 1 |
| 9 | AF332558 | Bcl-2 binding component 3 | PUMA | 4 | 1 |
| 11 | U45879 | Inhibitor of apoptosis protein 2 | IAP-2 | 4 | 1 |
| 12 | U37546 | Baculovirus IAP repeat-containing 3, c-IAP2 protein | MIHC | 3.5 | 1.5 |
| Genome stability/DNA repair | |||||
| 6 | NM_000489 | Alpha thalassemia/mental retardation syndrome X-linked | RAD54 | 2 | 1 |
| 33 | NM_000400 | Excision repair cross-complementing rodent repair, group 2 | ERCC2 | 3 | 4 |
| 37 | NM_000124 | Excision repair cross-complementing rodent repair, group 6 | CSB | 3 | 1 |
| 55 | U03911 | MutS homolog 2 (E.coli) | MSH2 | 7 | 1 |
| 57 | NM_000179 | MutS homolog 6 (E.coli) | MSH6 | 7 | 5 |
| 75 | NM_002879 | Human DNA damage repair and recombination protein RAD52 | RAD52 | 4 | 2 |
The fold induction represents the ratio of the intensity of each gene hybridized with the RNA isolated from the cells at high density or low density normalized to the intensity of the housekeeping gene. Each array was processed in an identical manner and the number represents an average of triplicate experiments. Standard deviations are within 20%. Each gene is demarcated by the position of gene in the array, the gene bank accession number, the description of gene and the common name. Genes are grouped by their distinct functional categories.
To independently evaluate the transcription level of MSH2 protein, western blot analysis was performed. Using MSH2-specific antibodies, analysis showed similar expression profiles of MSH2 modulated by cell density (Fig. 4C). The protein level was much higher in sparse cultures and gradually decreased with increasing cell density, confirming the gene expression data.
Correlation between MSH2 activity and ODN-mediated gene repair in human hepatic cells
To further investigate whether reduced levels of chromosomal gene repair in sparse populations of hepatocytes are affected by the induction of MMR activity, we generated several stable MMR-deficient VA13-EGFP− cell lines that expressed human MSH2-shRNA. MSH2 is a central component of the MMR pathway that targets mismatches arising during DNA replication, homologous recombination (HR), and in response to genotoxic stresses (O'Brien and Brown, 2006). It is crucial for the repair of all mismatched lesions, whereas other MSH proteins, MSH3 and MSH6, modulate the function of MSH2, depending on the different lesion types or developmental stages.
Four derivatives of pSureSilencing shRNA plasmid were constructed that expressed shRNAs targeted to four different regions of MSH2 mRNA; nucleotides 1491–1511 (Clone C1), 2723–2743 (Clone C2), 2485–2505 (Clone C3), and 1911–1931 (Clone C4) of the 3145-nt coding sequence. The constructs were nucleofected into VA13-EGFP− cells and stable neomycin-resistant clones were selected with G418 for 3 weeks. Although, constitutive MMR suppression could potentially affect genome stability in hepatocytes, leading to a mutator phenotype, such an approach was found to be suitable for the proposed experimental settings. The levels of MSH2 mRNA and protein in selected neomycin-resistant clones at sparse density were examined by RT-PCR, western blot, and cDNA array analyses (Fig. 5A–C). In all cases, three of four generated shRNA constructs were highly effective and significantly reduced MSH2 expression. The stable cell lines referred to as VA13-EGFP−/C2, VA13-EGFP−/C3, and VA13-EGFP−/C4 had MSH2 mRNA and protein levels reduced by 70%–80% (Fig. 5A and B). Although MSH2 protein levels were decreased, low levels of protein were still present, indicating that residual MMR complexes, heterodimers of MSH2/MSH6 or MSH2/MSH3, could still be formed in knockdown cells. In contrast, the MSH2 expression level was unchanged in stable neomycin-resistant VA13-EGFP−/NC cells transfected with empty NC vector. Moreover, the Clone 1 shRNA construct failed to produce any inhibitory effect in VA13-EGFP−/C1 cells. A cDNA array analysis was performed to confirm MSH2 inhibition of mRNA levels and assess the cellular response to altered MMR activity. As expected, levels of MSH2 expression were reduced in clonal populations C2, C3, and C4 and were consistent with RT-PCR and western blotting data. Interestingly, array profiles in knockdown cultures demonstrated only modest change in overall gene expression, as shown by reduced intensity of hybridization signals (Fig. 5C). Moreover, transcription profile was similar to that obtained with RNA samples isolated from dense cultures (Fig. 4B). The expression of MSH6, but not MSH3, was notably downregulated, indicating that repair of single base–base mispairs and smaller insertion/deletion mispairs was substantially suppressed in MSH2 knockdown cells. Also, expression of mitotic kinesin-like protein (MKLP1), which plays an essential role in assembly and function of the mitotic spindle and cell division, was dramatically downregulated.
FIG. 5.
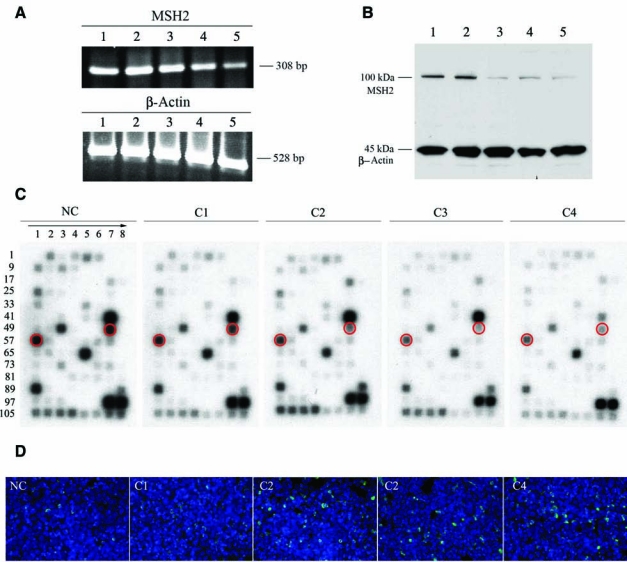
(A) The RT-PCR analysis of MSH2 gene expression in VA13-EGFP− cells stably expressing four different shRNAs (C1, C2, C3, and C4), which were specifically designed to target MSH2 mRNA. NC depicts cells transduced with empty pSureSilencing vector. The housekeeping gene (β-actin) was used as a loading control. (B) Immunodetection of MSH2 with monoclonal antihuman MSH2 antibodies in VA13-EGFP− cell lines expressing specific shRNAs: NC (Lane 1), C1 (Lane 2), C2 (Lane 3), C3 (Lane 4), and C4 (Lane 5). β-actin (loading control) was codetected on the blot with monoclonal antihuman β-actin antibodies. (C) cDNA gene expression profiles in VA13-EGFP− cell lines expressing specific shRNAs. Location of MSH2 and MSH6 hybridization signals is shown in red circles. (D) Chromosomal correction by ODNs in VA13-EGFP− cells transduced with MSH2 shRNAs. Cells with a corrected EGFP gene were detected 72 hours following transfection with EGFP-1 ODN using fluorescence microscopy as described in text.
For chromosomal targeting, sparse cultures of hepatocytes with constitutively downregulated MSH2 activity were transfected with correcting EGFP-1 ODN as described above. As a control, VA13-EGFP−/NC and VA13-EGFP−/C1 cell lines with unaffected MSH2 activity were used. In all cases, gene correction was detected 72 hours after transfection by fluorescence microscopy. To allow a more accurate comparison between transfection and detection of correction events, cells were harvested from the original plates and the same number of cells (5 × 105) from each individual transfection was deposited on glass slides by cytospin, followed by counting of EGFP expressing cells. As shown in Fig. 5D and Table 3, corrected EGFP+ cells were detected in all cell lines regardless of MSH2 status. However, correction frequencies were 20–35 times higher in MSH2 knockdown VA13-EGFP−/C2, VA13-EGFP−/C3, and VA13-EGFP−/C4 cells, but not in VA13-EGFP−/NC and VA13-EGFP−/C1 cells. Furthermore, correction rates were significantly higher than those in dense cultures with unaffected MSH2 activity. Overall, these data indicate that MMR activity has in fact a negative effect on ODN-mediated gene repair. Improved ODN-mediated gene repair is achieved under conditions of suppressed MMR activity.
Table 3.
The Frequency of Chromosomal Gene Correction by ODNs in VA13-EGFP−Cells Transduced With MSH2 shRNAs
| Cell line | Targeting frequencya |
|---|---|
| VA13-EGFP−/NC | (2.1 ± 1.4) × 10−5 |
| VA13-EGFP−/C1 | (2.3 ± 1.1) × 10−5 |
| VA13-EGFP−/C2 | (8.5 ± 1.4) × 10−4 |
| VA13-EGFP−/C3 | (4.5 ± 1.5) × 10−4 |
| VA13-EGFP−/C4 | (9.0 ± 1.5) × 10−4 |
The frequency of gene correction was determined among the results obtained from five independent transfection experiments by dividing the number of cells with corrected phenotype by the number of initially targeted cells (5 × 105).
Discussion
Using mutated neomycin and EGFP reporter genes, we generated transgenic human hepatocyte cell lines stably expressing reporter targets to evaluate the feasibility of chromosomal gene repair by ODN. Developed systems allowed reliable detection and quantification of correction events and permitted a robust analysis of cells that underwent gene modification. We have demonstrated that ODNs harboring a centrally located mismatch-forming nucleotide are capable of mediating permanent gene correction with relatively low frequencies, ranging from 2.5 × 10−5 to 1.0 × 10−4, as confirmed by phenotypic changes and DNA sequence analysis for designed base alteration at the target site. Our results are in good agreement with previously published data obtained on targeting by ODN in different somatic cells and mouse ES cells (Igoucheva et al., 2004a). However, we found that only 60% of EGFP corrected cells were able to produce viable clones. Several groups have reported similar results, with viability of corrected cells expressing EGFP ranging from 2% to 60% (Radecke et al., 2004; Olsen et al., 2005). The variation in viability appears to correlate with the copy number of the mutant EGFP, and specific cell type. For example, only 2% of corrected CHO-K1 cells with 30–60 copies of mutant EGFP per genome were viable (Olsen et al., 2005), compared with 60% of 293T cells with a single mutant EGFP gene (Radecke et al., 2004). Our previous experiments also support the notion that the clonal expansion of corrected cells varies according to the targeted gene and cell types. Using clonal analysis, we demonstrated that an ODN corrected a point mutation in the endogenous tyrosinase gene, resulting in an inheritable restoration of enzymatic activity, melanin synthesis, and pigmentation changes in albino melanocytes (Alexeev et al., 2002). We were able to clone more than 60% of these pigmented cells. Our clonal experiments in ES cells also showed that the number of LacZ corrected cells was similar to the number of viable LacZ positive colonies obtained by limiting dilution meaning that most of the mutant LacZ corrected ES cells expanded to make viable colonies (Pierce et al., 2003). Very recently, Murphy et al. (2007) showed that targeting ES-Neos cells with a correcting ODN resulted in about 50–100 Neor clones from the initial 1 × 106 ES cells.
It is still unclear why some cells do not divide following ODN-mediated gene correction. It is possible that a small number of cells exist that are poised to escape cellular DNA damage surveillance and these cells have a higher probability not only of successful gene repair but also of accumulating deleterious DNA alterations. The persistent presence of a large amount of ODNs in these cells might trigger repetitive DNA repair events that eventually lead to the apoptosis of ODN-repair competent cells. This situation might be quite analogous to the apoptosis of MMR-competent cells that is triggered by the repetitive but failed repair of a DNA lesion containing methylated bases, such as O-6-methylguanine (Stojic et al., 2004). As a consequence, MMR-competent cells are 100-fold more sensitive to killing by methylating agents than MMR-deficient cells.
The frequency of gene alteration was highly dependent on the mitotic activity of the cells, indicating that the proliferative status is an important factor for successful targeting in human hepatocytes. Site-directed nucleotide exchange was highly dependent on cell density, as shown by increased gene repair rates in populations of dense hepatocytes. During active division, cell cycle progression is constantly monitored by DNA repair systems to ensure that cells with DNA damage do not replicate. Comparative analysis of gene expression profiles in sparse and dense VA13 populations revealed that mitotically active VA13 cells had elevated levels of MSH2 and MSH6 mRNA transcripts, which is indicative of active MMR that primarily operates on DNA containing mismatches or small insertions/deletion loops. It is interesting to note that some components of MMR-dependent cell cycle checkpoint signaling, including ataxia telangiectasia mutated (ATM), Rad-3-related (ATR), and Chk1, which are involved in sensing and responding to replication stress and DNA damage, were upregulated (Zhou and Elledge, 2000). By contrast, growth-arrested hepatocytes showed significantly less overall gene expression, in particular, levels of MSH2 and MSH6 mRNAs, suggesting that MMR could be implicated in oligonucleotide targeting.
The role of MMR in ODN-directed gene repair was addressed in VA13-EGFP− cells constitutively expressing shRNAs targeted to MSH2 mRNA. Analysis of cDNA expression profiles in knockdown hepatic cells showed that suppression of MSH2 negatively affected the expression of its associated partner MSH6. It has been shown that cells lacking one member in a heteroduplex involved in MMR also express low levels of the other partner, suggesting that MMR protein stability is coupled tightly to the stability of its cognate partner (Chang et al., 2000). Suppression of MSH2 allowed effective oligonucleotide-mediated gene repair as shown by increased correction frequencies in cells with knockdown protein, supporting the notion that MMR can negatively affect gene repair induced by ODN action.
In principle, the ODN-directed gene repair was postulated to occur in two phases, DNA strand pairing and DNA repair. During the strand paring phase, an ODN is assimilated to the target DNA during active transcription, leading to the formation of a transiently open DNA duplex (D-loop structure) (Igoucheva et al., 2004a, 2004b). Once alignment has occurred, a mismatch is formed between the ODN and one of the target strands. Because the D-loop contains a mismatch, proteins involved in DNA MMR have been suggested to participate in the recognition and processing of mismatches in the transiently formed D-loop. However, data accumulated from several species and different systems indicate that MMR does not play an important role in ODN-directed repair and may in fact have an antagonistic effect. In E.coli, some mismatches created by ODN and the target DNA were repaired more efficiently than others in MMR-proficient E.coli strains (Li et al., 2003). However, overall rates of ODN-mediated gene modification were similar between mismatch competent and deficient E.coli strains. In yeast, the frequency of gene correction either did not change appreciably or actually increased in the absence of MMR activities of MutS and MutL homolog proteins (Maguire and Kmiec, 2007). In mammalian cells, several conflicting results have been reported regarding the participation of MMR proteins in gene repair. The absence of MutS homolog 2 protein (Msh2) reduced the level of gene correction in mammalian cell extracts. By contrast, the mouse ES cells lacking activity of Msh2 or Msh3 showed much greater modification rates than did the wild-type ES cells (Dekker et al., 2003; Dekker et al., 2006). A large increase in repair rates seen in MSH2−/− mouse ES cells, in comparison to the wild-type ES cells, indicated an antagonistic interaction between recombinase and MMR proteins. Such opposed effect of MMR and recombination can be attributed to interactions between recombinase and MMR proteins (Evans and Alani, 2000; Pichierri et al., 2001). The Msh2 protein, a sensor for DNA lesions, may block or reverse the attempted heteroduplex formation by recombinase that arises inappropriately at the stalled replication fork.
Very recently, we proposed an alternative model, according to which transient formation of a D-loop triggers the repair of the ODN mismatch by proteins involved in the nucleotide excision repair (NER) and transcription-coupled repair (TCR; Igoucheva et al., 2006a). Both pathways are the most versatile repair systems in terms of recognition of DNA lesions (van Hoffen et al., 2003). Our results clearly showed that components of the endonuclease complexes participating in NER and TCR, namely excision repair cross-complementing rodent repair deficiency complementation group 4 protein (encoded by ERCC4) and XPG-complementing protein (encoded by ERCC5), facilitate ODN-directed gene alteration. These findings suggested that a primary pathway for ODN-mediated gene modification involves some of the proteins participating in NER and TCR, but not MMR (Igoucheva et al., 2006a). Further studies on mechanistic aspects of gene repair will enable us to elucidate factors that influence repair rates and will greatly benefit the design of rational approaches toward increasing the efficiency of ODN-mediated gene repair. Moreover, the development of new approaches to detect and isolate virtually any cells that have undergone ODN-directed sequence alteration will allow ODN-mediated gene targeting to become broadly applicable.
In conclusion, the site-directed modification of hepatic genomic DNA by ODNs offers a significant alternative to traditional gene viral-based replacement strategies for liver disease. Because the majority of hepatocytes in the adult liver stay quiescent and divide infrequently, such cells are resistant to viral transduction. On the other hand, oligonucleotide-based technology seems to be very advantageous for in vivo applications because oligonucleotides can be administered locally or systemically, and successful targeting in hepatocytes will not require active cell division. Moreover, gene correction by ODN is permanent, thereby, eliminating the need for multiple treatments.
Acknowledgment
This work was supported in part by grants from the National Institute of Arthritis, Musculoskeletal and Skin Diseases (RO3 AR051134) to OI and the National Institute on Alcohol Abuse and Alcoholism (RO1 AA015421 and R37 AA010968) to ER.
References
- ALEXEEV V. IGOUCHEVA O. YOON K. Simultaneous targeted alteration of the tyrosinase and c-kit genes by single-stranded oligonucleotides. Gene Ther. 2002;9:1667–1675. doi: 10.1038/sj.gt.3301862. [DOI] [PubMed] [Google Scholar]
- BANDYOPADHYAY P. MA X. LINEHAN-STIEERS C. KREN B.T. STEER C.J. Nucleotide exchange in genomic DNA of rat hepatocytes using RNA/DNA oligonucleotides. Targeted delivery of liposomes and polyethyleneimine to the asialoglycoprotein receptor. J. Biol. Chem. 1999;274:10163–10172. doi: 10.1074/jbc.274.15.10163. [DOI] [PubMed] [Google Scholar]
- CHANG D.K. RICCIARDIELLO L. GOEL A. CHANG C.L. BOLAND C.R. Steady-state regulation of the human DNA mismatch repair system. J. Biol. Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- CLEMENS D.L. HALGARD C.M. MILES R.R. SORRELL M.F. TUMA D.J. Establishment of a recombinant hepatic cell line stably expressing alcohol dehydrogenase. Arch. Biochem. Biophys. 1995;321:311–318. doi: 10.1006/abbi.1995.1400. [DOI] [PubMed] [Google Scholar]
- DEKKER M. BROUWERS C. AARTS M. VAN DER TORRE J. DE VRIES S. VAN DE VRUGT H. TE RIELE H. Effective oligonucleotide-mediated gene disruption in ES cells lacking the mismatch repair protein MSH3. Gene Ther. 2006;13:686–694. doi: 10.1038/sj.gt.3302689. [DOI] [PubMed] [Google Scholar]
- DEKKER M. BROUWERS C. TE RIELE H. Targeted gene modification in mismatch-repair-deficient embryonic stem cells by single-stranded DNA oligonucleotides. Nucleic Acids Res. 2003;31:e27. doi: 10.1093/nar/gng027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGELHARDT J.F. YE X. DORANZ B. WILSON J.M. Ablation of E2A in recombinant adenoviruses improves trans-gene persistence and decreases inflammatory response in mouse liver. Proc. Natl. Acad. Sci. USA. 1994;91:6196–6200. doi: 10.1073/pnas.91.13.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS E. ALANI E. Roles for mismatch repair factors in regulating genetic recombination. Mol. Cell. Biol. 2000;20:7839–7844. doi: 10.1128/mcb.20.21.7839-7844.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERRY N. HEARD J.M. Liver-directed gene transfer vectors. Hum. Gene Ther. 1998;9:1975–1981. doi: 10.1089/hum.1998.9.14-1975. [DOI] [PubMed] [Google Scholar]
- HERZ J. GERARD R.D. Adenovirus-mediated transfer of low density lipoprotein receptor gene acutely accelerates cholesterol clearance in normal mice. Proc. Natl Acad. Sci. USA. 1993;90:2812–2816. doi: 10.1073/pnas.90.7.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERZOG R.W. Recent advances in hepatic gene transfer: more efficacy and less immunogenicity. Curr. Opin. Drug Discov. Devel. 2005;8:199–206. [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. PRYCE M. YOON K. Transcription affects formation and processing of intermediates in oligonucleotide-mediated gene alteration. Nucleic Acids Res. 2003;31:2659–2670. doi: 10.1093/nar/gkg360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. SCHARER O. YOON K. Involvement of ERCC1/XPF and XPG in oligodeoxynucleotide-directed gene modification. Oligonucleotides. 2006a;16:94–104. doi: 10.1089/oli.2006.16.94. [DOI] [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. YOON K. Targeted gene correction by small single-stranded oligonucleotides in mammalian cells. Gene Ther. 2001;8:391–399. doi: 10.1038/sj.gt.3301414. [DOI] [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. YOON K. Mechanism of gene repair open for discussion. Oligonucleotides. 2004a;14:311–321. doi: 10.1089/oli.2004.14.311. [DOI] [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. YOON K. Oligonucleotide-directed mutagenesis and targeted gene correction: a mechanistic point of view. Curr. Mol. Med. 2004b;4:445–463. doi: 10.2174/1566524043360465. [DOI] [PubMed] [Google Scholar]
- IGOUCHEVA O. ALEXEEV V. YOON K. Differential cellular responses to exogenous DNA in mammalian cells and its effect on oligonucleotide-directed gene modification. Gene Ther. 2006b;13:266–275. doi: 10.1038/sj.gt.3302643. [DOI] [PubMed] [Google Scholar]
- IGOUCHEVA O. YOON K. Gene targeting by oligonucleotides in keratinocytes. Methods Mol. Biol. 2005;289:287–302. doi: 10.1385/1-59259-830-7:287. [DOI] [PubMed] [Google Scholar]
- KAY M.A. Hepatic gene therapy for haemophilia B. Haemophilia. 1998;4:389–392. doi: 10.1046/j.1365-2516.1998.440389.x. [DOI] [PubMed] [Google Scholar]
- KREN B.T. BANDYOPADHYAY P. CHOWDHURY N.R. CHOWDHURY J.R. STEER C.J. Oligonucleotide-mediated site-directed gene repair. Methods Enzymol. 2002;346:14–35. doi: 10.1016/s0076-6879(02)46047-8. [DOI] [PubMed] [Google Scholar]
- KREN B.T. BANDYOPADHYAY P. STEER C.J. In vivo site-directed mutagenesis of the factor IX gene by chimeric RNA/DNA oligonucleotides. Nat. Med. 1998;4:285–290. doi: 10.1038/nm0398-285. [DOI] [PubMed] [Google Scholar]
- KREN B.T. COLE-STRAUSS A. KMIEC E.B. STEER C.J. Targeted nucleotide exchange in the alkaline phosphatase gene of HuH-7 cells mediated by a chimeric RNA/DNA oligonucleotide. Hepatology. 1997;25:1462–1468. doi: 10.1002/hep.510250626. [DOI] [PubMed] [Google Scholar]
- LI X.T. COSTANTINO N. LU L.Y. LIU D.P. WATT R.M. CHEAH K.S. COURT D.L. HUANG J.D. Identification of factors influencing strand bias in oligonucleotide-mediated recombination in Escherichia coli. Nucleic Acids Res. 2003;31:6674–6687. doi: 10.1093/nar/gkg844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGUIRE K.K. KMIEC E.B. Multiple roles for MSH2 in the repair of a deletion mutation directed by modified single-stranded oligonucleotides. Gene. 2007;386:107–114. doi: 10.1016/j.gene.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY B.R. MOAYEDPARDAZI H.S. GEWIRTZ A.M. DIAMOND S.L. PIERCE E.A. Delivery and mechanistic considerations for the production of knock-in mice by single-stranded oligonucleotide gene targeting. Gene Ther. 2007;14:304–315. doi: 10.1038/sj.gt.3302866. [DOI] [PubMed] [Google Scholar]
- O'BRIEN V. BROWN R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis. 2006;27:682–692. doi: 10.1093/carcin/bgi298. [DOI] [PubMed] [Google Scholar]
- OLSEN P.A. RANDOL M. KRAUSS S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–551. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- PICHIERRI P. FRANCHITTO A. PIERGENTILI R. COLUSSI C. PALITTI F. Hypersensitivity to camptothecin in MSH2 deficient cells is correlated with a role for MSH2 protein in recombinational repair. Carcinogenesis. 2001;22:1781–1787. doi: 10.1093/carcin/22.11.1781. [DOI] [PubMed] [Google Scholar]
- PIERCE E.A. LIU Q. IGOUCHEVA O. OMARRUDIN R. MA H. DIAMOND S.L. YOON K. Oligonucleotide-directed single-base DNA alterations in mouse embryonic stem cells. Gene Ther. 2003;10:24–33. doi: 10.1038/sj.gt.3301857. [DOI] [PubMed] [Google Scholar]
- RADECKE F. RADECKE S. SCHWARZ K. Unmodified oligodeoxynucleotides require single-strandedness to induce targeted repair of a chromosomal EGFP gene. J. Gene Med. 2004;6:1257–1271. doi: 10.1002/jgm.613. [DOI] [PubMed] [Google Scholar]
- ROE T. REYNOLDS T.C. YU G. BROWN P.O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOJIC L. BRUN R. JIRICNY J. Mismatch repair and DNA damage signaling. DNA Repair (Amst) 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- VAN HOFFEN A. BALAJEE A.S. VAN ZEELAND A.A. MULLENDERS L.H. Nucleotide excision repair and its interplay with transcription. Toxicology. 2003;193:79–90. doi: 10.1016/j.tox.2003.06.001. [DOI] [PubMed] [Google Scholar]
- XIAO W. BERTA S.C. LU M.M. MOSCIONI A.D. TAZELAAR J. WILSON J.M. Adeno-associated virus as a vector for liver-directed gene therapy. J. Virol. 1998;72:10222–10226. doi: 10.1128/jvi.72.12.10222-10226.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOON K. IGOUCHEVA O. ALEXEEV V. Expectations and reality in gene repair. Nat. Biotechnol. 2002;20:1197–1198. doi: 10.1038/nbt1202-1197b. [DOI] [PubMed] [Google Scholar]
- ZHOU B.B. ELLEDGE S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]