

Abstract
This study aimed to investigate the trajectories over time of health status and health service use in Rett syndrome by mutation type. Data were obtained from questionnaires administered over six years to 256 participants from the Australian Rett Syndrome Database. Health status (episodes of illness and medication load) and health service use (general practitioner and specialist visits and hospital stays) were summarized into composite scores with Principal Component Analysis. Linear and mixed regression models examined effects of mutation type and other variables on these scores over time. For some mutations (such as p.R255X, p.R168X) health status was poorer at a younger age and improved over time, while for p.R133C it was better at a younger age and deteriorated with time. For those with p.R133C health service use was lowest at a younger age and highest at 25 years. With other mutations, such as p.R255X, p.R270X, p.R294X, C terminal and p.R306C, health service use was higher at a younger age, but dropped off considerably by 25 years of age. Health service use generally declined in parallel with deterioration in health status, although this pattern differed by mutation type, demonstrating important variability in the course of Rett syndrome.
Background
Rett syndrome is a neurodevelopmental disorder with a cumulative incidence of 1.09 per 10,000 females by the age of 12 years (Laurvick et al., 2006). Children with Rett syndrome generally do not appear to have any problems at birth, but development subsequently regresses, with loss of hand function and communication skills usually evident by 18 months.
The association between Rett syndrome and mutations in the methyl-CpG binding protein 2 (MECP2) gene was first identified in 1999 (Amir et al., 1999). Mutations have been reported in 73 -96% of classical Rett syndrome cases in various studies with the percentage increasing as techniques improve (Colvin et al., 2003; Renieri et al., 2003). More than 200 different pathogenic MECP2 mutations have now been identified, with eight accounting for just over two thirds of mutation positive cases (Bebbington et al., 2008; Colvin et al., 2004). In a study combining Australian and international cases, mutations at the C terminal (CT), early truncating (ET) mutations and large deletions (LD) accounted for another fifth of mutation positive cases (Young et al., 2008). Amongst mutation types there is considerable phenotypic variation. Some mutations, such as p.R270X and p.R255X (Bebbington et al., 2008; Colvin et al., 2004; Huppke, Held, Handefeld, Engel, & Laccone, 2002) and p.R168X (Neul et al., 2008) have been shown to be associated with a severe clinical presentation and others, such as p.R294X and p.R133C, with a milder phenotype (Bebbington et al., 2008; Colvin et al., 2004; Leonard et al., 2003).
Genotype- phenotype relationships have previously been investigated (Bebbington et al., 2008; Colvin et al., 2004; Schanen et al., 2004) using information about the regression period, current functioning and comorbidities (e.g., epilepsy and scoliosis), often in the form of severity scales (Amir et al., 2000; Kerr et al., 2001; Monros et al., 2001). Given the known clinical progression in Rett syndrome (Hagberg, Witt-Engerström, Opitz, & Reynolds, 1986), the relationship between phenotype and genotype may be confounded by age. This has been accounted for in at least one study (Bebbington et al., 2008). However, no research has investigated the change in composite health status by mutation type over time. Is it possible that the health status of those affected by mutations with an apparently mild phenotype and better functioning will later deteriorate quite markedly? Conversely, might the health status of those with apparently severe mutations and very limited functioning have a less aggressive trajectory?
The only study, to our knowledge, that has been undertaken on the association between health service use and other factors is our earlier study showing that use was highest in younger cases and lowest in cases with milder phenotypes (Moore et al., 2005). Some relationships with specific genotypes were also identified. The aim of the present study was to examine the health status and health service use of females with Rett syndrome over time and identify any variation by age and by mutation using data from the Australian Rett Syndrome Database (ARSD).
Methods
Data Source
The Australian Rett Syndrome Database (ARSD) is an ongoing population-based register of Australian Rett syndrome participants born since 1976 (Laurvick et al., 2006). Cases (~15 per birth year) are identified most commonly either by clinicians through the Australian Paediatric Surveillance Unit (APSU) (Gazarian et al., 1999) or by families via the parent support group (Leonard & Bower, 1998). Since 2000, follow-up questionnaires have been administered every two years to Australian study families (Jian et al., 2006). Data collection procedures and this study have been approved and monitored by the Princess Margaret Hospital for Children Human Ethics Committee. There have now been four follow-up questionnaires completed by the parents or carers in the years 2000, 2002, 2004 and 2006, collecting demographic data and information on functional ability, medical conditions and healthcare service use. The data provide a longitudinal profile of the individual’s clinical history (over a period up to six years) – particularly in relation to episodes of illness, use of health services (doctors’ visits and hospitalizations) and use of medication. Data were available for the 256 cases whose parents or carers had completed at least one of the four follow-up questionnaires. The number of questionnaires completed on each participant depended on the year of recruitment and on whether they remained alive. Thus, four questionnaires were completed for 91 (35.5%) participants. Of these, 44 (17.2%) participants (many of whom were more recently recruited) had one questionnaire, 62 (24.2%) had two, and 59 (23.1%) three questionnaires. These cases yielded a total of 713 observations on health status and health service use, which form the basis of this analysis. Age at the time of questionnaire completion was grouped as follows: 0 to less than 5 years; 5 to less than 10 years; 10 to less than 15 years; 15 to less than 20 years; 20 to less than 25 years and 25 years and over.
Health status
Four specific aspects of health were used to assess health status: epilepsy, gastro-intestinal problems, nutritional problems and episodes of illness. We selected the first three because we found that the range of medication use in case subjects related almost entirely to these three areas. These four aspects of health status, along with scoliosis, are the major co-morbidities in Rett syndrome (Weaving, Ellaway, Gecz, & Christodoulou, 2005). We measured the impact of scoliosis through health service encounters (see below).
A severity code for epilepsy was based on use of antiepileptic drugs (AEDs). AEDs were coded by the number of drugs taken, and by the type of drug. First-line medications (carbamazepine, phenobarbitone, ethosuximide, sodium valproate and phenytoin) were given a score of 1. All other AEDs were categorised as second line drugs and given a higher score (1.5). Cases with any two AEDs were scored 2; any three AEDs were scored 3 and so on. Short-term rescue medications were not included. Based on the information about reflux and/or constipation management requirements, each individual was given a gastrointestinal (GI) score from 0 to 10 (0 = nil GI problems, 10 = severe). A score from 0–4 was given for nutritional difficulty based on questionnaire information related to eating ability and need for nutritional support: 0= nil difficulty; 4= Percutaneous Endoscopic Gastrostomy (PEG) in-situ.
A categorical variable was calculated based on quartiles of the distribution of all episodes of illness in the year preceding completion of each questionnaire. Episodes of illness were defined here as cold/flu, tonsillitis, pneumonia, bronchitis, episodes of asthma, ear infection, urinary tract infection (UTI), and other. Each individual was assigned a score of 1 to 4 with 1 representing no or 1 episode of illness in an average year, 2 representing more than 1 visit and less than 2.5 in an average year, 3 representing more than 2.5 and less than 5 visits in an average year and 4 being the maximum representing 5 or more episodes of illness in an average year.
Measures of epilepsy, gastrointestinal problems, nutritional status and illness episodes were combined into a single composite score “health status” using the first principal component from a Principal Component Analysis (PCA) (Stata, 1996–2006). The first component had an Eigenvalue of 1.361 and Rho of 0.3402. The eigenvectors for three of the four measures in Health Status were more equally weighted for epilepsy severity (eigenvector = 0.521), gastrointestinal problems (eigenvector = 0.552) and nutritional status (eigenvector = 0.599). Episodes of illnesses contributed least to the composite (eigenvector = 0.257).
Health Services
A composite score for health service use in the year preceding the completion of each questionnaire was derived using four separate variables: number of nights spent in hospital, number of day hospital visits, number of GP visits and number of specialist visits, during that year. These four variables were firstly recoded into categories (approximately quartiles) to make their measurement scales similar. The PCA procedure was then used again to combine them into a single composite from the first principal component. The first component had an Eigenvalue of 1.649 and Rho of 0.4122. The eigenvectors for three of the four measures in Health Services were more equally weighted for GP visits (eigenvector = 0.453 and hospitalizations (day stays: eigenvector = 0.458; night stays: eigenvector = 0.465)). However, specialist visits weighted more heavily (eigenvector = 0.608).
Data Analysis
Analyses by mutation type were based on 163 cases with the eight most common mutations, C terminal and early truncating mutations and large deletions as previously classified (Table 1) (Young et al., 2008). Those with no mutation (n=55) or not tested (n=16) were excluded as were 22 cases with less frequently reported mutations. Seven participants in the included mutation groups (two with p.270X and one each with an early truncating, large deletion, p.R168X, p.R106W and p.T158M) died prior to the onset of the follow-up component (Table 1). Seventeen participants died during the study including 3/12 (25%) of those with a large deletion, 3/17 (17.6%) of those with p.R270X and 3/14 (21.4%) of those with p.R306C. There were no deaths amongst those with C terminal deletions, p.R133C, p.R255X and p.R294X. Information on the cause of death was only available for 8/17 deaths in 6/8 from family report and in two from death certificates. Pneumonia or respiratory infection accounted for four deaths. In a further two, the cause was stated to be aspiration pneumonia. One death was thought to be associated with a seizure and another died in the course of palliative treatment on account of her multiple comorbidities. Although deaths were more common in those with certain mutations (p.R270X, p.R306C and large deletions) there were no statistically significant relationships.
Table 1.
Distribution of common mutations in those who did and did not enter follow-up component and in those who died during the follow-up.
| MECP2 mutation type | Died prior to follow-up component | Did not enter follow-up component for other reasons | Actually in “health” study i.e., follow-up component |
Died during the follow-up period |
||
|---|---|---|---|---|---|---|
| n | % | n | % | |||
| C-terminal deletion | 0 | 1 | 21 | 11.4 | 0 | 0.0 |
| Early truncating | 1 | 2 | 7 | 3.8 | 2 | 12.5 |
| Large deletion | 1 | 3 | 12 | 14.1 | 2 | 12.5 |
| p.R106W | 1 | 1 | 6 | 3.3 | 0 | 0.0 |
| p.R133C | 0 | 1 | 13 | 7.1 | 0 | 0.0 |
| p.R168X | 1 | 2 | 20 | 10.9 | 2 | 12.5 |
| p.R255X | 0 | 0 | 14 | 7.6 | 0 | 0.0 |
| p.R270X | 2 | 0 | 17 | 9.2 | 3 | 18.8 |
| p.R294X | 0 | 1 | 18 | 9.8 | 0 | 0.0 |
| p.R306C | 0 | 0 | 14 | 7.6 | 3 | 18.8 |
| p.T158M | 1 | 0 | 21 | 11.4 | 1 | 6.3 |
| Other | 0 | 3 | 22 | 12.0 | 3 | 18.8 |
| Total | 7 | 14 | 185 | 100.0 | 16 | 100.0 |
Newer p-values: χ2 p-value for died w/o follow-up vs. actually in “health” study: 0.448, χ2 p-value for (died w/o follow-up and did not return data) vs. actually in “health” study: 0.228, χ2 p-value for in “health” study vs. died during follow-up: 0.219.
Mean scores for health status and health service use and their confidence intervals were estimated using the Stata procedure Regress with the cluster option and adjust. The cluster option accounts for the multiple observations over the four questionnaires, while adjust was used to adjust the means for age differences within each mutation type. In order to examine trends in health status and health service use by age, a mixed model was specified using the Stata procedure, xtmixed, a multilevel mixed-effects linear regression model. The model was fitted using maximum likelihood estimation. Change in health status by age group was investigated, as well as whether this varied by mutation type by adding interaction terms. Trends by age group were graphed for each mutation type.
Results
Health status composite generally deteriorated (i.e. the score increased) with age, while health service use decreased with age (Figure 1).
Figure 1.
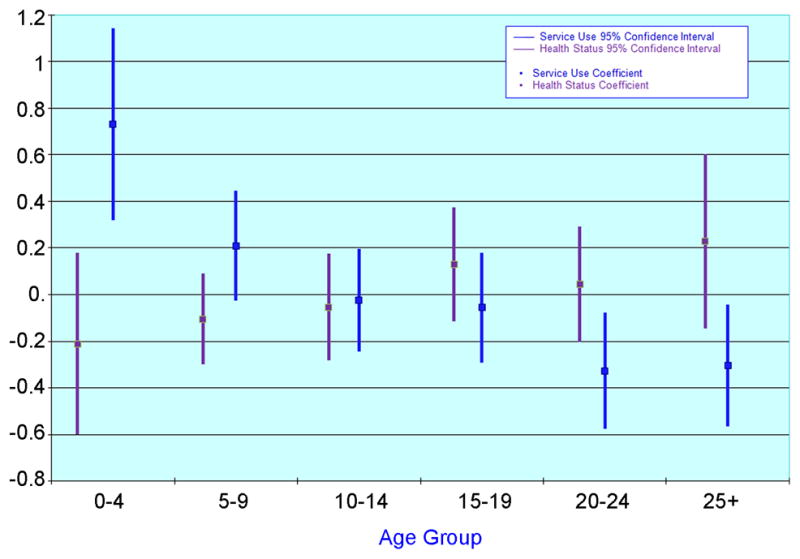
Health Status Composite and Health Service Use Composite by age group
Health status was poorest (most positive adjusted scores) in cases with early truncating mutations and p.R255X and best (most negative adjusted scores) in those with p.R133C, p.R294X and p.R306C mutations (Table 2). The adjusted score for health service use was highest for p.R133C, p.R255X and p.T158M (all three were of a comparable level) and lowest for p.R168X (Table 2). Confidence intervals for all estimates were wide and overlapping.
Table 2.
Health status and health service use composite scores.
| Fixed effects | Health status composite | 95% Confidence interval [LB, UB] | Health service use composite | 95% Confidence interval [LB, UB] |
|---|---|---|---|---|
| C terminal | −0.236 | [−0.565, 0.093] | −0.114 | [−0.609, 0.381] |
| Early truncating | 0.216 | [−0.756, 1.188] | 0.060 | [−0.504, 0.624] |
| Large deletion | 0.061 | [−0.424, 0.547] | 0.088 | [−0.395, 0.570] |
| p.R106W | 0.003 | [−0.459, 0.464] | −0.073 | [−0.470, 0.323] |
| p.R133C | −0.459 | [−0.969, 0.050] | 0.153 | [−0.392, 0.698] |
| p.R168X | −0.148 | [−0.593, 0.298] | −0.329 | [−0.656, −0.002] |
| p.R255X | 0.205 | [−0.497, 0.906] | 0.273 | [−0.348, 0.894] |
| p.R270X | 0.026 | [−0.473, 0.526] | 0.081 | [−0.502, 0.664] |
| p.R294X | −0.349 | [−0.702, 0.004] | −0.026 | [−0.371, 0.320] |
| p.R306C | −0.424 | [−0.868, 0.020] | −0.258 | [−0.722, 0.206] |
| p.T158M | 0.137 | [−0.390, 0.664] | 0.147 | [−0.281, 0.574] |
There was variability in initial health status, with some (e.g., p.R255X) being poorer at a younger age, and some (e.g., p.R133C) being better at a younger age. From this starting point, health status for some mutation types improved (i.e. scores decreased) with age (e.g., p.R168X, p.R255X) and some deteriorated (i.e. scores increased) (Figure 2).
Figure 2.

Health Status Composite by age and mutation type
Health services use also varied by mutation type and age (Figure 3). Those with p.R133C had the highest health service composite score at the age of 25 years, but in the youngest age group their use was lowest. With many mutations, such as p.R255X, p.R270X, p.R294X, C terminal deletions and p.R306C, there was a high level of health service use at a younger age, but this dropped off significantly by the age of 25 years to little or no use. For p.R133C, large deletions, early truncating and p.R168X, health service use increased with age.
Figure 3.

Health Service Use Composite by age and mutation type
We then examined the change in health status use over time by comparing the interaction term coefficients and the age coefficient (0.086 for health status, Table 3). The two mutations, p.R168X (coefficient -0.093, p=0.039) and p.R255X (coefficient -0.100, p=0.028) decreased in health status with age, that is, these cases improved in health status over time.
Table 3.
Fixed effects and interactions with time for health status and health service use composite scores.
| Fixed effects | Health status composite | 95% Confidence interval [LB, UB] | p-Value | Health service use composite | 95% Confidence interval [LB, UB] | p-Value |
|---|---|---|---|---|---|---|
| Age | 0.086 | [0.013, 0.159] | 0.021 | 0.062 | [−0.021, 0.144] | 0.142 |
| C terminal | −0.035 | [−0.122, 0.052] | 0.429 | −0.109 | [−0.206, −0.012] | 0.028 |
| Early truncating | −0.062 | [−0.175, 0.505] | 0.279 | −0.045 | [−0.167, 0.077] | 0.466 |
| Large deletion | 0.006 | [−0.105, 0.116] | 0.921 | −0.042 | [−0.166, 0.081] | 0.501 |
| p.R106W | −0.070 | [−0.184, 0.043] | 0.224 | −0.081 | [−0.205, 0.043] | 0.200 |
| p.R133C | Baseline | Baseline | ||||
| p.R168X | −0.093 | [−0.181, −0.005] | 0.039 | −0.073 | [−0.172, 0.025] | 0.143 |
| p.R255X | −0.100 | [−0.189, −0.011] | 0.028 | −0.121 | [−0.221, −0.021] | 0.018 |
| p.R270X | −0.042 | [−0.132, 0.049] | 0.367 | −0.120 | [−0.221, −0.020] | 0.019 |
| p.R294X | −0.042 | [−0.133, 0.049] | 0.365 | −0.111 | [−0.212, −0.010] | 0.032 |
| p.R306C | −0.061 | [−0.156, 0.035] | 0.216 | −0.139 | [−0.245, −0.032] | 0.011 |
| p.T158M | −0.021 | [−0.108, 0.066] | 0.635 | −0.079 | [−0.176, 0.017] | 0.108 |
Likelihood ratio test for including the interaction between age and mutation: illness LR χ2 (10) = 12.22, p-value 0.2704. Service use LR χ2 (10) = 12.04, p-value.
For the composite of health service use (age coefficient 0.062), those with p.R255X mutations (coefficient -0.121, p=0.018), C-terminal deletions (-0.109, p=0.028), p.R270X (−0.110, p=0.019), p.R294X (−0.110, p=0.032) or p.R306C (−0.138, p=0.011) all showed a decreasing use of health services with age. In contrast, use of health services increased with age for participants with p.R133C, early truncating mutations, or large deletions.
Discussion
We found a gradual deterioration in health status through childhood, with stabilisation from 15 years of age onwards. Health service use, in contrast, was greatest in those under 5 years and gradually decreased with age.
Those with p.R133C had the best health status overall and those with p.R255X and early truncating mutations worst, although there were no statistically significant differences. Health service use was also highest in those with p.R133C and p.R255X but lowest in p.R168X – again there were no statistically significant differences. For mutations other than p.R255X and p.R168X, health status generally deteriorated with age, and for p.R133C, health status was especially good at a young age. Despite the apparent general deterioration with age for health status, health service use also appeared to decrease with age for the majority of mutations.
To our knowledge no other research has been able to investigate the health status trajectory in Rett syndrome using a longitudinal data collection. Previous research investigating genotype phenotype relationships has been based on a clinical profile at one point in time. Deterioration over time has been a hallmark of Rett syndrome, as outlined in the original “staging” system, describing 29 classical cases in four different phases of disease profile (Hagberg et al., 1986). However it has not been known whether this trajectory might differ by mutation type. Until recently, the data available on age-related changes in Rett syndrome were limited to those derived from the early Swedish work (Witt-Engerström, 1992) carried out in the period prior to any knowledge of the genetic basis of Rett syndrome and those based on our own, earlier, cross-sectional data (Colvin et al., 2003). However in the past couple of years two studies have specifically examined the disorder profile into adulthood of Rett syndrome in general and of particular genotypes (Halbach et al., 2008; Smeets, Chenault, Curfs, Schrander-Stumpel, & Frijns, 2009). The first, a questionnaire study on 53 adults identified the major morbidities as neurological, respiratory and behavioural (Halbach et al., 2008). The second identified specifically milder disorder profiles for those with p.R133C, p.R306C and C-terminal deletions (Smeets et al., 2009).
The relationship of phenotype to genotype has probably best been captured in our analysis of the 637 cases in the international (InterRett) database in 2005 (Bebbington et al., 2008). This confirmed the work of others in showing that those with p.R270X and p.R255X, the mutations in the nuclear localising signal region of the Transcription Repression Domain, were the most severe. The mildest mutations were p.R133C and p.R294X also consistent with our earlier Australian data (Colvin et al., 2004). In the international analysis, C terminal deletions were found to be associated with a mild phenotype, corroborating other research (Smeets et al., 2005; Smeets et al., 2009), as was p.R306C, also shown by Schanen et al. (Schanen et al., 2004). In a large recently published study C-terminal deletions were found to be less severe than all other MECP2 mutations but not as mild as the p.R133C and p.R294X mutations (Bebbington et al., 2010). However they were more likely than other mutations to be associated with a normal head circumference and weight than other mutations (Bebbington et al., 2010).
In this present study, health status was poorest overall in those with p.R255X and early truncating mutations and best overall in those with p.R133C, p.R294X, and p.R306C, consistent with these previous results. Another more recently published series including 236 mutation positive cases with “typical” Rett syndrome, found p.R168X to be the most severe of the common mutations (Neul et al., 2008). However this study opted to exclude cases who were atypical because they were either milder or more severe than the classical cases and may not have met the revised criteria (Hagberg, Hanefeld, Percy, & Skjeldal, 2002). This could have resulted in the exclusion of cases with the p.R270X mutation that are more likely to have a severe phenotype.
Using severity scales to measure phenotypic status, research has been undertaken on this Australian cohort in the past (Colvin et al., 2004) and, more recently, on an international dataset (Bebbington et al., 2008). These severity scales, which had generally been developed for the purpose of comparing genotype-phenotype relationships, took into account the developmental trajectory at regression as well as current functioning. In contrast in this study, health status was measured according to frequency of illnesses complemented by information drawn mostly from medication used to manage epilepsy, gastro-intestinal and nutritional problems. Therefore, neither information about the regression period, nor functional ability, (even if scoliosis was associated with poor mobility), was used, but rather the focus was on the medical burden of the disorder. We are not aware of any other research which has attempted to measure the current health burden of Rett syndrome or any scales used to do so. The data used in this study represent the experiences of illnesses, morbidity, medication load and health service use as reported by families. The scales that we developed and integrated into a number of composite scores using PCA were based on information we had collected on health burden related to major co-morbidities in Rett syndrome – epilepsy, gastro-intestinal problems, nutritional difficulties and scoliosis. The latter is the most severe orthopaedic complication in Rett syndrome, affecting three quarters of subjects by the age of 13 years (Ager et al., 2006). Its influence on health burden would be accounted for through the number of specialist visits, GP visits, and hospital stays generated by its management. We also incorporated a measure of illness frequency into the health status composite. We acknowledge possible limitations associated with family self-report and the use of a PCA-derived variable, but it would be extremely difficult to collect data from hospital and doctor records across the whole of Australia over the time period in question.
Any improvement of the symptoms of Rett syndrome resulting from interventions such as clinical trials will have to be measured in the presence of the general trajectory of the Rett syndrome. As such, measurement of health status in individuals over time is an important aspect of research. A strength of this study is its use of longitudinal data and mixed effects models to account for multiple time-points per person to investigate this under-researched aspect of the condition. Moreover this study has also been able to use statistical techniques to develop a measure of the burden of illness and comorbidity in this population-based dataset. However, since, by necessity, we have 12 different mutation groups, this dataset, although one of the largest population-based samples, still has relatively few cases per mutation group, thus reducing the power to detect statistically significant differences in health burden, service use and trajectory. While principal components analysis has been used in this data set to combine the different aspects of comorbidity burden creating an overall score that can be used for the trajectory analysis, this has the disadvantage of making the effects reported difficult to translate into clinical differences.
The pattern of health service use in this study was, interestingly, the reverse of health status in that it decreased with age, whilst, as one would expect, medical needs increased. The major exception was the p.R133C mutation, where health service use increased substantially with age. As shown previously, (Bebbington et al., 2008; Leonard et al., 2003) this mutation has a milder phenotype and is associated with later onset of symptoms and regression. However, the relative increased use of health services with age suggests that this comparative advantage may not be maintained over time. Although, in general, health status deteriorated over time, the trend was in the opposite direction for two mutations (p.R168X and p.R255X). In recent research using video data (Downs et al., 2008), we found that the motor abilities of participants with p.R255X were better than anticipated from current literature.
Because subjects entered this cohort study at varying periods after birth and some died prior to administration of the follow-up questionnaires, survivor bias could have been operating. We have already shown that those with the p.R270X mutation appear to be at increased risk of early mortality (Jian et al., 2005). This is also reflected in our data which show that those with this mutation were less likely to be in the study (two died prior to the onset of follow-up) and more likely to die during the study (three died at this time). Our analysis was restricted to participants who survived to 2000 and whose family completed at least one follow-up questionnaire. The trajectory associated with a particular mutation (e.g., p.R270X) might therefore appear better than expected because the most severe cases with that mutation have died and the surviving group has a relatively milder phenotype.
The paradoxical relationship between health status and service use is interesting but perhaps not surprising as it could reflect “the inverse care law” i.e. that “the availability of good medical care tends to vary inversely with the need for it in the population served” (Tudor Hart, 2000). In a previous analysis using Australian data collected in 2000, it was noted that medical appointments were less frequent in those whose mothers had the lowest level of education (Moore et al., 2005). In that same analysis specialist, but not general practitioner visits, were most frequent in the youngest age group (Moore et al., 2005). It is plausible that around the time of diagnosis the child sees more specialists and that this drops off with age. On the other hand, given that in this disorder functional ability becomes more impaired, scoliosis develops (Ager et al., 2006) and growth becomes more compromised with age (Oddy et al., 2007), it is concerning that service use decreases. This could also reflect a belief by families that medical care has less to offer their children as they grow older. However we also know that some young women with Rett syndrome move from the family home into residential accommodation. It is possible that, once in this situation there is more difficulty in obtaining access to health services. We do know that there is considerable concern, in Australia and elsewhere, about the inadequacy of health services for young people with a disability as they move into adulthood (Binks, Barden, Burke, & Young, 2007).
Conclusion
In contrast to the increase in laboratory research, since 1999, there has been little examining the course of this disorder and its relation to genotype. In keeping with earlier studies, which characterised the natural history as a series of stages (Hagberg et al., 1986), we have now demonstrated a deterioration in health status over time but we have also shown that this varies according to genotype. What is disturbing is the apparent drop-off in use of health services in parallel with the clinical deterioration. Although it is possible that this pattern may be reflective of that occurring in other developmental disorders, the burden of Rett syndrome is substantial with 71% survival rate by the age of 25 years (Freilinger et al., 2010). We suggest the need for more research focussing on the clinical management of this complex condition taking into account the variation by genotype.
Acknowledgments
The authors would like to acknowledge the funding of the major aspects of the Australian Rett Syndrome program by the National Institutes of Health (5R01HD043100-05) and also the National Medical and Health Research Council (NHMRC) under project grant 303189 for the clinical aspects. HL is funded by NHMRC Program Grant 353514, CB by NHMRC Fellowship 353628.
We thank the Australian clinicians who have reported cases and the families for their ongoing support and participation in our study. We also acknowledge the support of the Rett Syndrome Association of Australia, the Australian Paediatric Surveillance Unit and the Rett Syndrome Australian Research Fund. We acknowledge the assistance of Dr Philippa Carter and Matthew Mulroy in coding of the medication data.
We are also grateful for the genotyping of the Australian Rett syndrome cases by Dr Mark Davis, Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Perth, Australia, and Dr John Christodoulou, Dr Linda Weaving and Ms Sarah Williamson, Western Sydney Genetics Program, the Children’s Hospital at Westmead, Sydney, New South Wales, Australia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ager S, Fyfe S, Christodoulou J, Jacoby P, Schmitt L, Leonard H. Predictors of scoliosis in Rett syndrome. Journal of Child Neurology. 2006;21:809–813. doi: 10.1177/08830738060210091501. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Annals of Neurology. 2000;47:670–679. [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nature Genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bebbington A, Leonard H, Ben Zeev B, Anderson A, Ravine D, Fyfe S, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international dataset. Neurology. 2008;70:868–875. doi: 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- Bebbington A, Percy A, Christodoulou J, Ravine D, Ho G, Jacoby P, et al. Updating the profile of C-terminal MECP2 deletions in Rett syndrome. Journal of Medical Genetics. 2010;47:242–248. doi: 10.1136/jmg.2009.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binks JA, Barden WS, Burke TA, Young NL. What do we really know about the transition to adult-centered health care? A focus on cerebral palsy and spina bifida. Archives of Physical Medical Rehabilitation. 2007;88:1064–1073. doi: 10.1016/j.apmr.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Colvin L, Fyfe S, Leonard S, Schiavello T, Ellaway C, de Klerk N, et al. Describing the phenotype in Rett syndrome using a population database. Archives of Disease in Childhood. 2003;88:38–43. doi: 10.1136/adc.88.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin L, Leonard H, de Klerk N, Davis M, Weaving L, Williamson S, et al. Refining the phenotype of common mutations in Rett syndrome. Journal of Medical Genetics. 2004;41:25–30. doi: 10.1136/jmg.2003.011130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J, Bebbington A, Jacoby P, Masall M, McIlroy O, Fyfe SD, et al. Mobility profile in Rett syndrome as determined by video analysis. Neuropediatrics. 2008;39:205–210. doi: 10.1055/s-0028-1104575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilinger M, Bebbington A, Lanator I, de Klerk N, Dunkler D, Seidl R, et al. Survival with Rett syndrome: comparing Rett’s original sample with data from the Australian Rett Syndrome Database. Developmental Medicine and Child Neurology. 2010 doi: 10.1111/j.1469-8749.2010.03716.x. in press. [DOI] [PubMed] [Google Scholar]
- Gazarian M, Williams K, Elliott E, Chant K, Longbottom H, Mellis C, et al. Evaluation of a national surveillance unit. Archives of Disease in Childhood. 1999;80:21–27. doi: 10.1136/adc.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. European Journal of Pediatric Neurology. 2002;6:293–297. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Witt-Engerström I, Opitz JM, Reynolds JF. Rett syndrome: a suggested staging system for describing impairment profile with increasing age towards adolescence. American Journal of Medical Genetics Part A. 1986;25:47–59. doi: 10.1002/ajmg.1320250506. [DOI] [PubMed] [Google Scholar]
- Halbach NS, Smeets EE, Schrander-Stumpel CT, van Schrojenstein Lantman de Valk HH, Maaskant MA, Curfs LM. Aging in people with specific genetic syndromes: Rett syndrome. American Journal of Medical Genetics Part A. 2008;146A:1925–1932. doi: 10.1002/ajmg.a.32361. [DOI] [PubMed] [Google Scholar]
- Huppke P, Held M, Handefeld F, Engel W, Laccone F. Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics. 2002;33:63–68. doi: 10.1055/s-2002-32365. [DOI] [PubMed] [Google Scholar]
- Jian L, Archer HL, Ravine D, Kerr A, de Klerk N, Christodoulou J, et al. p.R270X MECP2 mutation and mortality in Rett syndrome. European Journal of Human Genetics. 2005;13:1235–1238. doi: 10.1038/sj.ejhg.5201479. [DOI] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, et al. Predictors of seizure onset in Rett syndrome. Journal of Pediatrics. 2006;149:542–547. doi: 10.1016/j.jpeds.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Kerr AM, Nomura Y, Armstrong D, Anvret M, Belichenko PV, Budden S, et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain & Development. 2001;23:208–211. doi: 10.1016/s0387-7604(01)00193-0. [DOI] [PubMed] [Google Scholar]
- Laurvick C, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. Rett syndrome in Australia: a review of the epidemiology. Journal of Pediatrics. 2006;148:347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Leonard H, Bower C. Is the girl with Rett syndrome normal at birth? Developmental Medicine and Child Neurology. 1998;40:115–121. [PubMed] [Google Scholar]
- Leonard H, Colvin L, Christodoulou J, Schiavello T, Williamson S, Davis M, et al. Patients with the R133C mutation: is their phenotype different from patients with Rett syndrome with other mutations? Journal of Medical Genetics. 2003;40:E52. doi: 10.1136/jmg.40.5.e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monros E, Armstrong J, Aibar E, Poo P, Canos I, Pineda M. Rett syndrome in Spain: mutation analysis and clinical correlations. Brain & Development. 2001;23:S251–S253. doi: 10.1016/s0387-7604(01)00374-6. [DOI] [PubMed] [Google Scholar]
- Moore H, Leonard H, de Klerk N, Robertson I, Fyfe S, Christodoulou J, et al. Health service use in Rett syndrome. Journal of Child Neurology. 2005;20:42–50. doi: 10.1177/08830738050200010701. [DOI] [PubMed] [Google Scholar]
- Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddy WH, Webb KG, Baikie G, Thompson SM, Reilly S, Fyfe SD, et al. Feeding experiences and growth status in a Rett syndrome population. Journal of Pediatric Gastroenterology and Nutrition. 2007;45:582–590. doi: 10.1097/MPG.0b013e318073cbf7. [DOI] [PubMed] [Google Scholar]
- Renieri A, Meloni I, Longo I, Ariani F, Mari F, Pescucci C, et al. Rett syndrome: the complex nature of a monogenic disease. Journal of Molecular Medicine. 2003;81:346–354. doi: 10.1007/s00109-003-0444-9. [DOI] [PubMed] [Google Scholar]
- Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. American Journal of Medical Genetics Part A. 2004;126:129–140. doi: 10.1002/ajmg.a.20571. [DOI] [PubMed] [Google Scholar]
- Smeets E, Terhal P, Casaer P, Peters A, Midro A, Schollen E, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. American Journal of Medical Genetics Part A. 2005;132:117–120. doi: 10.1002/ajmg.a.30410. [DOI] [PubMed] [Google Scholar]
- Smeets EE, Chenault M, Curfs LM, Schrander-Stumpel CT, Frijns JP. Rett syndrome and long-term disorder profile. American Journal of Medical Genetics Part A. 2009;149A:199–205. doi: 10.1002/ajmg.a.32491. [DOI] [PubMed] [Google Scholar]
- Stata. Stata Statistical Software (Version 9th) College Station, TX, USA: StataCorp LP; 1996–2006. [Google Scholar]
- Tudor Hart J. Three decades of the inverse care law. British Medical Journal. 2000;320:18–19. [PubMed] [Google Scholar]
- Weaving LS, Ellaway CJ, Gecz J, Christodoulou J. Rett syndrome: clinical review and genetic update. Journal of Medical Genetics. 2005;42:1–7. doi: 10.1136/jmg.2004.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt-Engerström I. Age-related occurrence of signs and symptoms in the Rett syndrome. Brain & Development. 1992;14:S11–S20. [PubMed] [Google Scholar]
- Young DJ, Bebbington A, Anderson A, Ravine D, Ellaway C, Kulkarni A, et al. The diagnosis of autism in a female: could it be Rett syndrome? European Journal of Pediatrics. 2008;167:661–669. doi: 10.1007/s00431-007-0569-x. [DOI] [PubMed] [Google Scholar]