

Abstract
With an in vitro system that used a luminescent strain of Klebsiella pneumoniae to assess bacterial metabolic activity in near-real-time, we investigated the dynamics of complement-mediated attack in healthy individuals and in patients presenting to the emergency department with community-acquired severe sepsis. A novel mathematical/statistical model was developed to simplify light output trajectories over time into two fitted parameters, the rate of complement activation and the delay from activation to the onset of killing. Using Factor B–depleted serum, the alternative pathway was found to be the primary bactericidal effector: In the absence of B, C3 opsonization as measured by flow cytometry did not progress and bacteria proliferated near exponentially. Defects in bacterial killing were easily demonstrable in patients with severe sepsis compared with healthy volunteers. In most patients with sepsis, the rate of activation was higher than in normal subjects but was associated with a prolonged delay between activation and bacterial killing (P < 0.05 for both). Theoretical modeling suggested that this combination of accentuated but delayed function should allow successful bacterial killing but with significantly greater complement activation. The use of luminescent bacteria allowed for the development of a novel and powerful tool for assessing complement immunology for the purposes of mechanistic study and patient evaluation.
Keywords: alternative complement pathway, theoretical models, nonlinear dynamics, sepsis
CLINICAL RELEVANCE.
Complement abnormalities are a recognized but poorly understood part of the human response to overwhelming infection. In this article we describe a method for evaluating complement's primary function (i.e., killing bacteria), present a strategy for analyzing the results of such observations, and note functional abnormalities in the serum taken from septic patients.
The complement cascade fills a crucial niche in early host defense against bacterial infection. Activating by fully innate means (i.e., through the alternative and mannose-binding lectin pathways) or in conjunction with acquired humoral immunity (via the classical pathway), the system contributes to defense by enzymatically labeling microorganisms for phagocytosis through C3 opsonization, by generating a series of anaphylatoxins that alert cellular defenses to the presence of a pathogen, and, in some instances, by directly killing the invading cell by permeabilizing its cell wall with membrane attack complex (C5b6789n or MAC).
During experimental or human sepsis, globally assessing the complement system is typically done in one of two ways. Most often, traces of previous activation are sought by measuring concentrations of complement proteins (such as C3, which falls with activation) or species elaborated during activation such as C3a, C5a, or soluble MAC (1). Less commonly, function is directly assessed. This is typically done by measuring complement hemolytic activity, with the canonical example being the CH50 method. CH50 is known to fall in severe human sepsis and multiorgan failure (2). How best to interpret this capacity (i.e., the complement-mediated cytolysis of an enucleate and other-species blood cell fully opsonized by a third species' IgG) in the setting of an overwhelming infection is not self-evident.
A more relevant functional assay of the complement system's performance in the presence of a pathogen could prove useful for pathophysiological studies and understanding clinical sepsis. Recently, an assay strategy for evaluating the capacity of serum to kill bacteria has been described that uses light output from luminescent bacteria (3–5). The assay is straightforward to perform and allows easy evaluation of experimental conditions, such as specific complement factor depletion. A shortcoming of the technique is the relative complexity of the experimental readout, namely a time series of luminescence measurements that evolve nonlinearly during host attack. This problem has limited the interpretation of reports using the technique in two ways: (1) Extracting understanding of complement function by the shape of assay luminescence curves is not intuitive, and (2) although assay results statistically may be handled by nonparametric means, such as repeated measures ANOVA (which makes no assumptions regarding the shape of the curves), a parametric approach that fits functionally meaningful parameters to each patient's results has not been available.
In the current study, we have extended this serum bactericidal assay using a strain of luminescent Klebsiella pneumoniae, with a specific goal of considering this assay as a means of quantifying patient antibacterial capacity in the setting of severe infection and sepsis. Klebsiella is an important pathogen in human respiratory and systemic illness and has been an important tool in previous studies of the dynamics of host–pathogen interactions in rodent models (6–9).
Our objectives were to confirm the role of human complement in killing this organism in vitro and to quantify the relative contributions of each of the activation pathways in this regard, including the relative timing of C3 opsonization compared with the timing of bactericidal activity, and to compare the bacterial killing capacity of healthy volunteers and patients presenting to an emergency department with community-acquired severe sepsis, with a specific intention of developing a parametric means of comparing individuals that would provide some functional insight into any differences that were noted between patient groups.
Fundamental to these efforts was the development of a dynamical model of complement-mediated bacterial killing that would permit parameterization of luminescence curves for theoretical and direct statistical purposes. Once complex bacterial light output histories could be reduced to a small number of parameters, statistical comparisons between various experimental conditions and between patients were possible, and more theoretical predictions could be made regarding the implications of the functional changes we observed in the serum of patients with sepsis.
MATERIALS AND METHODS
Bacterial Strains and Growth Conditions
K. pneumoniae Xen 39 (Caliper Life Sciences, Hopkinton, MA) is a constitutively luminescent strain and was used in all experiments. Before use, organisms were streaked overnight from cryopreserved stock onto Luria Bertani agar, then grown to mid-log growth in lysogeny broth media at 37°C the day of experimentation. Cells were washed and turbidometrically quantified as previously noted (10).
Luminescence Determinations
Organisms (2 × 104) were added to 100 μl of serum or other test media. These were maintained in a 37°C heat block in room air. Light output was recorded using a single-channel luminometer (Sirius FB12; Zylux, Inc., Huntsville, AL). This quantity of bacteria produced relative light measurements of approximately 5,000 units. Preliminary experiments were performed to confirm that the assay was well within the linear range of the device; bacterial inocula of 106 produced identical results. Luminescence measurements were taken as frequently as feasible; in most instances, one measurement was taken every 8 minutes. All experiments were performed at least in triplicate.
Assays in Complement-Depleted Sera
Sera immunodepleted of C1q (classical pathway deficient), factor B (alternative pathway deficient), or C5 (membrane attack complex deficient), as well as the associated positive control human serum were obtained from Quidel (San Diego, CA). Mannose (100 mM) was used to block mannose-binding lectin activity (10).
Detection of Constitutive Anti-Klebsiella IgM and IgG
Mid-log growth bacteria were opsonized with serum at 4°C for 1 hour and washed three times in phosphate-buffered saline. Bacteria were then incubated with FITC-labeled goat anti-huIgG or anti-huIgM (Jackson Immunoresearch, West Grove, PA) for 1 hour, washed, and examined for fluorescence using standard flow cytometry (Cytomics 500; Becton-Dickinson).
Flow Cytometric Determination of Complement C3 Opsonization Dynamics
Bacteria were added to serum prewarmed to 37°C and removed after 1, 2, 4, 6, or 8 minutes into ice–cold saline with EDTA to quench complement activation. Organisms were stained with FITC sheep anti-huC3 (Accurate Chemical and Scientific Corp., Westbury, NY) and analyzed by standard flow cytometry.
Samples from Severely Septic Patients and Healthy Control Subjects
Serum samples were taken from patients presenting to the emergency department with severe sepsis and evidence of hypoperfusion defined by (1) suspected or confirmed infection, (2) any two of four criteria of systemic inflammatory response, and (3) systolic blood pressure less than 90 mm Hg or MAP less than 65 after 20 ml/kg crystalloid (septic shock) or a whole blood lactate of mM or greater (severe sepsis). These individuals were part of a recently described prospective study of complement activation in early sepsis (1). Samples were drawn at the time of enrollment. Patients receiving fluoroquinolone antibiotics before enrollment were excluded from our analysis because of the adverse effect of this class of antibiotics on the assay (see Results). The study was reviewed by the institutional review board of the University of Michigan and Carolinas Medical Center, and written informed consent was obtained from each participant.
Mathematical Model of Bacterial Killing
Complement-mediated killing was characterized as having three steps: (1) activation; (2) a delay, during which membrane attack complexes were formed and inserted and cellular machinery was disabled; and (3) bacterial death, in which luminescence ceased. These concepts were incorporated in a system of delay differential equations,
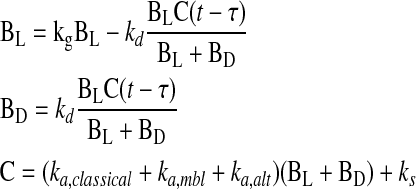 |
where BL, BD, and C are the number of live and dead bacteria and the amount of activated complement, respectively; kg and ks are empirically determined rates of bacterial growth and spontaneous complement activation via C3 “tick over;” ka is the rate of cascade activation via the classical, mannose-binding lectin, and alternative pathways (per minute); kd is the rate of complement mediated killing (per min); and τ is the delay between activation and killing (in minutes). The growth constant, kg, was experimentally determined in our laboratory to be 0.0104/min for K. pneumoniae Xen 39. The rate constant, ks, which corresponds to the rate of spontaneous tick over of the complement system (needed to initiate activation in this model), was set to 2.5 × 10−5/min, matching the accepted half-life of C3 in serum of approximately 200 hours (11). The use of a time delay, t, allows the many individual known complement reactions between activation and bacterial killing to be lumped in such a way that focus could be drawn on the immediately observable and measurable outcomes. To estimate the three remaining parameters for any patient, nonlinear least-squares curve fitting was performed against luminescence data. Specifically, the lsqcurvefit function of Matlab 2008a (Mathworks, Natick, MA) was used in conjunction with the delay differential equation solver dde23 s to generate parameter estimates. Both functions were used with default settings.
Model Sensitivity Analysis
The influence of model parameters on model behavior was considered using formal sensitivity analysis, as we have described previously (12). Details of this methodology and its results are presented in the online supplement.
RESULTS
The dynamics of complement-mediated killing of K. pneumoniae are shown in Figure 1A. In general, K. pneumoniae inoculated with normal human serum proliferated for approximately 1 hour, after which their metabolic activity, as indicated by luminescence diminished such that by 2 hours, bacteria encountering normal serum were no longer detectably luminescent. Quantitative culture confirmed that loss of detectable luminescence of a 2 × 104 colony-forming unit inoculum corresponded to greater than 99% reduction in colony-forming units. All three pathways participated in this response, although interruption of the MBL or classical pathway resulted only in delayed killing. Depletion of factor B removed the ability of serum to eliminate the organism. Absence of the alternative pathway was indistinguishable from the absence of membrane attack complex (i.e., in C5-depleted serum). To statistically comment on these observations, the luminescent histories in Figure 1A were modeled using a delay differential equation incorporating estimated bacterial proliferation and three components of host defense: complement activation, bacterial killing, and a necessary delay between these two processes, during which membrane attack complex was formed and inserted and during which the bacterial metabolic machinery necessary for luminescence was shut down. Doing so allowed direct comparison of apparent complement activation rates associated with each complement activation pathway (Figure 1B).
Figure 1.

Contributions of complement pathways to serum-mediated killing. (A). Bacterial luminescence during incubation with normal and various deficient sera. Serum depleted of C1q (classical pathway) or inhibited with mannose (mannose-binding lectin pathway) produced defects in the progression of killing. However, serum deficient in Factor B (alternative pathway) allowed exponential bacterial growth and was not distinguishable from C5-depleted, and therefore membrane-attack-complex–deficient, serum. (B). Fitted values of complement activation rate of data in A, as described in the text. n = 3 for each experimental condition in panels A and B. (C and D) Presence of anti-Klebsiella IgG and IgM in pooled normal human serum by flow cytometry. Bacteria incubated with normal human serum or phosphate-buffered saline were probed by flow cytometry for surface adsorbed IgG and IgM. Ample amounts of anti-Klebsiella IgG and IgM were present, indicating that the limited role of C1q noted in panels A and B was not a result of insufficient antibodies to activate the classical pathway. Cytometry results are representative of triplicate experiments.
Because the classical pathway requires complement-fixing antibodies to fully activate, we considered the possibility that C1q depletion's modest impact on bacterial killing was due to an absence of available antibodies. This was not the case. Extensive amounts of IgG and IgM were detectable, by flow cytometry, on the surface of organisms incubated with normal human serum (Figures 1C and 1D).
Based on these results, we next examined, by bacterial flow cytometry, the timing and extent of C3 opsonization by normal serum and serum deficient of the classical or the alternative pathway (Figure 2). Organisms were exposed to normal serum or serum depleted of C1q or factor B and stained with FITC-labeled anti-C3. For normal and C1q-deficient serum, evidence of C3 coverage was detectable within 2 minutes of exposure; this was extensive by 8 minutes. However, factor B–depleted serum showed no progression of bacterial coverage. These data, together with the dynamics shown in Figure 1, indicate that serum killing of K. pneumoniae is a process characterized by rapid, alternative pathway–driven activation and opsonization (within minutes of contact) followed by bacterial death within hours.
Figure 2.

Timing of C3 opsonization of Klebsiella pneumoniae. Organisms were incubated with normal or variously depleted sera. Aliquots were removed at the time points shown, stained with FITC-labeled antihuman C3, and analyzed by flow cytometry. In normal human serum (upper series), C3 coverage was detectable within 2 minutes of exposure and was well underway by 8 minutes. Serum lacking classical pathway activity (by C1q depletion, middle series) showed delayed onset but similar coverage by the last measured time point. Serum lacking alternative pathway activity (by Factor B depletion, lower series) showed no progression of opsonization over time. Results are representative of three replicate studies and are scaled such that the area under each curve is equal to 1.0 (i.e., they are scaled as probability density functions).
In studying this system, our aim in part was to establish a means of quantifying complement bactericidal function in patients. Because of a strong clinical emphasis to deliver antibiotics as quickly as possible to acutely ill patients, it is highly likely in clinical studies of complement function that patients may receive antibiotics before enrollment. It was therefore imperative to understand the impact of several clinically relevant antibiotics on assay performance. Formal antibiotic susceptibility testing was performed on the strain of K. pneumoniae used, and the effects of three antibiotics (levofloxacin, piperacillin/tazobactam, and ceftriaxone) were examined in the killing assay at 100 times their mean inhibitory concentration. Only levofloxacin interfered with the assay (Figure 3), producing rapid loss of luminescence compared with normal serum. Other antibiotics, to which the organism had laboratory-confirmed susceptibility, had no effect on the assay, indicating that the inhibitory or cidal effects of these agents had a slower onset than complement-mediated killing. Based on these results, subsequent analysis of patient samples was limited to those having not received a fluoroquinolone antibiotic for 2 weeks before enrollment and blood sampling.
Figure 3.

Impact of clinically relevant antibiotics on luminescence. Bacteria were inoculated with serum and 100 times the mean inhibitory concentration of levofloxacin or piperacillin and tazobactam, representative members of two classes of broad-spectrum antibiotics likely to be encountered in clinical samples. Levofloxacin produced bacterial killing much faster than serum, whereas piperacillin and tazobactam did not accelerate killing beyond that seen with complement alone. Results represent means ±SD of three to five replicates. Note that the SD bars in these plots are largely obscured by value icons.
We studied complement-mediated bacterial killing in 20 adult patients presenting to the emergency department with community-acquired severe sepsis (1). These patients were aged 55 ± 18 years and had infectious sources including pneumonia (n = 7), urinary tract (n = 5), intraabdominal (n = 3), skin (n = 1), and primary bacteremia (n = 1). Three patients had no source definitively identified. Three patients died of their illness. Representative luminescence waveforms from four subjects (one healthy control subject and three patients with severe sepsis) are shown in Figure 4A. Although there was intersample variability in the morphology of raw outputs, assays among patients with sepsis in general took one of the three forms shown—normal, accelerated activation, or prolonged delay—before the onset of bacterial clearance.
Figure 4.

Analysis of bacterial killing capacity in healthy volunteers and patients with sepsis. (A) Representative luminescence tracings of four subjects, one healthy and three severely septic, illustrating the differences in patterns seen. Tracing labels carry over to the scatter plot in B. (B) Plot of all observed activation rates and delays among the 30 subjects studied. (C and D). Box plots representing the 5th, 25th, 50th, 75th, and 95th percentile of parameter estimates for activation rate (per minutes) and delay (in minutes) between healthy (n = 10) and patients with severe sepsis (n = 20). Dots represent values outlying more than 2 SD from the sample mean. The groups were statistically different by t tests, although the results were unexpected: Most septic patients showed evidence of faster activation than the control subjects, suggesting up-regulation of complement effects or loss of regulatory function in the pathway.
We used a delay differential equation model to capture the key features of these complex assay outputs. The model included seven rate constants and a delay term. The rate of bacterial growth, kg, was determined experimentally on the bench. The rate of spontaneous complement activation (i.e., tick-over) was taken from the literature. The activation rates for the classical, MBL, and alternative pathways could not be determined individually in patient samples and were therefore lumped into a single activation rate, ka, that could be fit by least squares regression. The killing rate, kd, was fixed at the average estimated value across all patients (0.009/min) based on model sensitivity analysis (see below and the online supplement). This simplification produced a two-parameter problem wherein the lumped rate of complement activation, ka, and the delay between activation and killing, τ, were estimated for each patient sample.
A scatter plot of estimated observed activation rate and delay is shown for all subjects in Figure 4B. Activation rates and delays among healthy control subjects were relatively tightly clustered, although significant scatter of these values among patients with severe sepsis was noted. Statistical comparison of activation rate (Figure 4C) and delay before killing (Figure 4D) showed significant differences (P < 0.05 for each) between the two study groups. Although variation was seen among patients with sepsis, these patients in general showed evidence of accelerated activation but delayed effectiveness in bacterial killing.
We performed a formal parameter sensitivity analysis to better understand the theoretical contribution of each of our model parameters to the evolution of light output, and thus bacterial proliferation, over time. In Figure 5A, the impact of a 100% increase in any of the model parameters is plotted against the expected change in final assay luminescence that such a parameter change would produce. This gives a quantitative sense of the relative importance of each model term. The rate of bacterial growth, kg, was the dominant parameter, followed by the rates of activation, ka, and killing, kd. The same strategy was used to compare the impact of the differences in activation rate and delay between patients with sepsis and healthy subjects. In the gray bar in Figure 5A, we show the predicted effect of the combined accelerated activation but delayed killed seen in patients with sepsis in clearing bacteria from the assay system during a 2-hour experiment. In short, the abnormalities we identified in patients with sepsis would not be predicted on average to produce a detectable abnormality in bacterial killing at 2 hours after activation.
Figure 5.

(A) Theoretical sensitivity of bacterial luminescence and (B) extent of complement activation to various conditions based on the mathematical model described in the text. Arrows represent the direction and magnitude (in fold-change) of a doubling of any of the parameters used. (A) Doubling the rate of complement-mediated killing or of complement activation results in a predicted near-halving of remaining luminescent bacteria at 120 minutes after exposure. (B) Increasing the killing rate would yield a reduction in the amount of complement needed to accomplish the task, whereas increasing the rate of activation would be associated with almost double the amount of activation. In both panels, the gray boxes show the predicted impact on (A) bacterial killing and (B) complement activation on the differences in ka and τ noted between patients with sepsis and healthy subjects in our study. The model predicts that septic serum produces the same amount of killing as healthy serum after 2 hours of incubation but requires approximately 1.5 times the amount of complement activation to carry this out.
In Figure 5B, we consider more theoretically the impact of our model on the amount of complement activation that would take place during an episode of complement-mediated killing. Although we did not experimentally record complement activation in these experiments, the model does keep track of the theoretical, albeit nondimensional, extent of activation over time. Changes in this variable therefore may serve as a hypothesis generating feature of our analytical approach. As with bacterial proliferation, the extent of complement activation that occurs during killing is predominantly a function of the rate of bacterial growth. However, unlike what was observed in Figure 5A, the complement activation rate should be a much stronger determinant of total complement production than is the rate of bacterial killing. Furthermore, our model predicts that the impact of accelerated activation and delayed killing seen in patients with sepsis should be associated with greater complement activation than would be observed in healthy individuals confronted with the same bacterial challenge (Figure 5B, gray bar). Confirmation of these predictions awaits a technique capable of detecting the small amount of complement split products elaborated upon contact with a modest, clinically realistic bacterial challenge.
DISCUSSION
In this report we have significantly extended a real-time technique for assessing complement-mediated bacterial killing. Against the strain of K. pneumoniae studied, complement rapidly surface-deposited C3. Killing followed only after a considerable lag; although opsonization, as confirmed by flow cytometry, was well underway within 10 minutes of exposure, eradication required almost 2 hours. This response was primarily a result of the alternative pathway; depletion of the classical pathway or blocking of the MBL pathway delayed but did not stop bacterial inactivation. Serum killed this susceptible organism more rapidly than did β-lactam antibiotics but not as swiftly as did a fluoroquinolone.
Significant functional differences were seen in patients with sepsis. These changes did not indicate simple loss of function in the setting of serious infection. Rather, the kinetic activation rate necessary to explain the luminescence trajectories were significantly faster than normal in most patients. The explanation for this observation is not clear. It is possible that during sepsis, there is increased synthesis of the complement components necessary to activate on bacterial surfaces. We have preliminarily noted this in a murine model of Klebsiella pneumonia, wherein the abundance of complement and complement receptor mRNA increased in the liver and lung rapidly after the onset of sepsis (data not shown). Alternatively, it is possible that widespread activation of complement leads to exhaustion of regulatory proteins, including factors H and I. The dynamic interplay between consumption and production of pro- and anticomplement proteins during sepsis remains to be determined.
We believe the described method for quantifying complement function using real-time tracking of bacterial viability offers an important research tool in evaluating host response to bacterial pathogens. There are important limitations nevertheless. First, the strain studied is not a virulent one. It is fully susceptible to complement-mediated killing by human and mouse serum. In mice, it is rapidly cleared from the blood stream (within 5 min), and our laboratory has been unable to establish an LD50 dose due to its low virulence (7). As a result, studies of activity against this strain do not fully capture host–pathogen interactions likely to be encountered during infection, such as the presence of blocking polysaccharide O-antigen and capsule usually encountered on Klebsiella and other gram-negative organisms (8).
We used this technique to evaluate clinical samples of patients with severe sepsis. Because we did not collect convalescent serum from these patients, it is not possible to establish whether the complement functional differences seen were a result of being severely infected or were rather a preexisting defect predisposing some individuals to serious infection. We suspect the former to be the case, but this question remains unanswered.
We also acknowledge the limitations of the mathematical model used to permit statistical analysis of assay output. As with any statistical technique, a mathematical idea that matches the trend of the phenomenon has been imposed on data generated by a hidden process. The theoretical constructs of activation rate and delay thus serve a useful purpose for generating parameters amenable to statistical comparisons among and between patients and other experimental groups. They furthermore provide clues suggesting subsequent experiments that may enhance our understanding of the process of complement-mediated killing and refine the mathematical description of that process.
Acknowledgments
The authors thank Barbara Smith and Alex Jacobsen for their invaluable assistance in developing the assay and in recruiting patients for study.
This work was funded by NIGMS grants GM069438 (J.G.Y, D.M.B.) and GM076652 (A.E.J.) and by NSF grant no. 06-541 (P.W.N.).
Originally Published in Press as DOI: 10.1165/rcmb.2009-0292OC on December 11, 2009
Author Disclosure: D.B. has received sponsored grants from National Institute for General Medical Sciences (NIGMS) ($100,000+) for the current project and from National Science Foundation (NSF) ($100,000+), Department of Energy ($100,000+), and Department of Defense ($100,000+) for unrelated projects. D.B. has received industry-sponsored grants from Biolife, LLC ($10,000-$50,000) and from Rapid Biosense, LLC ($10,000-$50,000) and has received sponsored grants from NIGMS ($100,000+) for the current project and from NSF ($100,000+) for unrelated projects. A.J. has received industry-sponsored grants from Critical Biologics ($50,000-$100,000) and Hutchinson Technology ($10,001-$50,000) and has received sponsored grants from NIGMS ($100,000+) for the current project. P.N. has received sponsored grants from Burroughs Wellcome ($100,000+) for the current project and from NSF ($100,000+) for unrelated projects. M.T. has received industry-sponsored grants from Biolife, LLC ($10,000-$50,000) and Rapid Biosense, LLC ($10,000-$50,000) and has received sponsored grants from NIGMS ($100,000+) for the current project and NSF ($100,000+) for unrelated projects. S.Y. has received industry-sponsored grants from NSF ($10,000-$50,000) for the current project. J.Y. has received industry-sponsored grants from Biolife, LLC ($10,000-$50,000) and Rapid Biosense, LLC ($10,000-$50,000) and has received sponsored grants from NIGMS ($100,000+) for the current project and from NSF ($100,000+) for unrelated projects. None of the other authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Younger J, Bracho D, Chung H, Lee M, Rana G, Sen A, Jones A. Complement activation in emergency department patients with severe sepsis. Acad Emerg Med 2010;17:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goya T, Morisaki T, Torisu M. Immunologic assessment of host defense impairment in patients with septic multiple organ failure: relationship between complement activation and changes in neutrophil function. Surgery 1994;115:145–155. [PubMed] [Google Scholar]
- 3.Deryabin DG, Polyakov EG. Effect of human serum on bioluminescence of natural and recombinant luminescent bacteria. Bull Exp Biol Med 2004;138:276–279. [DOI] [PubMed] [Google Scholar]
- 4.Deryabin DG, Polyakov EG. Conditions that influence bacterial luminescence in the presence of blood serum. Microbiology 2005;74:159–163. [PubMed] [Google Scholar]
- 5.Deryabin DG, Polyakov EG. On-line determination of serum bactericidal activity using recombinant luminescent bacteria. Bull Exp Biol Med 2006;142:234–238. [DOI] [PubMed] [Google Scholar]
- 6.Ben-David I, Price S, Bortz D, Greineder C, Cohen S, Bauer A, Jackson T, Younger J. Dynamics of intrapulmonary growth in a murine model of repeated microaspiration. Am J Respir Cell Mol Biol 2005;33:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung H, Cartwright M, Thompson A, Bortz D, Jackson T, Younger J. Dynamical system analysis of Staphylococcus epidermidis bacteremia. Shock 2008;30:518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lugo J, Price S, Miller J, Ben-David I, Weinberg J, Mancuso P, Younger J. Lipopolysaccharide o-antigen promotes persistent murine bacteremia. Shock 2007;27:186–191. [DOI] [PubMed] [Google Scholar]
- 9.Shankar-Sinha S, Valencia G, Janes B, Rosenberg J, Whitfield C, Bender R, Standiford T, Younger J. Klebsiella pneumoniae o-antigen contributes to bacteremia and lethality during murine pneumonia. Infect Immun 2004;72:1423–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Younger J, Shankar-Sinha S, Mickiewicz M, Brinkman A, Younkin E, Sarma J, Standiford T, Zetoune F, Ward P. Murine complement interactions with pseudomonas aeruginosa and their effects during acute pneumonia. Am J Respir Cell Mol Biol 2003;29:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pangburn M, Muller-Eberhard H. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of c3a. Ann N Y Acad Sci 1983;421:291–298. [DOI] [PubMed] [Google Scholar]
- 12.Bortz D, Nelson P. Sensitivity analysis of a nonlinear lumped parameter model of HIV infection dynamics. Bull Math Biol 2004;66:1009–1026. [DOI] [PubMed] [Google Scholar]