

Abstract
Although an immune dysfunction and the involvement of infectious agents in the pathophysiology of schizophrenia are discussed since decades, the field never came into the mainstream of research. In schizophrenia a blunted type-1 immune response seems to be associated with a dysbalance in the activation of the enzyme indoleamine 2,3-dioxygenase (IDO) and in the tryptophan - kynurenine metabolism resulting in increased production of kynurenic acid in schizophrenia. This is associated with an imbalance in the glutamatergic neurotransmission, leading to an NMDA antagonism in schizophrenia. The immunological effects of antipsychotics rebalance partly the immune imbalance and the overweight of the production of the kynurenic acid. This immunological imbalance results in an inflammatory state combined with increased prostaglandin E2 (PGE2) production and increased cyclo-oxygenase-2 (COX-2) expression. COX-2 inhibitors have been tested in clinical trials, pointing to favourable effects in schizophrenia.
Keywords: Schizophrenia, major depression, kynurenine, inflammation, therapy
Introduction
Pathophysiological studies in schizophrenia were focussed on disturbances of the dopaminergic neurotransmission over decades without convincing results, antipsychotic antidopaminergic drugs still show unsatisfactory therapeutic effects. New concepts in the biological research of schizophrenia are required.
Recently, genetic data from multiple large cohorts of patients were published in ‘Nature’ showing that different gene loci located on chromosome 6p22.1 are the most probable susceptibility genes for schizophrenia (Shi et al., 2009; Stefansson et al., 2009; Purcell et al., 2009).
The region includes several genes of interest, which are related to the immune function. The strongest evidence for association was observed in or near a cluster of histone protein genes which could be relevant through their roles in regulation of DNA transcription or repair, i.e. in epigenetics (Costa et al., 2007), or their direct role in antimicrobial defence (Kawasaki and Iwamuro, 2008). Moreover, several genes of the HLA complex, which regulate the immune function and already earlier have been discussed to be involved in schizophrenia, are located in these regions (Fellerhoff et al., 2007).
Although an immune dysfunction and the involvement of infectious agents in the pathophysiology of schizophrenia are discussed since decades, the field never came into the mainstream of research. These genetic findings and further recent interesting observations, however, may contribute shifting research into the direction of immunological alterations and inflammation as cause for schizophrenia.
Infectious agents such as cytomegalo-, influenza-, borna-virus, Chlamydia, toxoplasma gondii and many others have been discussed to be involved in schizophrenia, but results from animal models of schizophrenia indicate that not a certain virus or other infectious agent but the immune response determines the risk for schizophrenia: Immune stimulants such as lipopolysaccharides or poly I:C, which mimick a bacterial or viral infection both lead to typical behaviour alterations in the offsprings of animals (Zuckerman and Weiner, 2005; Winter et al., 2009). In humans it could be shown that increased maternal levels of the pro-inflammatory cytokine Interleukin-8 (IL-8) during pregnancy are associated with an increased risk for schizophrenia in the offspring – whatever the reason for increased IL-8 was (Brown, 2006).
There is no doubt that the dopaminergic neurotransmission plays an important role in the pathophysiology of schizophrenia. Although the role of dopamine in schizophrenia has intensely been studied, the exact underlying pathological mechanisms are still unclear.
Dopaminergic hyperfunction in the limbic system and dopaminergic hypofunction in the frontal cortex are discussed to be the main neurotransmitter disturbances. Recent research provides further insight that glutamatergic hypofunction might be the cause for this dopaminergic dysfunction in schizophrenia (Swerdlow et al., 2009). The function of the glutamatergic system is closely related to the immune system and to the tryptophan-kynurenine metabolism, which both seem to play a key role in the pathophysiology of schizophrenia (Müller and Schwarz, 2007b; Müller and Schwarz, 2007a).
The immune response and the type-1 and type-2 polarization
The innate immune system is the phylogenetic oldest part of the immune response, e.g. natural killer (NK-) –cells and monocytes as the first barrier of the immune system being part of it. The adaptive immune response with the antibody producing B-lymphocytes, the T-lymphocytes and their regulating ‘immunotransmitters’, the cytokines, is the specifically acting component of the immune system. Cytokines regulate all types and all cellular components of the immune system including the innate immune system. Helper T-cells are of two types, T-helper-1 (TH-1) and T-helper-2 (TH-2). TH-1 cells produce the characteristic ‘type-1’ activating cytokines such as Interleukin-2 (IL-2) and Interferon-γ (IFN-γ). However, since not only TH-1 cells but also certain monocytes/macrophages (M1) and other cell-types produce these cytokines, the immune response is named type-1 immune response. The humoral – antibody producing - arm of the adaptive immune system is mainly activated by the type-2 immune response. TH-2 or certain monocytes/macrophages (M2) produce mainly IL-4, IL-10, and IL-13 (Mills et al., 2000). Another terminology separates the cytokines into pro-inflammatory and anti-inflammatory ones. Pro-inflammatory cytokines such as tumor-necrosis-factor-α(TNF-α) and IL-6 are primarily secreted from monocytes and macrophages, activating other cellular components of the inflammatory response. While TNF-α is an ubiquitous expressed cytokine mainly activating the type-1 response, IL-6 also activates the type-2 response including the antibody production. Anti-inflammatory cytokines such as IL-4 and IL-10 help to down-regulate the inflammatory immune response.
The type-1 immune system promotes the cell-mediated immune response directed against intracellular pathogens, whereas the type-2 response helps B-cell maturation and promotes the humoral immune response including the production of antibodies directed against extracellular pathogens. Type-1 and type-2 cytokines antagonize each other in promoting their own type of response, while suppressing the immune response of the other, therefore the term ‘polarized’ is used.
Inflammation in schizophrenia
Infection during pregnancy in mothers of off-springs later developing schizophrenia has been repeatedly described, in particular in the second trimester (Brown et al., 2004; Buka et al., 2000). As opposed to any single pathogen, the immune response, itself, of the mother may be related to the increased risk for schizophrenia in the offspring (Zuckerman and Weiner, 2005). Indeed, increased IL-8 levels of mothers during the second trimenon were associated with an increased risk for schizophrenia in the offspring (Brown et al., 2004). A five-fold increased risk for developing psychoses later on, however, was detected after infection of the CNS in early childhood (Brown et al., 2004; Gattaz et al., 2004). These data were confirmed in recent studies (Koponen et al., 2004; Brown, 2008; Dalman et al., 2008).
Signs of inflammation were found in schizophrenic brains (Körschenhausen et al., 1996), and the term ‘mild localized chronic encephalitis’ to describe a slight but chronic inflammatory process in schizophrenia was proposed (Bechter, 2001).
Type-1 and type-2 immune response in schizophrenia
A well established finding in schizophrenia is the decreased in vitro production of IL-2 and IFN-γ (Wilke et al., 1996; Müller et al., 2000), reflecting a blunted production of type-1 cytokines. Decreased levels of neopterin, a product of activated monocytes/macrophages, also point to a blunted activation of the type-1 response (Sperner-Unterweger et al., 1999a). The decreased response of lymphocytes after stimulation with specific antigens reflects a reduced capacity for a type-1 immune response in schizophrenia, as well (Müller et al., 1991). ICAM-1 is a type-1 related protein and a cell-adhesion molecule expressed on macrophages and lymphocytes. Decreased levels of the soluble (s) inter-cellular adhesion molecule-1 (ICAM-1), as found in schizophrenia, also represent an under-activation of the type-1 immune system (Schwarz et al., 2000). Decreased levels of the soluble TNF-receptor p55 – mostly decreased when TNF-α is decreased – were observed, too (Haack et al., 1999). A blunted response of the skin to different antigens in schizophrenia was observed before the era of antipsychotics (Molholm, 1942). This finding could be replicated in unmedicated schizophrenic patients using a skin test of the cellular immune response (Riedel et al., 2007). However, there are some conflicting results indicating possibly increased levels of type-1 cytokines in schizophrenia (Bresee and Rapaport, 2009). The latest meta-analysis showed dominant pro-inflammatory changes in schizophrenia but not the Th2 cytokines (Potvin et al., 2008). After including antipsychotic medication effects into the analysis – these effects on immune parameters are discussed below – only increases of IL-1 receptor antagonist serum levels and of IL-6 serum levels were found. The results of this meta-analysis support the view, that antipsychotic medication is a confounding factor in the analysis of the immune system in schizophrenia, up-regulating type-1 immune markers. Therefore studies of the immune system should regard the antipsychotic medication.
One study showed an increased of the type-1 cytokine IFN-γ in unmedicated schizophrenics (Kim et al., 2004) but methodological concerns have to be raised because the majority of samples of patients and controls were under the detection limit of IFN-γ. Several type-1 parameters, hypothesized to be down-regulated in schizophrenia, were not included into the metaanalysis, because only a few studies have been performed in unmedicated patients.
Moreover, several other signs of activation of the type-2 immune response are described in schizophrenia, including the increased Th2 type of lymphocytes in the blood (Sperner-Unterweger et al., 1999b), the increased production of Immunoglobulin E (IgE) and an increase of IL-10 serum levels (Schwarz et al., 2001; van Kammen et al., 1997). In the CSF, IL-10 levels were found to be related to the severity of the psychosis (van Kammen et al., 1997).
The key cytokine of the type-2 immune response is IL-4. Increased levels of IL-4 in the CSF of juvenile schizophrenic patients have been reported (Mittleman et al., 1997), which indicates that the increased type-2 response in schizophrenia is not only a phenomenon of the peripheral immune response.
Different stages of immune-dysfunction in schizophrenia?
Several reports described increased serum IL-6 levels in schizophrenia (Cazzullo et al., 1998). IL-6 serum levels might be especially high in patients with an unfavourable course of the disease (Lin et al., 1998) and in patients with a long duration of the disease (Ganguli et al., 1994). On the other hand, a significant relationship between the soluble IL-6R levels in the CSF and the paranoid-hallucinatory symptoms in schizophrenia have been reported (Müller et al., 1997a). A decreased level of the IL-6-complex functionally inhibiting molecule soluble gp130 in the CSF were detected, too, pointing a functional increase of IL-6 in schizophrenia (own unpublished results). IL-6 is a product of activated monocytes and some authors refer it as a marker of the type-2 immune response, although it is acting as a pro-inflammatory cytokine, too. The data show, however, that the immune response in schizophrenia is confounded by factors partly disease-inherent such as duration of disease, chronicity, prevailing symptoms or therapy response, partly other factors such as antipsychotic medication, smoking etc. The relationship between the long duration of disease, therapy resistance and high IL-6 levels point to differential immune processes possibly taking place in different stages of schizophrenia. It can be hypothesized that the blunted type-1 response is found primarily in early stages of schizophrenia, while a chronic pro-inflammatory stage with an overweight of the type-2 response including high IL-6 levels may predominate in later stages. Since IL-6 also induces autoimmunity, in later stages an autoimmune process may additionally play a role. The autoimmune hypothesis of schizophrenia has been discussed over decades (Knight, 1984; Hanson and Gottesman, 2005).
The view of different immune stages can also be underlined from the view of therapeutic studies: as it is discussed later, cyclo-oxygenase-2 inhibitors have therapeutic effects in early stages of schizophrenia, but seemingly only marginal effects in late stages.
Therapeutic mechanisms and the type-1/type-2 imbalance in schizophrenia
In-vitro studies show that the blunted IFN-γ production becomes normalized after therapy with neuroleptics (Wilke et al., 1996). An increase of ‘memory cells’ (CD4+CD45RO+ cells) - one of the main sources of IFN-γ production - during anti-psychotic therapy with neuroleptics was observed by different groups (Müller et al., 1997c). Additionally, an increase of sIL-2R - the increase reflects an increase of activated, IL-2 bearing T-cells - during anti-psychotic treatment was described (Müller et al., 1997b). The reduced sICAM-1 levels show a significant increase during short term anti-psychotic therapy (Schwarz et al., 2000) and the ICAM-1 ligand leucocyte function antigen-1 (LFA-1) shows a significantly increased expression during anti-psychotic therapy (Müller et al., 1999). The increase of TNF-α and TNF-α receptors during therapy with clozapine was observed repeatedly (Pollmächer et al., 2001). Moreover, the blunted reaction to vaccination with salmonella typhii was not observed in patients medicated with anti-psychotics (Ozek et al., 1971). An elevation of IL-18 serum levels was described in medicated schizophrenics (Tanaka et al., 2000). Since IL-18 plays a pivotal role in the type-1 immune response, this finding is consistent with other descriptions of type-1 activation during antipsychotic treatment.
Regarding the type-2 response, several studies point out that anti-psychotic therapy is accompanied by a functional decrease of the IL-6 system (Maes et al., 1997; Müller et al., 2000). These findings provide further evidence that antipsychotics have a ‘balancing’ effect on cytokines.
Divergent effects of the role of type-1/type-2 immune activation are associated with different effects to the kynurenine metabolism in schizophrenia
The only known naturally occurring NMDA receptor antagonist in the human CNS is kynurenic acid (KYNA). KYNA is one of the at least three neuroactive intermediate products of the kynurenine pathway. Kynurenine (KYN) is the primary major degradation product of tryptophan (TRP). While the excitatory KYN metabolites 3-hydroxykynurenine (3HK) and quinolinic acidolinic acid (QUINOLINIC ACID) are synthesized from KYN en route to NAD, KYNA is formed in a dead end side arm of the pathway Fig. (1) (Schwarcz and Pellicciari, 2002).
Fig (1).
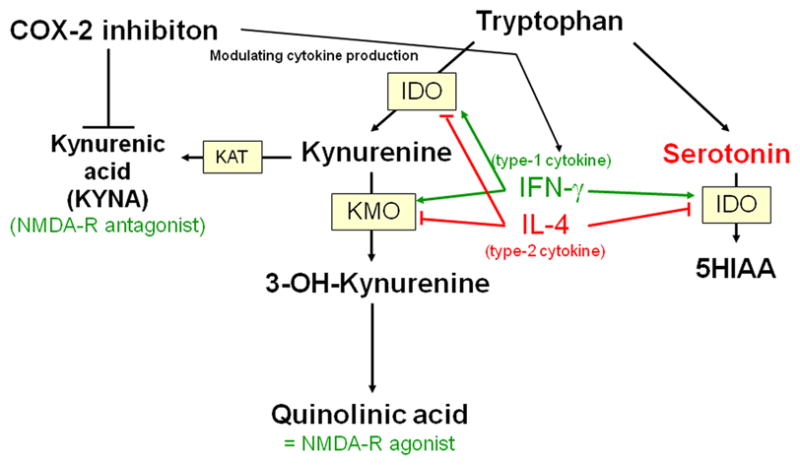
Pathways of the tryptophan/kynurenine metabolism to the NMDA receptor antagonist kynurenic acid and to the NMDA receptor agonist quinolinic acid.
KYNA acts both, as a blocker of the glycine co-agonistic site of the NMDA receptor and as a non-competitive inhibitor of the α7 nicotinic acetylcholine receptor (Hilmas et al., 2001).
The production of KYN metabolites is partly regulated by IDO and tryptophan 2,3-dioxygenase (TDO). Both enzymes catalyze the first step in the pathway, the degradation from tryptophan to kynurenine. Type-1 cytokines, such as IFN-γ and IL-2 stimulate the activity of IDO (Grohmann et al., 2003).
There is a mutual inhibitory effect of TDO and IDO: a decrease in TDO activity occurs concomitantly with IDO induction, resulting in a coordinate shift in the site (and cell types) of tryptophan degradation (Takikawa et al., 1986). While it has been known for a long time that IDO is expressed in different types of CNS cells, TDO was thought for many years to be restricted to liver tissue. It is known today, however, that TDO is also expressed in CNS cells, probably restricted to astrocytes (Miller et al., 2004).
The type-2 or Th-2 shift in schizophrenia may result in a down-regulation of IDO through the inhibiting effect of Th2 cytokines. TDO, on the other hand, was shown to be over-expressed in post mortem brains of schizophrenic patients (Miller et al., 2004). The type-1/type-2 imbalance with type-2 shift is therefore associated with over-expression of TDO.
Additionally, the type-1/type-2 imbalance is associated with the activation of astrocytes and an imbalance in the activation of astrocytes/microglial cells (Aloisi et al., 2000). The functional overweight of astrocytes may lead to a further accumulation of KYNA.
Indeed, a study referring to the expression of IDO and TDO in schizophrenia showed exactly the expected results. An increased expression of TDO compared to IDO was observed in schizophrenic patients and the increased TDO expression was found, as expected, in astrocytes, not in microglial cells (Miller et al., 2004).
Imaging studies – support for the inflammation hypothesis?
Inflammatory changes, such as demyelinating plaques in MS or in acute viral encephalitis do not present themselves in neuroimaging studies of schizophrenia or depression. There is, however, a progressive loss of brain-volume in schizophrenia.
In schizophrenia, there is no doubt that a smaller volume of the CNS can be observed already during the first episode and a progressive loss of the CNS volume including gray matter occurs during the further course of the disease especially in schizophrenics with a poor outcome (Gogtay et al., 2008; Steen et al., 2006). A relationship between the volume loss and an increased genetic risk for higher production of the immune marker IL-1 was described (Meisenzahl et al., 2001), as well as morphological changes of brain volume and increased IL-6 CSF levels in acute schizophrenia (Garver et al., 2003).
With the positron emission tomography, a ligand (PK 11195) for microglial activation which is increased in inflammatory processes in the CNS can be estimated (Versijpt et al., 2003). In schizophrenia, increased expression of PK11195 as hint for an inflammatory process could be shown (van Berckel et al., 2008). Another PET-study using the same ligand PK11195 could replicate the finding of an increased expression of this marker of an inflammatory process in patients during an acute exacerbation of schizophrenia. This increase was in particular found in the hippocampus of the patients (Doorduin et al., 2009).
Following the hypothesis of an ongoing immune process, it would be expected that the brain volume reductions, regularly found in schizophrenia, are not only due to a neurodevelopmental disturbance (Jakob and Beckmann, 1986), but also directly preceding the first episode of schizophrenia and, being further progressive. In fact, the Edinburgh High Risk Study, recently showed that a marked reduction of the inferior temporal gyrus over the time preceded the first onset of schizophrenia (Job et al., 2006) and the progressive loss of brain volume has repeatedly been demonstrated (e.g. (Chakos et al., 2005)).
Volume loss might be the result of different pathological processes, other than inflammatory causes may play a role. Nevertheless, the results of these studies are encouraging and further studies should focus on the relationship between inflammatory markers in the blood, CSF, CNS and the volume loss.
Astrocytes, microglia, and type-1/type-2 response
The cellular sources for the polarized immune response in the CNS are astrocytes and microglia cells. Microglial cells, deriving from peripheral macrophages, secrete preferably type-1 cytokines such as IL-12, while astrocytes inhibit the production of IL-12 and ICAM-1 and secrete the type-2 cytokine IL-10 (Aloisi et al., 1997). Therefore, the type-1/type-2 imbalance in the CNS seems to be represented by the imbalance in the activation of microglial cells and astrocytes, although it has to be taken into consideration that the production of cytokines by astrocytes and microglial cells depends from the activation conditions. The view of an over-activation of astrocytes in schizophrenia is supported by the finding of increased levels of S100B -a marker of astrocyte activation – independent of the medication state of the schizophrenic patients (Rothermundt et al., 2004). Microglia activation, however, was only found in a small percentage of schizophrenics and is speculated to be a medication effect (Bayer et al., 1999). A type-1 immune activation as an effect of neuroleptic treatment has repeatedly been observed.
The tryptophan- kynurenine metabolism in schizophrenia
In contrast to microglial cells which produce quinolinic acidolinic acid, astrocytes play a key role in the production of KYNA in the CNS. Astrocytes are the main source of KYNA (Heyes et al., 1997). The cellular localization of the kynurenine metabolism is primarily in macrophages and microglial cells, but also in astrocytes (Kiss et al., 2003). KMO, however, a critical enzyme in the kynurenine metabolism, is absent in human astrocytes (Guillemin et al., 2001). Accordingly, it has been described that astrocytes cannot produce the product 3-hydroxykynurenine (3-HK) but they are able to produce large amounts of early kynurenine metabolites, such as KYN and KYNA (Guillemin et al., 2001). This supports the observation that inhibition of KMO leads to an increase of the KYNA production in the CNS (Chiarugi et al., 1996). The complete metabolism of kynurenine to quinolinic acidolinic acid is observed mainly in microglial cells, only a small amount of quinolinic acid is produced in astrocytes via a side-arm of the kynurenine metabolism. Therefore, due to the lack of kynurenine-hydroxylase (KYN-OHse), in case of high tryptophan breakdown to KYN, KYNA may accumulate in astrocytes.
A second key-player in the metabolisation of 3-HK are monocytic cells infiltrating the CNS. They help astrocytes in the further metabolism to quinolinic acidolinic acid (Guillemin et al., 2001). However, the low levels of sICAM-1 (ICAM-1 is the molecule that mainly mediates the penetration of monocytes and lymphocytes into the CNS) in the serum and in the CSF of nonmedicated schizophrenic patients (Schwarz et al., 2000) and the increase of adhesion molecules during antipsychotic therapy indicate that the penetration of monocytes may be reduced in nonmedicated schizophrenic patients (Müller et al., 1999).
COX-2 Inhibition as therapeutic approach in schizophrenia
Animal studies show that COX-2 inhibition can lower the increase of the pro-inflammatory cytokines IL-1β, TNF-α, and of PGE2, but it can also prevent clinical symptoms such as anxiety and cognitive decline, which are associated with this increase of pro-inflammatory cytokines (Casolini et al., 2002). Moreover, treatment with the COX-2 inhibitor celecoxib – but not with a COX-1 inhibitor - prevented the dysregulation of the hypothalamus-pituitary-adrenal-axis, in particular the increase of cortisol, one of the biological key features associated with depression (Casolini et al., 2002; Hu et al., 2005). This effect can be expected because PGE2 stimulates the HPA-axis in the CNS (Song and Leonard, 2000) and PGE2 is inhibited by COX-2 inhibition. Moreover, the functional effects of IL-1 in the CNS – sickness behavior being one of these effects – were also shown to be antagonized by treatment with a selective COX-2 inhibitor (Cao et al., 1999).
Additionally, COX-inhibition provokes differential effects on the kynurenine metabolism: while COX-1 inhibition increases the levels of KYNA, COX-2 inhibition decreases them (Schwieler et al., 2005). Therefore, psychotic symptoms and cognitive dysfunctions, observed during therapy with COX-1 inhibitors, were assigned to the COX-1 mediated increase of KYNA. The reduction of KYNA levels - by a prostaglandin-mediated mechanism – might be an additional mechanism to the above described immunological mechanism for therapeutic effects of selective COX-2 inhibitors in schizophrenia (Schwieler et al., 2005).
Indeed, in a prospective, randomized, double-blind study of therapy with the COX-2 inhibitor celecoxib add-on to risperidone in acute exacerbation of schizophrenia, a therapeutic effect of celecoxib was observed (Müller et al., 2002). Immunologically, an increase of the type-1 immune response was found in the celecoxib treatment group (Müller et al., 2004a). The finding of a clinical advantage of COX-2 inhibition, however, could not be replicated in a second study. Further analysis of the data revealed that the outcome depends on the duration of the disease (Müller et al., 2004b). This observation is in accordance with results from animal studies showing that the effects of COX-2 inhibition on cytokines, hormones, and particularly on behavioural symptoms are dependent on the duration of the preceding changes and the time-point of application of the COX-2 inhibitor (Casolini et al., 2002). In subsequent clinical studies following a similar randomized double-blind placebo controlled add-on design of 400 milligram celecoxib to risperidone (in one study risperidone or olanzapine) in partly different patients populations, similar positive results of cyclo-oxygenase inhibition could be obtained: in a Chinese population of first manifestation schizophrenics (Zhang et al., 2006), and in an Iranian sample of chronic schizophrenics (Akhondzadeh et al., 2007). In continuously ill schizophrenics, however, no advantage of celecoxib could be found (Rapaport et al., 2005). In an own further study using the COX-2 inhibitor celecoxib add-on the amisulpride in first manifestation schizophrenics, a statistical significant beneficial effect of celecoxib compared to placebo was observed (Müller et al, submitted). Since all other add-on studies used risperidone as the basic antipsychotic, it was important to show that the effect of celecoxib is not dependent from risperidone. However, it is a weakness that until now all studies had the add-on design, while monotherapy with a COX-2 inhibitor in early stages of schizophrenia would be required from a scientific point of view. In schizophrenia, COX-2 inhibition showed beneficial effects preferentially in early stages of the disease, the data regarding chronic schizophrenia are controversial, possibly in part due to methodological concerns. Given that an inflammatory process is involved in the pathophysiology of schizophrenia, it cannot be expected that a short term intervention of five or six weeks would reverse a chronic inflammatory process which takes place over years. Moreover, during the process of chronification, a neurobiological neurodegeneration process may take place secondary to inflammation including the involvement of the tryptophan-kynurenine pathway, which by itself additionally provokes neurodegeneration (Myint et al., 2007). Those long-term processes, however, might not be influenced by short-term intervention with a COX-2 inhibitor. The data are still preliminary and further research has to be performed, e.g. with other COX-2 inhibitors.
Conclusion and outlook
The possible influence of an immunological process for the pathogenesis of schizophrenia resulting in inflammation has long been neclected. Increasing evidence for a role of proinflammatory cytokines in schizophrenia, the strong influence of pro- and anti-inflammatory cytokines on the tryptophan/kynurenine metabolism and, related to that mechanism, the influence of cytokines on the glutamatergic neurotransmission, the results of imaging studies, genetic findings, and last not least the therapeutic effect of anti-inflammatory drugs support the view that psychoneuroimmunology inflammation rightly came recently more into the focus of schizophrenia research. It had to be regarded on the other hand that immunological research is susceptible for artefacts, interfering variables such as medication, smoking, stress, sleep, and others play an important role and cannot always been controlled. This can be shown at the example of stress: Stress is not only according to the ‘vulnerability-stress-model’ a condition sine qua non in schizophrenia – it is also a confounding factor for research of the immune system and inflammatory processes. Similarly for neuro-imaging studies: Volume loss might be the result of different pathological processes, other than inflammatory causes may play a role. Nevertheless, the results of these studies are encouraging and further studies should focus on the relationship between inflammatory markers in the blood, CSF, and volume loss of the CNS. Moreover, the influence of different stages in schizophrenia might have been neclected, too. It is discussed, that schizophrenia is a syndrome with different underlying pathological processes. Inflammation, however, also includes different stages and processes ranging from an acute to a chronic inflammation, including an autoimmune process.
These considerations show, that a lot of further research is necessary to clarify the role of the immune system in schizophrenia, but recent results including therapeutic progress encourages further emphasis for this fascinating field.
Table. Studies of COX-2 inhibitors in schizophrenia.
Overview on studies using a cyclo-oxygenase-2 inhibitor for antipsychotic therapy in schizophrenia.
| Authors | Diagnosis | Course and duration | Duration of trial | N | Study design | Concomitant drug | COX-2 inhibitor | Outcome |
|---|---|---|---|---|---|---|---|---|
| Zhang et al, 2006 (Zhang et al., 2006) | schizophrenia | First manifestation | 12 weeks | 40 | double-blind, randomized placebo-controlled add-on | Risperidone (flexible dose) | Celecoxib 400 mg/day | significant advantage of the COX-2 inhibitor |
| Müller et al., 2002 (Müller et al., 2002) | schizophrenia | Not specified mean 5,9 y | 5 weeks | 50 | double-blind, randomized placebo-controlled add-on | Risperidone (flexible dose) | Celecoxib 400 mg/day | significant advantage of the COX-2 inhibitor |
| Rappard and Müller, 2004 (Rappard and Müller, 2004) | schizophrenia | ≤ 10 years | 11 weeks | 270 | double-blind, randomized placebo-controlled add-on | Risperidone (flexible dose) | Celecoxib 400 mg/day | non advantage of the COX- 2inhibitor |
| Rapaport et al, 2005 (Rapaport et al., 2005) | schizophrenia | Continuously ill mean 20 y | 8 weeks | 38 | double-blind, randomized placebo-controlled add-on | Risperidone Olanzapine or (constant dose) | Celecoxib 400 mg/day | non advantage on the COX-2 inhibitor |
| Akhondzadeh et al, 2007 (Akhondzadeh et al., 2007) | schizophrenia | Chronic type (active phase) | 8 weeks | 60 | double-blind, randomized placebo-controlled add-on | Risperidone (fixed dose) | Celecoxib 400 mg/day | significant advantage of the COX-2 inhibitor |
Reference List
- Akhondzadeh S, Tabatabaee M, Amini H, Ahmadi Abhari SA, Abbasi SH, Behnam B. Celecoxib as adjunctive therapy in schizophrenia: a double-blind, randomized and placebo-controlled trial. Schizophr Res. 2007;90:179–185. doi: 10.1016/j.schres.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Penna G, Cerase J, Menendez IB, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol. 1997;159:1604–1612. [PubMed] [Google Scholar]
- Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141–147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. 1999;271:126–128. doi: 10.1016/s0304-3940(99)00545-5. [DOI] [PubMed] [Google Scholar]
- Bechter K. Mild encephalitis underlying psychiatric disorders - A reconsideration and hypothesis exemplified on Borna disease. Neurol Psychiatry Brain Res. 2001;9:55–70. [Google Scholar]
- Bresee C, Rapaport MH. Persistently increased serum soluble interleukin-2 receptors in continuously ill patients with schizophrenia. Int J Neuropsychopharmacol. 2009;12:861–865. doi: 10.1017/S1461145709000315. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. The risk for schizophrenia from childhood and adult infections. Am J Psychiatry. 2008;165:7–10. doi: 10.1176/appi.ajp.2007.07101637. [DOI] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Buka SL, Goldstein JM, Seidman LJ, Tsuang MT. Maternal recall of pregnancy history: accuracy and bias in schizophrenia research. Schizophr Bull. 2000;26:335–350. doi: 10.1093/oxfordjournals.schbul.a033457. [DOI] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Ozaki M, Watanabe Y. Lipopolysaccharide injected into the cerebral ventricle evokes fever through induction of cyclooxygenase-2 in brain endothelial cells. J Neurosci. 1999;19:716–725. doi: 10.1523/JNEUROSCI.19-02-00716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res. 2002;68:337–343. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- Cazzullo CL, Scarone S, Grassi B, Vismara C, Trabattoni D, Clerici M, Clerici M. Cytokines production in chronic schizophrenia patients with or without paranoid behaviour. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:947–957. doi: 10.1016/s0278-5846(98)00059-1. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Schobel SA, Gu H, Gerig G, Bradford D, Charles C, Lieberman JA. Duration of illness and treatment effects on hippocampal volume in male patients with schizophrenia. Br J Psychiatry. 2005;186:26–31. doi: 10.1192/bjp.186.1.26. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Carpenedo R, Moroni F. Kynurenine disposition in blood and brain of mice: effects of selective inhibitors of kynurenine hydroxylase and of kynureninase. J Neurochem. 1996;67:692–698. doi: 10.1046/j.1471-4159.1996.67020692.x. [DOI] [PubMed] [Google Scholar]
- Costa E, Dong E, Grayson DR, Guidotti A, Ruzicka W, Veldic M. Reviewing the role of DNA (cytosine-5) methyltransferase overexpression in the cortical GABAergic dysfunction associated with psychosis vulnerability. Epigenetics. 2007;2:29–36. doi: 10.4161/epi.2.1.4063. [DOI] [PubMed] [Google Scholar]
- Dalman C, Allebeck P, Gunnell D, Harrison G, Kristensson K, Lewis G, Lofving S, Rasmussen F, Wicks S, Karlsson H. Infections in the CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am J Psychiatry. 2008;165:59–65. doi: 10.1176/appi.ajp.2007.07050740. [DOI] [PubMed] [Google Scholar]
- Doorduin J, De Vries EFJ, Willemsen ATM, de Groot JC, Dierck RA, Klein HC. Schizophrenia related psychosis is associated with focal neuroinflammation: a (11C)-PL11195 positron emission tomography study. Neurol Psychiatry Brain Res . 2009;(1):14. [Google Scholar]
- Fellerhoff B, Laumbacher B, Mueller N, Gu S, Wank R. Associations between Chlamydophila infections, schizophrenia and risk of HLA-A10. Mol Psychiatry. 2007;12:264–272. doi: 10.1038/sj.mp.4001925. [DOI] [PubMed] [Google Scholar]
- Ganguli R, Yang Z, Shurin G, Chengappa KN, Brar JS, Gubbi AV, Rabin BS. Serum interleukin-6 concentration in schizophrenia: elevation associated with duration of illness. Psychiatry Res. 1994;51:1–10. doi: 10.1016/0165-1781(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Garver DL, Tamas RL, Holcomb JA. Elevated interleukin-6 in the cerebrospinal fluid of a previously delineated schizophrenia subtype. Neuropsychopharmacology. 2003;28:1515–1520. doi: 10.1038/sj.npp.1300217. [DOI] [PubMed] [Google Scholar]
- Gattaz WF, Abrahao AL, Foccacia R. Childhood meningitis, brain maturation and the risk of psychosis. Eur Arch Psychiatry Clin Neurosci. 2004;254:23–26. doi: 10.1007/s00406-004-0431-3. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Lu A, Leow AD, Klunder AD, Lee AD, Chavez A, Greenstein D, Giedd JN, Toga AW, Rapoport JL, Thompson PM. Three-dimensional brain growth abnormalities in childhood-onset schizophrenia visualized by using tensor-based morphometry. Proc Natl Acad Sci U S A. 2008;105:15979–15984. doi: 10.1073/pnas.0806485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 2003;24:242–248. doi: 10.1016/s1471-4906(03)00072-3. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001;78:842–853. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- Haack M, Hinze-Selch D, Fenzel T, Kraus T, Kuhn M, Schuld A, Pollmacher T. Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res. 1999;33:407–418. doi: 10.1016/s0022-3956(99)00021-7. [DOI] [PubMed] [Google Scholar]
- Hanson DR, Gottesman II. Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet. 2005;6:7. doi: 10.1186/1471-2350-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Chen CY, Major EO, Saito K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem J. 1997;326:351–356. doi: 10.1042/bj3260351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Wang X, Pace TW, Wu H, Miller AH. Inhibition of COX-2 by celecoxib enhances glucocorticoid receptor function. Mol Psychiatry. 2005;10:426–428. doi: 10.1038/sj.mp.4001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob H, Beckmann H. Prenatal developmental disturbances in the limbic allocortex in schizophrenics. J Neural Transm. 1986;65:303–326. doi: 10.1007/BF01249090. [DOI] [PubMed] [Google Scholar]
- Job DE, Whalley HC, McIntosh AM, Owens DG, Johnstone EC, Lawrie SM. Grey matter changes can improve the prediction of schizophrenia in subjects at high risk. BMC Med. 2006;4:29. doi: 10.1186/1741-7015-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets. 2008;8:195–205. doi: 10.2174/1871526510808030195. [DOI] [PubMed] [Google Scholar]
- Kim YK, Myint AM, Lee BH, Han CS, Lee HJ, Kim DJ, Leonard BE. Th1, Th2 and Th3 cytokine alteration in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:1129–1134. doi: 10.1016/j.pnpbp.2004.05.047. [DOI] [PubMed] [Google Scholar]
- Kiss C, Ceresoli-Borroni G, Guidetti P, Zielke CL, Zielke HR, Schwarcz R. Kynurenate production by cultured human astrocytes. J Neural Transm. 2003;110:1–14. doi: 10.1007/s00702-002-0770-z. [DOI] [PubMed] [Google Scholar]
- Knight JG. Is schizophrenia an autoimmune disease? A review. Methods Find Exp Clin Pharmacol. 1984;6:395–403. [PubMed] [Google Scholar]
- Koponen H, Rantakallio P, Veijola J, Jones P, Jokelainen J, Isohanni M. Childhood central nervous system infections and risk for schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2004;254:9–13. doi: 10.1007/s00406-004-0485-2. [DOI] [PubMed] [Google Scholar]
- Körschenhausen DA, Hampel HJ, Ackenheil M, Penning R, Müller N. Fibrin degradation products in post mortem brain tissue of schizophrenics: a possible marker for underlying inflammatory processes. Schizophr Res. 1996;19:103–109. doi: 10.1016/0920-9964(95)00073-9. [DOI] [PubMed] [Google Scholar]
- Lin A, Kenis G, Bignotti S, Tura GJ, De JR, Bosmans E, Pioli R, Altamura C, Scharpe S, Maes M. The inflammatory response system in treatment-resistant schizophrenia: increased serum interleukin-6. Schizophr Res. 1998;32:9–15. doi: 10.1016/s0920-9964(98)00034-6. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–858. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G, Maag K, Hegerl U, Hahn K, Moller HJ. Association of an interleukin-1beta genetic polymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry. 2001;158:1316–1319. doi: 10.1176/appi.ajp.158.8.1316. [DOI] [PubMed] [Google Scholar]
- Miller CL, Llenos IC, Dulay JR, Barillo MM, Yolken RH, Weis S. Expression of the kynurenine pathway enzyme tryptophan 2,3-dioxygenase is increased in the frontal cortex of individuals with schizophrenia. Neurobiol Dis. 2004;15:618–629. doi: 10.1016/j.nbd.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. [Google Scholar]
- Mittleman BB, Castellanos FX, Jacobsen LK, Rapoport JL, Swedo SE, Shearer GM. Cerebrospinal fluid cytokines in pediatric neuropsychiatric disease. J Immunol. 1997;159:2994–2999. [PubMed] [Google Scholar]
- Molholm HB. Hyposensitivity to foreign protein in schizophrenic patients. Psychiatr Quarterly. 1942;16:565–571. [Google Scholar]
- Müller N, Ackenheil M, Hofschuster E, Mempel W, Eckstein R. Cellular immunity in schizophrenic patients before and during neuroleptic treatment. Psychiatry Res. 1991;37:147–160. doi: 10.1016/0165-1781(91)90072-w. [DOI] [PubMed] [Google Scholar]
- Müller N, Dobmeier P, Empl M, Riedel M, Schwarz M, Ackenheil M. Soluble IL-6 receptors in the serum and cerebrospinal fluid of paranoid schizophrenic patients. Eur Psychiatry. 1997a;12:294–299. doi: 10.1016/S0924-9338(97)84789-X. [DOI] [PubMed] [Google Scholar]
- Müller N, Empl M, Riedel M, Schwarz M, Ackenheil M. Neuroleptic treatment increases soluble IL-2 receptors and decreases soluble IL-6 receptors in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 1997b;247:308–313. doi: 10.1007/BF02922260. [DOI] [PubMed] [Google Scholar]
- Müller N, Riedel M, Ackenheil M, Schwarz MJ. Cellular and humoral immune system in schizophrenia: a conceptual re-evaluation. World J Biol Psychiatry. 2000;1:173–179. doi: 10.3109/15622970009150588. [DOI] [PubMed] [Google Scholar]
- Müller N, Riedel M, Dehning S, Spellmann I, Müller-Arends A, Cerovecki A, et al. Is the therapeutic effect of celecoxib in schizophrenia depending from duration of disease? Neuropsychopharmacology. 2004a;29:176. [Google Scholar]
- Müller N, Riedel M, Hadjamu M, Schwarz MJ, Ackenheil M, Gruber R. Increase in expression of adhesion molecule receptors on T helper cells during antipsychotic treatment and relationship to blood-brain barrier permeability in schizophrenia. Am J Psychiatry. 1999;156:634–636. [PubMed] [Google Scholar]
- Müller N, Riedel M, Scheppach C, Brandstätter B, Sokullu S, Krampe K, Ulmschneider M, Engel RR, Möller HJ, Schwarz MJ. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am J Psychiatry. 2002;159:1029–1034. doi: 10.1176/appi.ajp.159.6.1029. [DOI] [PubMed] [Google Scholar]
- Müller N, Riedel M, Schwarz MJ, et al. Immunomodulatory effects of neuroleptics to the cytokine system and the cellular immune system in schizophrenia. In: Wieselmann G, editor. Current update in psychoimmunology. Wien, New York: Springer; 1997c. pp. 57–67. [Google Scholar]
- Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007a:1–13. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Müller N, Schwarz MJ. The immunological basis of glutamatergic disturbance in schizophrenia: towards an integrated view. J Neurotransmission. 2007b:269–280. doi: 10.1007/978-3-211-73574-9_33. [DOI] [PubMed] [Google Scholar]
- Müller N, Ulmschneider M, Scheppach C, Schwarz MJ, Ackenheil M, Möller HJ, Gruber R, Riedel M. COX-2 inhibition as a treatment approach in schizophrenia: immunological considerations and clinical effects of celecoxib add-on therapy. Eur Arch Psychiatry Clin Neurosci. 2004b;254:14–22. doi: 10.1007/s00406-004-0478-1. [DOI] [PubMed] [Google Scholar]
- Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord. 2007;98:143–151. doi: 10.1016/j.jad.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Ozek M, Toreci K, Akkok I, Guvener Z. [Influence of therapy on antibody-formation] Psychopharmacologia. 1971;21:401–412. doi: 10.1007/BF02419063. [DOI] [PubMed] [Google Scholar]
- Pollmächer T, Schuld A, Kraus T, Haack M, Hinze-Selch D. On the clinical relevance of clozapine-triggered release of cytokines and soluble cytokine-receptors] Fortschr Neurol Psychiatr . 2001;69(Suppl 2):S65–74. S65–S74. doi: 10.1055/s-2001-16533. [DOI] [PubMed] [Google Scholar]
- Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory Cytokine Alterations in Schizophrenia: A Systematic Quantitative Review. Biol Psychiatry. 2008;63:801–808. doi: 10.1016/j.biopsych.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport MH, Delrahim KK, Bresee CJ, Maddux RE, Ahmadpour O, Dolnak D. Celecoxib augmentation of continuously ill patients with schizophrenia. Biol Psychiatry. 2005;57:1594–1596. doi: 10.1016/j.biopsych.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Rappard F, Müller N. Celecoxib Add-on Therapy Does Not Have Beneficial Antipsychotic Effects Over Risperidone Alone in Schizophrenia. Neuropsychopharmacology. 2004;29(Suppl 1):S222. [Google Scholar]
- Riedel M, Spellmann I, Schwarz MJ, Strassnig M, Sikorski C, Möller HJ, Müller N. Decreased T cellular immune response in schizophrenic patients. J Psychiatr Res. 2007;41:3–7. doi: 10.1016/j.jpsychires.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Rothermundt M, Falkai P, Ponath G, Abel S, Burkle H, Diedrich M, Hetzel G, Peters M, Siegmund A, Pedersen A, Maier W, Schramm J, Suslow T, Ohrmann P, Arolt V. Glial cell dysfunction in schizophrenia indicated by increased S100B in the CSF. Mol Psychiatry. 2004;9:897–899. doi: 10.1038/sj.mp.4001548. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- Schwarz MJ, Chiang S, Müller N, Ackenheil M. T-helper-1 and T-helper-2 responses in psychiatric disorders. Brain Behav Immun. 2001;15:340–370. doi: 10.1006/brbi.2001.0647. [DOI] [PubMed] [Google Scholar]
- Schwarz MJ, Riedel M, Ackenheil M, Müller N. Decreased levels of soluble intercellular adhesion molecule-1 (sICAM-1) in unmedicated and medicated schizophrenic patients. Biol Psychiatry. 2000;47:29–33. doi: 10.1016/s0006-3223(99)00206-1. [DOI] [PubMed] [Google Scholar]
- Schwieler L, Erhardt S, Erhardt C, Engberg G. Prostaglandin-mediated control of rat brain kynurenic acid synthesis--opposite actions by COX-1 and COX-2 isoforms. J Neural Transm. 2005;112:863–872. doi: 10.1007/s00702-004-0231-y. [DOI] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Leonard BE. Fundamentals of psychoneuroimmunology. Chichester, New York: J Wiley and Sons; 2000. [Google Scholar]
- Sperner-Unterweger B, Miller C, Holzner B, Widner B, Fleischhacker WW, Fuchs D. Measurement of neopterin, kynurenine and tryptophan in sera of schizophrenic patients. In: Müller N, editor. Psychiatry, Psychoimmunology, and viruses. Wien, New York: Springer; 1999a. pp. 115–119. [Google Scholar]
- Sperner-Unterweger B, Whitworth A, Kemmler G, Hilbe W, Thaler J, Weiss G, Fleischhacker WW. T-cell subsets in schizophrenia: a comparison between drug-naive first episode patients and chronic schizophrenic patients. Schizophr Res. 1999b;38:61–70. doi: 10.1016/s0920-9964(98)00175-3. [DOI] [PubMed] [Google Scholar]
- Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry . 2006;188:510–8. 510–518. doi: 10.1192/bjp.188.6.510. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de FR, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St CD, Goldstein DB, Stefansson K, Collier DA. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J. The Effects of Memantine on Prepulse Inhibition. Neuropsychopharmacology. 2009 doi: 10.1038/npp.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa O, Yoshida R, Kido R, Hayaishi O. Tryptophan degradation in mice initiated by indoleamine 2,3-dioxygenase. J Biol Chem. 1986;261:3648–3653. [PubMed] [Google Scholar]
- Tanaka KF, Shintani F, Fujii Y, Yagi G, Asai M. Serum interleukin-18 levels are elevated in schizophrenia. Psychiatry Res. 2000;96:75–80. doi: 10.1016/s0165-1781(00)00196-7. [DOI] [PubMed] [Google Scholar]
- van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–822. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- van Kammen DP, McAllister-Sistilli CG, Kelley ME. Relationship between immune and behavioral measures in schizophrenia. In: Wieselmann G, editor. Current Update in Psychoimmunology. Wien, New York: Springer; 1997. pp. 51–55. [Google Scholar]
- Versijpt JJ, Dumont F, Van Laere KJ, Decoo D, Santens P, Audenaert K, Achten E, Slegers G, Dierckx RA, Korf J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study Eur Neurol. 2003;50:39–47. doi: 10.1159/000070857. [DOI] [PubMed] [Google Scholar]
- Wilke I, Arolt V, Rothermundt M, Weitzsch C, Hornberg M, Kirchner H. Investigations of cytokine production in whole blood cultures of paranoid and residual schizophrenic patients. Eur Arch Psychiatry Clin Neurosci. 1996;246:279–284. doi: 10.1007/BF02190280. [DOI] [PubMed] [Google Scholar]
- Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, Meyer U. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol. 2009;12:513–524. doi: 10.1017/S1461145708009206. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. A double-blind, placebo-controlled trial of celecoxib addes to risperidone in first-episode and drug-naive patients with schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2006;256(Suppl 2):50. [Google Scholar]
- Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]