


Abstract
The OGG1 gene encodes a highly conserved DNA glycosylase that repairs oxidized guanines in DNA. We have investigated the in vivo function of the Ogg1 protein in yeast mitochondria. We demonstrate that inactivation of ogg1 leads to at least a 2-fold increase in production of spontaneous mitochondrial mutants compared with wild-type. Using green fluorescent protein (GFP) we show that a GFP–Ogg1 fusion protein is transported to mitochondria. However, deletion of the first 11 amino acids from the N-terminus abolishes the transport of the GFP–Ogg1 fusion protein into the mitochondria. This analysis indicates that the N-terminus of Ogg1 contains the mitochondrial localization signal. We provide evidence that both yeast and human Ogg1 proteins protect the mitochondrial genome from spontaneous, as well as induced, oxidative damage. Genetic analyses revealed that the combined inactivation of OGG1 and OGG2 [encoding an isoform of the Ogg1 protein, also known as endonuclease three-like glycosylase I (Ntg1)] leads to suppression of spontaneously arising mutations in the mitochondrial genome when compared with the ogg1 single mutant or the wild-type. Together, these studies provide in vivo evidence for the repair of oxidative lesions in the mitochondrial genome by human and yeast Ogg1 proteins. Our study also identifies Ogg2 as a suppressor of oxidative mutagenesis in mitochondria.
INTRODUCTION
Mitochondria are the major sites of energy (ATP) production in the cell. Mitochondria also perform many other cellular functions, such as respiration and heme, lipid, amino acid and nucleotide biosynthesis. Mitochondria also maintain the intracellular homeostasis of inorganic ions and initiate programmed cell death (1–5). Mitochondria contain approximately 1000 proteins; however, only 13 of these proteins are encoded by human mitochondrial DNA (mtDNA). These 13 proteins constitute the essential subunits of the electron transport system (1–5). All other mitochondrial proteins are encoded by the nuclear genome, synthesized in the cytoplasm and transported into mitochondria. Proteins destined for mitochondria contain a mitochondrial localization signal (MLS) (5).
Mitochondria are the major source of endogenous reactive oxygen species (ROS) in cells as they contain the electron transport chain that reduces oxygen to water by addition of electrons during oxidative phosphorylation. Mitochondrial respiration accounts for about 90% of oxygen consumption in the cell (1–5). Each mitochondrion produces 107 ROS molecules/cell/day during normal oxidative phosphorylation (6,7). Human mtDNA, unlike nuclear DNA, contains no introns and has no protective histone proteins. These features of mtDNA make it more vulnerable than nuclear DNA to damage by ROSs produced within the mitochondria. Consistent with this finding, mtDNA appears to accumulate mutations at a frequency 10 times higher than that of nuclear DNA (8). Mutations in mtDNA are involved in the pathogenesis of a variety of diseases including cancer (1,9,10), heart disease (11), cardiomyopathies, diabetes, degenerative diseases such as Parkinson’s, Alzheimer’s and Huntington’s disease and other neurological disorders (1,12). Accumulation of somatic mutations of mtDNA also appears to be a feature of normal aging in all vertebrates thus far examined (1). The genetic changes observed in these diseases range from point mutations to deletions or insertions in the mtDNA (13–15).
ROSs produced in mitochondria can lead to over 100 different types of nucleotide base modifications in DNA (16). Among these, 7,8-hydroxy-2-deoxyguanine (8-hydroxyguanine) is the most abundant form of oxidized base. 8-hydroxyguanine is a mutagen because it mispairs with adenine during DNA replication, thereby causing guanine:cytosine (G:C) to thymine:adenine (T:A) transversions and vice versa (17,18). To prevent the mutagenic effect of 8-hydroxyguanine, the bacterium Escherichia coli contains a GO system. The bacterial GO system consists of three proteins: MutM (also known as Fpg protein), a DNA glycosylase/lyase that recognizes 8-hydroxyguanine:C and catalyzes the excision of 8-hydroxyguanine; MutY, a DNA glycosylase that recognizes 8-hydroxyguanine:A and catalyzes the excision of A; and MutT, a pyrophosphohydrolase that sanitizes the nucleotide precursor pool of 8-oxo-dGTP and inorganic pyrophosphate (17–21).
Yeast and other eukaryotic cells also contain DNA repair proteins that repair modified bases produced by oxidative damage. The yeast OGG1 gene encodes a DNA glycosylase that functionally complements the defect of the fpg E.coli mutant (22,23). Yeast Ogg1 excises 8-hydroxyguanine opposite cytosine but acts poorly on 8-hydroxyguanine opposite adenine or guanine (23). The yeast Ogg1 also contains AP lyase activity. Site-directed mutagenesis of Ogg1 lysine 241 indicates that this residue is essential for catalytic activity of the enzyme (24). The yeast ogg1 null mutant exhibits a mutator phenotype and is not sensitive to oxidative DNA damaging agents (22–24). The human homolog of yeast OGG1 (hOGG1) was cloned by several groups (25–31). The hOGG1 gene maps to chromosome 3p25, a region frequently deleted in many cancers (32). Base substitution mutation in hOGG1 has also been reported (32). Like the yeast protein, hOgg1 also catalyzes the cleavage of 8-hydroxyguanine and complements the mutator phenotype of the fpg strain of E.coli (28). Seven spliced variants of hOgg1 have been reported (33). However, two isoforms, types 1a and 2a, appear to be the major forms found in most human tissue (33). Both hOgg1 isoforms contain glycosylase and AP lyase activity (33). Like the yeast counterpart, these forms repair 8-oxo-G opposite C. The type 1a possesses a nuclear localization signal (NLS) at the C-terminus and localizes to the nucleus (33), while the hOgg1 type 2a localizes to mitochondria (33). Interestingly, hOgg1 is not the only DNA repair protein that localizes to mitochondria. Other proteins that are localized into the mitochondria include Mth1, Nth1, Ape1 as well as Udg1 that removes uracil incorporated into DNA (34–39).
Yeast Saccharomyces cerevisiae is an excellent eukaryotic model system to study DNA repair mechanisms because DNA repair pathways are highly conserved between human and yeast. Furthermore, yeast and human mitochondria resemble each other in structure and function. We have begun a systematic analysis of mtDNA repair in S.cerevisiae. In this paper we provide in vivo evidence that the inactivation of OGG1 in yeast leads to spontaneous mutations in the mitochondrial genome. Our analysis reveals that the Ogg1 N-terminus contains a distinct MLS. We also report that expression of hOGG1 suppresses both the spontaneous and induced mutations in the mitochondrial genome.
MATERIALS AND METHODS
Strains, media and reagents
The genotypes of yeast strains used in this study are presented in Table 1. Yeast strains were grown in YPD medium (1% yeast extract, 2% Bacto-peptone, 2% dextrose and 2% agar for plates). Additionally, yeast colonies were grown in YPG medium (1% yeast extract, 2% Bacto-peptone, 2% glycerol, 2% ethanol and 2% agar for plates) for detection of respiratory incompetent colonies. Synthetic complete (SC) medium (yeast nitrogen base without amino acids 0.67%, 2% dextrose, 0.2% drop-out mix and 2% agar for plates) lacking uracil was used for transformation of yeast with plasmids. SC medium containing 1 g/l of 5-fluoroorotic acid (40) was used to select ura– yeast segregants. All restriction enzymes and DNA modifying enzymes were obtained from Life Technologies. Hydrogen peroxide was purchased from Baker, adriamycin from Adra Chemical. Bacterial strain DH10B was used in all cloning steps. Luria–Bertani (LB) medium (1% yeast extract, 0.5% Bacto-tryptone, 1% NaC1 and 2% agar for plates) was used for the growth of E.coli. Ampicillin was added at 100 µg/ml to LB medium for the growth of plasmid-containing strains.
Table 1. The yeast strains used in this study.
| Strains |
Genotype |
Source |
| Y433 |
MATα ade2-101, lys 2-801, ura 3-52, 112, his3Δ200 |
R.Schiestl |
| YO433 |
Y433 ogg1-Δ::hisG:URA3:hisG |
R.Schiestl |
| YON1433 |
YO433 ntg1-Δ::URA3 |
This study |
| FY250 | MATα ura3-52, his3Δ200,leu2Δ1 trp1Δ63 | G.Verdine |
Plasmid construction
The pYES2–OGG1 plasmid, carrying the yeast OGG1 cDNA, was used (kindly provided by Robert Schiestl and John Davidson, Harvard School of Public Health, Boston, MA). The coding sequence was excised with BamHI and EcoRI, purified by gel electrophoresis and ligated into BamHI and EcoRI sites of the p426ADH vector. This vector is a 6.3 kb yeast vector containing the URA3 yeast-selectable marker (41). This construct was called p426ADH–OGG1. The hOGG1 coding region was purchased from Research Genetics. The coding sequence was excised with EcoRI and StuI, purified by gel electrophoresis and ligated into EcoRI and Xhol sites of the p426ADH vector. This plasmid was called p426ADH–hOGG1. Yeast wild-type strain (Y433) and its ogg1 null derivative (YO433) were transformed with the p426ADH–OGG1, p426ADH–hOGG1 and p426ADH vector by the lithium acetate method (42).
Measurement of mitochondrial mutants
Single yeast colonies were grown to saturation (for 24 h) in 5 ml YPD. Cells were washed once and resuspended in 200 µl of sterile water. Aliquots (100 µl) were plated on N3C (chloramphenicol) and N3E (erythromycin) plates (43). Erythromycin resistant (ER) and chloramphenicol resistant (CAPR) colonies were scored after incubation at 30°C for 5–6 days. Simultaneously, an aliquot of each of the 5 ml cultures was used to determine the number of viable cells using YPD plates.
Petite mutants, characteristically containing deletions in mtDNA, form small colonies and do not grow on media containing non-fermentable carbon sources such as glycerol (YPG medium) (44,45). Interestingly, ade2 yeast strains (and other adenine-requiring strains) produce red pigment and form red colonies. However, cells that lose the ability to respire do not accumulate red pigment and so form white colonies on YPD plates (44,45). We have tested the utility of this phenotype of the white colonies to analyze mitochondrial mutants and we consistently found that white colones were unable to grow on YPG plates. We therefore used the ade2 yeast strain Y433, and its derivative null strains for analysis of mitochondrial mutants. Saturated cultures were diluted, plated on YPD and incubated at 30°C for 3–4 days. The number of red (wild-type) and white (petite) colonies were scored. The wild-type and its ogg1 null derivative strain expressing the yeast or human OGG1 under the alcohol dehydrogenase (ADH1) promoter were grown to saturation in SC medium lacking uracil. They were then diluted and plated on YPD plates. Spontaneous mitochondrial mutants were scored by the color assay as described above.
Cell viability measurements
A single yeast colony was inoculated in 5 ml of YPD medium and allowed to grow overnight at 30°C. This culture was diluted in 10 ml of YPD to an optical density at OD600 of 0.2 and allowed to grow at 30°C to an OD600 of 0.6. Cells constitutively expressing yeast and human OGG1 or vector alone were grown in SC medium lacking uracil at all steps. Cells were pelleted and resuspended in sterile water containing adriamycin as described previously (46). After treatment the cells were diluted and plated on YPD plates. Colonies were counted after incubation for 3–4 days at 30°C.
Intracellular localization of Ogg1p
The construction of the pGFP–C–FUS plasmid has been described previously (47). The plasmid is a 6.3 kb vector containing the URA3 yeast selectable marker and carrying the MET25 promoter. The pGFP–C–FUS expression vector was employed for the generation of Ogg1p–GFP fusion. A 1.1 kb insert containing the entire OGG1 structural gene (1–375 amino acids) was generated by PCR from the pYES2–OGG1 plasmid using the primers 5′-CAGGGATCCATGAAAAGTGAGCTATGT-3′and 5′-CAGAAGCTTATCTATTTTTGCTTCTTTG-3′. To generate yeast Ogg1 lacking the MLS, 11 amino acids from the N-terminal (construct pGFPc–yOgg1ΔN11) were deleted by PCR using the primers 5′-CAGGGATCCATGTCTTATAAATTCGGC-3′ and 5′-CAGAAGCTTATCTATTTTTGCTTCTTTG-3′. The PCR products were digested with BamHI and HindIII and were subcloned into the BamHI and HindIII sites of the pGFP–C–FUS vector. To generate yOgg1 lacking the NLS the yeast OGG1 coding sequence was excised from pYES2–yOGG1 plasmid by digesting with BamHI and DraI, generating a 0.9 kb OGG1 fragment and thus 63 amino acids from the C-terminal (pGFPc–yOgg1ΔC63) protein. The resulting DNA fragment was ligated into BamHI and SmaI sites of the pGFP–C–FUS vector. The resulting plasmids, pGFPc–yOgg1ΔC63 and pGFPc–yOgg1–ΔN11 and pGFPc–yOGG1 were sequenced to verify that the PCR product matched the reported sequence. Yeast strain FY250 was transformed by the lithium acetate method (42) with either the in-frame GFP–Ogg1 fusion construct or the pGFP–C–FUS vector. Cells were grown on SC plates lacking uracil and methionine for 20 h. They were then resuspended in 20 µl of 10 mM MitoTracker dye (Molecular Probes). Following 2 min incubation at room temperature, cells were washed once with 1× PBS, pelleted and resuspended in 5 µl of 1× PBS. A small aliquot (5 µl) of the resulting cell suspension was mounted on glass slides. In order to visualize the DNA present in nuclear and mitochondrial compartments, cells were stained and then suspended in 20 µl of 50 ng/ml 4,6-diamido-2-phenylindole (DAPI; Molecular Probes). Fluorescence was examined using a green fluorescent protein (GFP) optimized filter and a DAPI optimized filter while the MitoTracker signal was examined through a Texa Red/Rhodamine filter. A Zeiss-Axiovert 135 TV inverted microscope equipped with a PXL camera (SENSYS Photometrics) was used to document results.
RESULTS
Increased frequency of petite mutants in the ogg1 strain
Specific point mutations in the rib2 and rib3 mitochondrial genes lead to erythromycin and chloramphenicol resistance. This property of the mitochondrial genome has been routinely employed to measure the frequency of mitochondrial mutants in yeast (48–52). This method showed no significant differences in the frequency of mitochondrial mutants between the wild-type and the ogg1 mutant (data not shown). We then used a colony color method as described in Materials and Methods. The colony color method revealed that inactivation of ogg1 gene causes at least a 2-fold induction of mitochondrial petite mutants (white) compared to the wild-type strain (Fig. 1). We tested the ability of white petite mutants to grow on YPG medium and found that none of the colonies did, suggesting that these colonies harbor a defect in mitochondrial metabolism (data not shown). These data suggest that inactivation of ogg1 leads to increased frequency of mitochondrial mutants.
Figure 1.

Frequency of spontaneous mitochondrial mutants: the wild-type (WT) and isogenic ogg1 null mutant (ogg1) were grown, the cells were washed, diluted in sterile water and plated. The ade2 auxotrophic wild-type strain forms red colonies on YPD (containing glucose) and grow on YPG (containing glycerol). If red colonies lose their mitochondrial function they turn white and appear as petite colonies that do not grow on YPG. The number of independent cultures used in the study is shown in parentheses.
Increased frequency of petite mutants results in ρ– and not ρ0
Mitochondrial petite colonies may arise because of a deletion mutation or a total loss of the mitochondrial genome (44). We therefore isolated mitochondrial DNA from several white petite mutants (chosen randomly) and digested with ApaI and analyzed by gel electrophoresis as described by Querol et al. (53). Figure 2 shows the digestion pattern of mtDNA isolated from wild-type cells and three different white petite mutants in ogg1 null background. It is evident from Figure 2 that petite mutants contained deletions in their mtDNA and none of them lost their entire mtDNA. Random isolates of ogg1 petite mutants also did not display total loss of mtDNA when examined by fluorescence microscopy with a DNA-specific dye, DAPI (data not shown). We conclude that inactivation of ogg1 does not result in the complete loss of mtDNA but leads to deletion of mtDNA.
Figure 2.
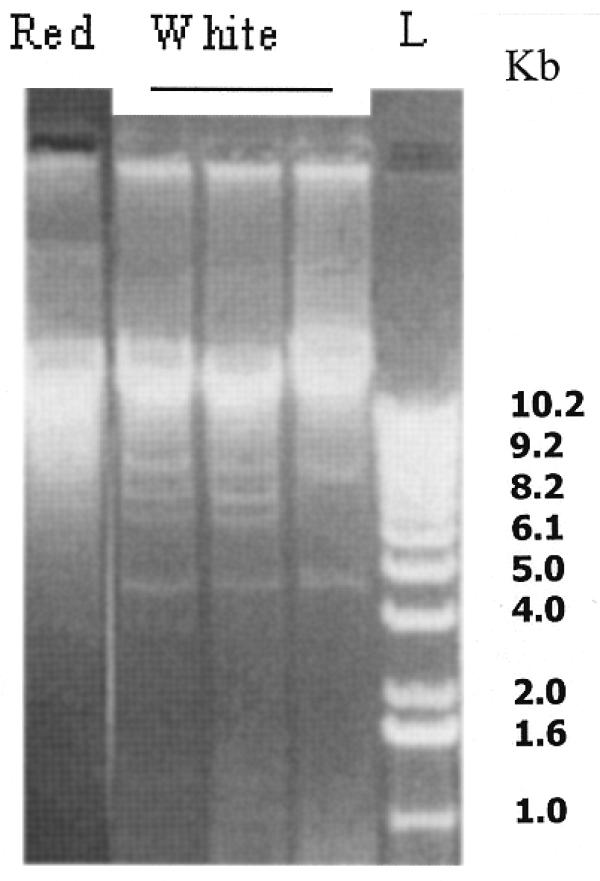
OGG1 inactivation leads to mutations in the mitochondrial genome. Mitochondrial DNA was isolated from red and white colonies and digested with ApaI. The digested mtDNA was run on a 1% agarose gel. L, lambda 1 kb ladder.
Ogg1 localizes to mitochondria
Our studies indicate that inactivation of ogg1 leads to an increased frequency of mitochondrial mutants suggesting that Ogg1 should be present in the mitochondria. To test the probable localization of Ogg1 in mitochondria, we constructed an in-frame Ogg1 fusion with GFP cDNA in a vector named pGFP–C–FUS (47). This vector allows the expression of the GFP fusion protein in yeast from the MET25 promoter (47). The MET25 promoter is turned on when methionine is absent from the medium. The yeast strain Y250 was transformed, the transformant selected and streaked on SD medium without methionine. After 2 days of growth, cells were examined under the microscope. We examined the localization of the three GFP–Ogg1 fusion constructs: GFPc–Ogg1ΔC63, GFPc–Ogg1Δ11N and GFPc–Ogg1 (containing the entire Ogg1 reading frame except the Ogg1 stop codon). Figure 3 shows representative images of yeast expressing the fusion protein. Figure 3A, panel 1 shows a pattern of fluorescence (MitoTracker Red) that coincides with the mitochondria (control). Panel 2 shows that the GFPc–Ogg1ΔC63 fusion protein localizes in the mitochondria. The GFPc–Ogg1ΔC63 image when merged with MitoTracker dye (control) shows a punctate pattern overlapping with mitochondria. When 11 amino acids from the N-terminus were deleted (GFPc–Ogg1ΔN11) mitochondrial localization was abolished. It is evident from the overlapping images stained with DAPI (that stains mitochondrial and nuclear DNA) that GFPc–Ogg1ΔN11 was localized to the nucleus (Fig. 3A and B). Interestingly, GFPc–Ogg1 containing the entire open reading frame was localized to the cytoplasm. We conclude that the Ogg1 protein contains two distinct localization signals: one MLS located at the N-terminus and one NLS located at the C-terminus.
Figure 3.

Subcellular localization of the Ogg1–GFP fusion protein. Yeast cells expressing pGFPc–Ogg1 fusion were grown as described in Materials and Methods. MitoTracker Red (Molecular Probe) was used to locate mitochondria in the cells (see MitoTracker panel). (A) The merged images show the cells expressing: pGFPc–yOGG1ΔC63, which is localized to mitochondria; pGFPc–yOGG1ΔN11, which is not localized to mitochondria and pGFPc–yOGG1, which is localized to the cytoplasm. (B) The merged image shows that cells expressing pGFPc–yOGG1ΔN11 are localized to the nucleus. The localization to the nucleus was monitored by DAPI staining of the DNA.
Ogg1 expression reduces the frequency of petite formation
The in vivo role of hOgg1 in mitochondria is not clear. To analyze the effects of expression of yeast and human Ogg1 in yeast mitochondria, we cloned OGG1 cDNA under the control of the ADH promoter. The wild-type strain Y433 was transformed with Ogg1–ADH and petite colonies were counted. Figure 4A demonstrates that petite formation is reduced in cells expressing Ogg1 compared to cells containing the vector alone. We also cloned hOgg1 under the ADH promoter and transformed the wild-type strain. Like yeast Ogg1, cells expressing hOGG1 produced significantly fewer petite colonies than the vector alone. The hOGG1 and yeast OGG1 were also expressed in the ogg1 null mutant (Fig. 4A). Figure 4A demonstrates that expression of Ogg1 in the null mutant reversed the effect of the ogg1 mutation in this strain.
Figure 4.


OGG1 expression suppresses the frequency of mitochondrial mutants. Yeast and human OGG1 cDNA was cloned under the control of the constitutive ADH promoter as described in Materials and Methods. Both wild-type (WT) and ogg1 null mutant (ogg1) were transformed and the frequency of mitochondrial mutants was determined by the colony color assay (see Materials and Methods). (A) Suppression of spontaneous mitochondrial mutants in wild-type and ogg1 null mutant cells expressing either yeast or human OGG1. (B) Suppression of adriamycin-induced mitochondrial mutants in wild-type cells expressing either yeast or human OGG1 (yOGG1 and hOGG1, respectively). Adriamycin concentration was 50 µg/ml. The number of independent cultures used in the study is shown in parentheses in (A).
Adriamycin is an anthracycline drug that is activated in mitochondria. Upon activation inside the mitochondria it produces ROSs (46). Adriamycin, therefore, is a potent inducer of petite mutants (data not shown; 46). We tested whether adriamycin-induced petite formation is decreased by expression of yeast Ogg1 and hOgg1. Figure 4B shows that, in response to adriamycin, cells that contain only the vector produce approximately 60% petite mutants. However, expression of yeast or hOgg1 in cells reduced the frequency of petite formation significantly. Taken together, these data suggest: (i) that the majority of spontaneous petite mutants in wild-type cells arise due to 8-hydroxyguanine in mtDNA; (ii) that 8-hydroxyguanine in mtDNA is repaired by yeast and human Ogg1 proteins; and (iii) that yeast and hOgg1 proteins repair oxidative lesions in mtDNA induced by adriamycin.
Suppression of petite formation in ogg1 ogg2 double mutant
Saccharomyces cerevisiae contains an Ogg1 homolog known as Ogg2 (17), also known as Ntg1 (36,54). Although Ogg2 localizes to mitochondria, its biological function is unknown (36,54). In order to characterize the role of Ogg2 in repairing damaged DNA in mitochondria, we constructed an ogg1 ogg2 double mutant strain. We measured the frequency of petite formation in this strain using the color assay. Figure 5 shows that inactivation of both ogg1 and ogg2 results in a reduced level of mitochondrial petite colonies in comparison to the ogg1 strain. These data suggest that ogg2 functions as a suppressor of oxidative DNA repair in mitochondria.
Figure 5.

Suppression of the mitochondrial mutation in the ogg1 ogg2 double mutant. The ogg1 ogg2 double knock-out strain was generated as described in Materials and Methods. Percentage of mitochondrial mutants was determined by the colony color assay. The number of independent cultures used in the study is shown in parentheses.
DISCUSSION
The mitochondrial genome of eukaryotic cells is extremely susceptible to damage due to constant exposure to significant amounts of ROSs produced endogenously by mitochondria as a by-product of oxidative phosphorylation. Organisms contain defence mechanisms to minimize the accumulation of ROS-induced damage. Recent biochemical and cell biological studies indicate the existence of base excision repair proteins in human mitochondria (1,33–35,37). Saccharomyces cerevisiae contains an Ogg1 homolog, named Ogg2 (17). Ogg2 repairs oxidative lesions and is localized to the mitochondria (36,54). However, You et al. (54) found no evidence for in vivo repair in mitochondria by Ogg2. Erythromycin and chloramphenicol-resistant colonies arise due to point mutations in mtDNA encoded rib2 or rib3 genes (49,50). This method has been frequently used by many investigators including You et al. to measure the frequency of mitochondrial mutants (48–52,54). We also used this method to analyze the effect of ogg1 inactivation on the frequency of mitochondrial mutants. Our analysis revealed no significant difference in frequency of mitochondrial mutants between the wild-type and the ogg1 mutant (data not shown). However, when we used the colony color method it revealed at least a 2-fold difference in the generation of mitochondrial mutants between the ogg1 and the wild-type strain. It is likely that the observed lack of difference between the mutant and the wild-type is due to a limited target size provided by the rib2 and rib3 genes and the required specific point mutations that render cells resistant. In comparison, colony color measures random mutation in the entire mitochondrial genome that provides a significant target size (85 kb) for mutagenesis. Thus the colony color method is more sensitive at detecting mitochondrial mutations and should be helpful in identifying other genes involved in repairing the mitochondrial genome.
The observed insignificant differences between the wild-type and ogg1 strain on the accumulation of antibiotic-resistant colonies suggest that a lack of Ogg1 does not result in increased point mutation in mtDNA. We found that ogg1 inactivation leads to deletion in mtDNA. Ogg1 repairs several lesions in addition to 8-hydroxyguanine. These include 2,6-diamino-4-hydroxy-5(N-methylforamamido) pyrimidine, 2,6-diamino-4-hydroxy-5-formamidopyridine (Fapy) as well as 7,8-hydroxy-8-oxoadenine placed opposite a cytosine or a 5-methyl cytosine (24). In the absence of Ogg1, it is likely that the chain terminating lesions such as Fapy are not repaired, which will lead to deletions in mtDNA. We do not yet know the complete spectrum of oxidative DNA lesions generated in the mitochondria but it is likely that the lesions similar to the ones generated in the nucleus are also generated in the mitochondria and are repaired by Ogg1 in both organelles. However, more experiments are required to address this possibility in vivo in the mitochondria.
The yeast Ogg1 contains a bipartite NLS located at the C-terminus between residues 341 and 362 (KKRK-X12-KQMKL) (55). Deletion of 63 amino acids from the C-terminus led to the exclusive localization of Ogg1 to mitochondria (Fig. 3). The N-terminus of Ogg1 contains features typical of an MLS. When 11 amino acids from the N-terminus were deleted, Ogg1 localization in the mitochondria was abolished. Instead, Ogg1 was localized in the nucleus because it contained the NLS at the C-terminus. These data suggest that Ogg1 contains two distinct localization signals, an MLS and an NLS, which are located at the N- and C-termini, respectively (Fig. 3). It has been suggested previously that in proteins with multiple organelle localization signals, the N-terminal signal usually dominates (33,35). Interestingly, this is not the case for yeast Ogg1. When yeast Ogg1 contained both the MLS and NLS, it was present throughout the cytoplasm (Fig. 3) indicating that neither signal dominates. However, the MLS in hOgg1 appears to be weak because hOgg1 containing both the MLS and NLS localizes to the nucleus (33).
We report that the constitutive expression of yeast and human Ogg1 protects yeast cells from spontaneous, as well as induced, oxidative damage to the mitochondrial genome. This study indicates that Ogg1 expression may be a rate-limiting step in repairing oxidative lesions in mitochondria. The reduced frequency of petite formation by yeast expressing hOgg1 suggests that human Ogg1 should be transported to mitochondria to repair any damage to the mitochondrial genome. Indeed, our data suggest that ectopically expressed hOgg1 accumulates in yeast mitochondria (data not shown). This observation is consistent with previous reports that MLSs from other species function in yeast cells (5,56).
The Ogg2 protein removes 8-hydroxyguanine paired with guanine (17). In addition, Ogg2 repairs thymine glycol, dihydrothymidine, dihydroxyuracil, 5-hydroxy-6-hydrothymine, 5-hydroxy-6-hydrouracil, 5-hydroxy-5-methyldantoin, 5-hydroxyuracil, 5-hydroxycytosine, Fapy–7MeG, Fapy G, Fapy A and abasic sites (54). We examined the frequency of petite formation in ogg1 single and ogg1 ogg2 double null mutants. Surprisingly, we found that inactivation of ogg2 together with ogg1 reduces the frequency of petite formation to the wild-type level. Interestingly, the ogg2 single mutant does not show a significant increase in frequency of spontaneous mitochondrial petite formation. It is important to note that the combined inactivation of ogg1 and ogg2 function reduces mutation in the nuclear genome (17). One possibility is that in the absence of both the Ogg1 and Ogg2 proteins, one or more alternative pathways are activated that repair oxidative DNA lesions in mitochondria. We propose a model in which Ogg2 may function as (i) a bypass suppressor or (ii) an epistasis suppressor (Fig. 6). In the first case, Ogg1 and Ogg2 function in a parallel pathway and the absence of one protein may increase the level of the other. As an example, a mutation that abolishes the function of CYC1 gene (eliminating the major form of cytochrome c) is suppressed by a mutation that increases the level of another isoform encoded by CYC7 gene (57). It is also possible that a mutation in Ogg2 may alter the specificity of a protein in a related but distinct pathway, so that it subsititutes for the function of Ogg1. In the second case, Ogg1 and Ogg2 may be in the same pathway and a null mutation in OGG2 gene restores the wild-type phenotype because it is epistatic to Ogg1. Suppression by epistasis is observed in many pathways in gene regulation, such as those controlling amino acid biosynthesis in S.cerevisiae (57). Experiments are underway to test this model.
Figure 6.

A proposed model describing the function of Ogg2 as a suppressor of mutation in the mitochondrial genome. See text for details.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Greg Verdine (Harvard University, Boston, MA), Robert Schiestl and John Davidson for ogg1 and other knock-out strains, and Dr Colin Garvie for a primer used in this study. We thank Dr Sue Jinks-Robertson for NTG1 knock-out plasmid. This work was supported by a grant from NIH RO1 09714-01 and the American Heart Association 9930223N. We thank Aditi Chaterjee and Anna Rasmussen for help with figures, Debkumar Pain for help with the model and Debkumar Pain and Kylie Keshav for critical reading of this manuscript.
References
- 1.Singh K.K. (1998) Mitochondrial DNA Mutations in Aging, Disease, and Cancer. Springer, New York, NY.
- 2.Schatz G. (1995) Mitochondria: beyond oxidative phosphorylation. Biochim. Biphys. Acta, 1271, 123–126. [DOI] [PubMed] [Google Scholar]
- 3.Tzagoloff A. (1982) Mitochondria. Plenum Press, New York, NY.
- 4.Scheffler I. (1999) Mitochondria. Wiley-Liss, New York, NY.
- 5.Mihara K. (2000) Targeting and insertion of nuclear-encoded preproteins into the mitochondrial outer membrane. Bioessays, 22, 364–371. [DOI] [PubMed] [Google Scholar]
- 6.Richter C. (1988) Do mitochondrial DNA fragments promote cancer and aging? FEBS Lett., 241, 467–473. [DOI] [PubMed] [Google Scholar]
- 7.Shigenaga M.K., Hagen,T.M. and Ames,B.N. (1994) Oxidative damage and mitochondrial decay in aging. Proc. Natl Acad. Sci. USA, 91, 10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grossman L.I. and Shoubridge,E.A. (1996) Mitochondrial genetics and human disease. Bioessays, 18, 983–991. [DOI] [PubMed] [Google Scholar]
- 9.Polyak K., Li,Y., Zhu,H., Lengauer,C., Willson,J.K., Markowitz,S.D., Trush,M.A., Kinzler,K.W. and Vogelstein,B. (1998) Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature Genet., 20, 291–293. [DOI] [PubMed] [Google Scholar]
- 10.Fliss M.S., Usadel,H., Caballero,O.L., Wu,L., Buta,M.R., Eleff,S.M., Jen,J. and Sidransky,D. (2000) Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science, 287, 2017–2019. [DOI] [PubMed] [Google Scholar]
- 11.Shoffner J.M. and Wallace,D.C. (1992) Heart disease and mitochondrial DNA mutations. Heart Disease Stroke, 1, 235–241. [PubMed] [Google Scholar]
- 12.Luft R. (1994) The development of mitochondrial medicine. Proc. Natl Acad. Sci. USA, 91, 8731–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang D., Takeshig,K., Sekiguchi,M. and Singh,K.K. (1998) In Singh,K.K. (ed.), Mitochondrial DNA Mutations in Aging, Disease, and Cancer. Springer, New York, NY, pp. 1–15.
- 14.Rassmussen L. and Singh,K.K. (1998) Genetic integrity of mitochondrial genome. In Singh,K.K. (ed.), Mitochondrial DNA Mutations in Aging, Disease, and Cancer. Springer, New York. NY, pp. 115–122.
- 15.Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 16.Jaruga P. and Dizdaroglu,M. (1996) Repair of products of oxidative DNA base damage in human cells. Nucleic Acids Res., 24, 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruner S.D., Nash,H.W., Lane,W.S. and Verdine,G.L. (1998) Repair of oxidatively damaged guanine in Saccharomyces cerevisiae by an alternative pathway. Curr. Biol., 8, 393–403. [DOI] [PubMed] [Google Scholar]
- 18.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 19.Michaels M.L. and Miller,J.H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 21.Kohno T., Shinmura,K., Tosaka,M., Tani,M., Kim,S.R., Sugimara,H., Nohmi,T., Kasai,H. and Yokota,J. (1998) Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene, 16, 3219–3225. [DOI] [PubMed] [Google Scholar]
- 22.Thomas D., Scot,A.D., Barbey,R., Padula,M. and Boiteux,S. (1997) Inactivation of OGG1 increases the incidence of G.C→T.A. transversions in Saccharomyces cerevisiae: evidence for endogenous oxidative damage to DNA in eukaryotic cells. Mol. Gen. Genet., 254, 171–178. [DOI] [PubMed] [Google Scholar]
- 23.Nash H.M., Bruner,S.D., Scharer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996). Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 24.Guibourt, N., Castaing,B., Van Der Kemp,A.P. and Boiteux,S. (2000) Catalytic and DNA binding properties of the Ogg1 protein of Saccharomyces cerevisiae: comparison between the wild type and the K241R and K241Q active-site mutant proteins. Biochemistry, 39, 1716–1724. [DOI] [PubMed] [Google Scholar]
- 25.Radicella J.P., Dhérin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roldan-Arjona T., Wei,Y.F., Carter,K.C., Klungland,A., Anselmino,C., Wang,R.P., Augustus,M. and Lindahl,T. (1997) Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aburatani H., Hippo,Y., Ishida,T., Takashima,R., Matsuba,C., Kodama,T., Takao,M., Yasui,A., Yamamoto,K. and Asano,M. (1997) Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res., 57, 2151–2156. [PubMed] [Google Scholar]
- 29.Arai K., Morishita,K., Shinmura,K., Kohno,T., Kim,S.R., Nohmi,T., Taniwaki,M., Ohwada,S. and Yopkota,J. (1997) Cloning of human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 30.Lu R., Nash,H.M. and Verdine,G.L. (1997) A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol., 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 31.Bjoras M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeburg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevillard S., Radicella,J.P., Levalois,C., Lebeau,J., Poupon,M.F., Oudard,S., Dutrillaux,B. and Boiteux,S. (1998) Mutations in OGG1, a gene involved in the repair of oxidative DNA damage, are found in human lung and kidney tumours. Oncogene, 16, 3083–3086. [DOI] [PubMed] [Google Scholar]
- 33.Nishioka K., Ohtsubo,T., Oda,H., Fujiwara,T., Kang,D., Sugimachi,K. and Nakabeppu,Y. (1999) Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell, 10, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinz K.G. and Bogenhagen,D.F. (1998) Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol. Cell. Biol., 18, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slupphaug G., Markussen,F.H., Olsen,L.C., Aasland,R., Aarsaether,N., Bakke,O., Krokan,H.E. and Helland,D.E. (1993) Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res., 11, 2579–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alseth I., Eide,L., Pirovano,M., Rognes,T., Seeberg,E. and Bjoras,M. (1999) The Saccharomyces cerevisiae homologues of endonuclease III from Escherichia coli, Ntg1 and Ntg2, are both required for efficient repair of spontaneous and induced oxidative DNA damage in yeast. Mol. Cell. Biol., 19, 3779–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fung H., Kow,Y.W., Van Houten,B., Taatjes,D.J., Hatahet,Z., Janssen,Y.M., Vacek,P., Faux,S.P. and Mossman,B.T. (1998) Asbestos increases mammalian AP-endonuclease gene expression, protein levels, and enzyme activity in mesothelial cells. Cancer Res., 58, 189–194. [PubMed] [Google Scholar]
- 39.Kang D., Nishida,J., Iyama,A., Nakabeppu,Y., Furuichi,M., Fujiwara,T., Sekiguchi,M. and Takeshige,K. (1995) Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem., 270, 14659–14665. [DOI] [PubMed] [Google Scholar]
- 40.Boeke J.D., Trueheart,J., Natsoulis,G. and Fink,G.R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol., 154, 164–175. [DOI] [PubMed] [Google Scholar]
- 41.Mumberg D., Muller,R., and Funk,M. (1995) Yeast vectors for controlled expression of heterologous proteins in different genetic back ground. Gene, 156, 119–122. [DOI] [PubMed] [Google Scholar]
- 42.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 43.Rickwood D. and Hames,B.D. (1988) In Campbell,I. And Duffus,J.H. (eds), Yeast: A Practical Approach. IRL Press, Oxford, pp. 185–247.
- 44.Dujon B. (1981) Mitochondrial genetics and functions. In Strathern,J.N., Jones,E.W. and Broach,J.R. (eds), The Molecular Biology of the Yeast Saccharomyces. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 505–635.
- 45.Reaume S.E. and Tatum,E.L. (1949) Spontaneous and nitrogen mustard-induced nutritional deficiencies in Saccharomyces cerevisiae. Arch. Biochem., 22, 331–338. [PubMed] [Google Scholar]
- 46.Kule C., Ondrejickova,O. and Verner,K. (1994) Doxorubicin, daunorubicin, and mitoxantrone cytotoxicity in yeast. Mol. Pharmacol., 46, 1234–1240. [PubMed] [Google Scholar]
- 47.Niedenthal R.K., Riles,L., Johnston,M. and Hegemann,J.H. (1996) Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast, 12, 773–786. [DOI] [PubMed] [Google Scholar]
- 48.Foury F. (1989) Cloning and sequencing of the nuclear gene MIP1 encoding the catalytic subunit of the yeast mitochondrial DNA polymerase. J. Biol. Chem., 264, 20552–20560. [PubMed] [Google Scholar]
- 49.Cui Z. and Mason,T. (1989) A single nucleotide substitution at the rib2 locus of the yeast mitochondrial gene for 21S rRNA confers resistance to erythromycin and cold sensitive ribosome assembly. Curr. Genet., 16, 273–279. [DOI] [PubMed] [Google Scholar]
- 50.Foury F. and Vanderstraeten,S. (1992) Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J., 11, 2717–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sor F. and Fukuhara,H. (1983) Complete DNA sequence coding for the large ribosomal RNA of yeast mitochondria. Nucleic Acids Res., 11, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chi N.-W. and Kolodner,R.D. (1994) Purification and characterization of MSH1, a yeast mitochondrial protein that binds to DNA mismatches. J. Biol. Chem., 269, 29984–29992. [PubMed] [Google Scholar]
- 53.Querol A., Bazzio,E., Huerta,T. and Ramon,D. (1992) Molecular monitoring of wine fermentations conducted by yeast strains. Appl. Environ. Microbiol., 58, 2948–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.You H.J., Swanson,R.L., Harrington,C., Corbett,A.H., Jinks-Robertson,S., Senturker,S., Wallace,S.S., Boiteux,S., Dizdaroglu,M. and Doetsch,P.W. (1999) Saccharomyces cerevisiae Ntg1p and Ntg2p: broad specificity N-glycosylases for the repair of oxidative DNA damage in the nucleus and mitochondria. Biochemistry, 38, 11298–11306. [DOI] [PubMed] [Google Scholar]
- 55.van der Kemp P., Thomas,D., Barbey,Y., DeOliveira,R. and Boiteux,S. (1996) Cloning and expression in E. coli of the OGG1 gene of S. cerevisiae which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methyl-formamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowler C., Alliotte,T., Van den Bulcke,M., Bauw,G., Vandekerckhove,J., Van Montagu,M. and Inze,D. (1989) A plant manganese superoxide dismutase is efficiently imported and correctly processed by yeast mitochondria. Proc. Natl Acad. Sci. USA, 86, 3237–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guarente L. (1993) Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet., 9, 362–366. [DOI] [PubMed] [Google Scholar]