

Abstract
We have investigated how the Arf gene product, p19Arf, is activated by Tgfβ during mouse embryo development to better understand how this important tumor suppressor is controlled. Taking advantage of new mouse models, we provide genetic evidence that Arf lies downstream of Tgfβ signaling in cells arising from the Wnt1-expressing neural crest and that the anti-proliferative effects of Tgfβ depend on Arf in vivo. Tgfβ1, -2, and -3 (but not BMP-2, another member of the Tgfβ superfamily) induce p19Arf expression in wild type mouse embryo fibroblasts (MEFs), and they enhance Arf promoter activity in ArflacZ/lacZ MEFs. Application of chemical inhibitors of Smad-dependent and -independent pathways show that SB431542, a Tgfβ type I receptor (TβrI) inhibitor, and SB203580, a p38 MAPK inhibitor, impede Tgfβ2 induction of Arf. Genetic studies confirm the findings; transient knockdown of Smad2, Smad3, or p38 MAPK blunt Tgfβ2 effects, as does Cre recombinase treatment of Tgfbr2fl/fl MEFs to delete Tgfβ receptor II. Chromatin immunoprecipitation reveals that Tgfβ rapidly induces Smads 2/3 binding and histone H3 acetylation at genomic DNA proximal to Arf exon 1β. This is followed by increased RNA polymerase II binding and progressively increased Arf primary and mature transcripts from 24 through 72 h, indicating that increased transcription contributes to p19Arf increase. Last, Arf induction by oncogenic Ras depends on p38 MAPK but is independent of TβrI activation of Smad 2. These findings add to our understanding of how developmental and tumorigenic signals control Arf expression in vivo and in cultured MEFs.
Keywords: p38 MAPK, SMAD Transcription Factor, Transcription Coactivators, Transforming Growth Factor β (TGFβ), Tumor Suppressor, Arf, Eye Development
Introduction
Arf is conserved in amniotes as a gene encoding p19Arf (p14ARF in humans), a tumor suppressor that exerts its effects by both p53-dependent and -independent mechanisms (1). Early studies, predominantly in cultured cells, showed that Arf is induced to check proliferation in sequentially passed MEFs3 in vitro (2, 3). Moreover, Arf expression is augmented in the presence of certain oncoproteins, like adenovirus E1A, Myc, E2F, Bcr-Abl, and RasV12 (3–7). These findings coupled with the initial failure to identify Arf expression in the developing mouse embryo (8) led to the concept that Arf acts as an oncogene sensor that is induced by cell autonomous mechanisms in response to inappropriate or excessive cell proliferation signals (9).
More recent observations point toward Arf regulators extending beyond oncogenic signals. For instance, mouse Arf expression increases with age in a variety of cells that have not suffered overt oncogenic stress (10). Closer evaluation of the developing mouse embryo shows Arf to be robustly expressed in a temporally and spatially restricted pattern in the developing hyaloid vessels and cornea in the eye and also in perivascular cells flanking the intra-embryonic umbilical vessels (11, 12). A developmental function of p19Arf is only clear in the pericyte-like cells in the hyaloid vessels of the primary vitreous, where it prevents overgrowth of the pericytes to prompt the developmentally timed regression of the hyaloid vessels (12). Homozygous Arf deletion results in primary vitreous hyperplasia, ocular lens opacification, retinal dysplasia, and blindness by 2 weeks of age (11, 12).
The expanded role of p19Arf in the embryo raises the question of how developmental signals operate and whether they overlap with oncogenic signals controlling Arf. Using a candidate-gene approach, transforming growth factor β2 (Tgfβ2), a member of the Tgfβ cytokine superfamily, was recently found to be critical for Arf expression at several sites in the developing mouse (13). Supporting the importance of this finding, mouse embryos lacking Tgfβ2 have primary vitreous hyperplasia similar to that observed in Arf−/− embryos (14, 15). Importantly, these observations can be replicated in vitro because exogenous Tgfβ2 enhances Arf expression in cultured MEFs and maintains a proliferation arrest in an Arf-dependent manner (13), thereby providing a model system to further investigate mechanisms.
Members of the Tgfβ superfamily frequently modulate the transcription of key target genes through Smad proteins, which directly transduce Tgfβ receptor activation to the nucleus. In addition, Smad-independent signaling through p38 MAPK, ERK, PI3K/Akt, and JNK provide alternative mechanisms of gene activation (16). In this manuscript we demonstrate that Tgfβ and p19Arf act on cells of the same lineage; that Arf is required for the anti-mitogenic effects of Tgfβ in vivo and that both Smad- and p38 MAPK-dependent mechanisms underlie Arf induction by Tgfβ.
EXPERIMENTAL PROCEDURES
Animals, Cell Lines, and Reagents
Arffl/+ (17), ArfGfp/+ (18), and ArflacZ/+ (13) mice were maintained in a mixed C57BL/6 × 129/Sv genetic background. Tgfβ2+/− mice (14) were purchased from The Jackson Laboratory (Bar Harbor, ME). Tgfbr2fl/fl mice (19) were obtained from A. Chytil and H. L. Moses (Vanderbilt University). AC-Tgfβ1 transgenic mice were previously described (20). Wnt1-Cre transgenic mice were purchased from The Jackson Laboratory (stock #003829). Primary MEFs from wild type (WT), ArflacZ/lacZ, and Tgfbr2fl/fl mice were obtained and cultivated as previously described (8). Animal studies were approved by the University of Chicago Animal Care and Use Committee.
Tgfβ1, -2, and -3 and BMP-2 were purchased from R&D Systems, Inc. (Minneapolis, MN). The siRNA targeting Smads 2 and 3, p38 MAPK, and control reagents were from Dharmacon, Inc. (Lafayette, CO). Anisomycin, SB203580, SP600125, U0126, and LY294002 (EMD Chemicals Inc; Gibbstown, NJ) and SB431542 (Tocris Bioscience; Ellisville, MO) were used in some experiments involving MEFs. β-Galactosidase in cultured cells was measured using a commercially available kit (Applied Biosystems; Foster City, CA). Adenovirus vectors encoding red fluorescent protein and Cre recombinase were produced in our laboratory using vectors provided by T. C. He (University of Chicago). Murine stem cell virus-based retrovirus vectors encoding human H-RASV12 were produced in our laboratory using vectors from Addgene (Cambridge, MA). Antibodies used in Western blotting experiments were directed against the following: Smad2/3, phospho-Smad2, phospho-p38 MAPK, phospho-Akt, phospho-p44/42 MAPK, and phospho-JNK (Cell Signaling Technology; Danvers, MA); TβrII and Hsc70 (Santa Cruz Biotechnology, Inc.; Santa Cruz, CA); p19Arf (Abcam Inc; Cambridge, MA).
Histology Studies
Female mice pregnant with embryonic day (E) 13.5 litters received BrdU (10 mg/g in PBS) by intraperitoneal injection 4 and 2 h before euthanasia using CO2. Whole embryos were fixed in 4% paraformaldehyde in PBS for 4 h at 4 °C and then equilibrated in 20% sucrose overnight at 4 °C. Fixed embryo heads were embedded in TBS Tissue Freezing Media (Fisher) before cryostat sectioning. Hematoxylin- and eosin-staining was performed using 5-μm sections as previously described (2, 3). For BrdU staining, 5-μm sections were blocked in 10% donkey serum, 0.1% Triton X-100, PBS at room temperature and then stained using sheep α-BrdU polyclonal antibody (1:100, Fitzgerald Industries International; Action, MA) at room temperature for 90 min. The primary antibody was detected with a Dylight 488-conjugated donkey anti-sheep secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Sections were mounted in VectaShield mounting media with DAPI (Vector Laboratories, Inc; Burlingame, CA) and visualized using a Leica DM IRB fluorescent microscope at 400× magnification. The fraction of DAPI-positive cells in the vitreous that were BrdU-positive was determined using at least three embryos from two or more different litters. Quantification was verified by two individuals who were blinded to the genotypes. Photomicrographs were obtained using an Optronics camera and MagnaFire 2.1C imaging software (Optronics, Goleta, CA).
Cell Culture, Western Blot Analysis, and β-Galactosidase Assay
Early passage wild type and ArflacZ/lacZ MEFs were treated with either Tgfβ1 (5 ng/ml), Tgfβ2 (5 ng/ml), Tgfβ3 (10 ng/ml), or BMP2 (150 ng/ml) or an equivalent volume of vehicle (4 mm HCl) for 1.5–72 h. Arf-null 10T1/2 cells were transduced with Gfp- or Arf-expressing retrovirus as negative and positive controls for Western blotting. In some experiments SB203580 (20 μm), SB431542 (10 μm), SP600125 (10 μm), U0126 (10 μm), and LY 294002 (5 μm) were applied to MEFs for 20 min before Tgfβ2 or vehicle was added to the culture media. In some studies, wild type MEFs or ArflacZ/lacZ MEFs were seeded at 50% confluence in 6- or 12-well plates 24 h before transfection using of siRNA targeting specific genes (or scrambled siRNA as a control) (0.5 μm) using DharmaFECT 2 (Dharmacon) according to the manufacturer's instructions. In some studies, Tgfbr2fl/fl MEFs were infected with adenovirus encoding Cre recombinase or red fluorescent protein as a control for 48 h before exposure to vehicle or Tgfβ2 for Western blotting for p19Arf. In some experiments, early passage wild type MEFs were infected with retrovirus encoding H-RASV12 and treated with SB203580 (20 μm) and SB431542 (10 μm) concurrently. For all studies Western blotting and β-galactosidase assays were performed in wild type and ArflacZ/lacZ MEFs, respectively, as previously described (13). Experimental findings were confirmed in at least two independent experiments, with quantitative data from β-galactosidase assays pooled from all representative experiments.
As a control for SB203580 activity, the p38 MAPK assay (Cell Signaling Technology) was used according to the manufacturer's instructions. Briefly, phosphorylated p38 MAPK was immunoprecipitated from the corresponding cell lysate, and its activity was determined by in vitro kinase assay measuring phosphorylation of its substrate ATF-2.
Chromatin Immunoprecipitation (ChIP)
For ChIP experiments, wild type MEFs (3 × 106/ChIP) were treated with Tgfβ2 (5 ng/ml) or vehicle for 1.5 h or 24 h. Cells were cross-linked and sonicated as previously described (13) and then subjected to immunoprecipitation using anti-Smad2/3 antibody (sc6033, Santa Cruz Biotechnology), anti-acetylated histone H3 (06-599, Millipore, Billerica, MA), or anti-RNA polymerase II (sc899, Santa Cruz). Goat IgG (AB-108-C, R&D Systems) and rabbit IgG (sc2027, Santa Cruz) were used as negative controls. Protein A/G-Sepharose beads (sc2003, Santa Cruz) were used to collect the protein-chromatin complexes. The beads were washed sequentially with low salt, high salt, LiCl, and TE buffers (Upstate ChIP kit, Millipore) and eluted in 0.1 m NaHCO3, 1% SDS. Cross-linking was reversed by incubation at 67 °C overnight, and the genomic DNA was extracted using Qiagen PCR purification kit. A total of 7% of the precipitated DNA and 1% input DNA was amplified by PCR using primer sets for different regions of Arf (distal, 5′-TTCCAGGCCTTGCCATCTTCCTAT-3′ (forward) and 5′-TGGTCTGGCTGCAGTAAAGTAGCA-3′ (reverse); proximal, 5′-AGATGGGCGTGGAGCAAAGAT-3′ (forward) and 5′-ACTGTGACAAGCGAGGTGAGAA-3′ (reverse); Nedcin (5′-GGTCCTGCTCTGATCCGAAG-3′ (forward) and 5′-GGGTCGCTCAGGTCCTTACTT-3′ (reverse)). Immunoprecipitated DNA was amplified and quantified using Fast SYBR Green Master mix and the StepOnePlus real-time PCR system (both from Applied Biosystems). Results are pooled from two or three individual experiments.
Quantitative RT-PCR for Arf Expression
Total RNA extraction and cDNA preparation were accomplished using RNeasy (Qiagen) and Superscript III RT (Invitrogen) according to the manufacturer's recommendations. Quantitative RT-PCR was performed using Fast SYBR Green Master mix and the StepOnePlus real-time PCR system (both from Applied Biosystems). The following gene-specific primers were used: mature Arf transcript, 5′-TTCTTGGTGAAGTTCGTGCGATCC-3′ (forward) and 5′-CGTGAACGTTGCCCATCATCATCA-3′ (reverse); primary Arf transcript, 5′-TGGCCATAGAGGTGAACCCTTCTT-3′ (forward) and 5′-ATCCTGCACCGAGAAAGCACTGAA-3′ (reverse); Gapdh mature transcript, 5′-TCAACAGCAACTCCCACTCTTCCA-3′ (forward) and 5′-ACCCTGTTGCTGTAGCCGTATTCA-3′ (reverse). Results are pooled from three separate experiments.
Statistical Analysis
Quantitative data are presented as the mean ± S.D. from three or more representative experiments. Statistical significance (p value <0.05) was calculated using Student's t test.
RESULTS
Arf Is Required for Anti-proliferative Effects of Tgfβ in Vivo
Arf expression is blunted in the developing eyes of Tgfβ2−/− embryos (13), and both Arf−/− and Tgfβ2−/− embryos have primary vitreous hyperplasia evident at embryonic day (E) 13.5 (13, 15, 21). These observations imply that Tgfβ2 lies “upstream” of Arf. Furthermore, they are consistent with the idea that p19Arf is required for the anti-proliferative effects of Tgfβ2 in the mouse eye. To formally test this hypothesis, we took advantage of a transgenic mouse line in which Tgfβ1 is expressed from the αA-crystallin promoter (here called AC-Tgfβ1 mice) (20). Indeed, previous studies demonstrated that transgenic expression of Tgfβ1 can rescue the Tgfβ2-dependent primary vitreous hyperplasia in E18.5 embryos, although the mechanism for the rescue has not been elucidated (22). This finding coupled with our prior observation that Tgfβ1 can activate the Arf promoter in cultured MEFs in a manner comparable with Tgfβ2 (13) allowed us to explore the epistatic relationship between Tgfβ2 and Arf in vivo.
Histological examination of eyes from E13.5 embryos revealed vitreous hyperplasia restricted to the ArfGfp/Gfp and Tgfβ2−/− embryos as compared with their heterozygous littermates (Fig. 1, A and B). Hyperplasia was associated with increased BrdU incorporation in cells in the primary vitreous (Fig. 1, C and D). Ectopic Tgfβ1 blunted the excess proliferation in AC-Tgfβ1+, Tgfβ2−/− embryos as compared with Tgfβ2−/− embryos (Fig. 1D, top panel). In contrast, the Arf−/− defect and deregulated cell proliferation was unchanged in E13.5 AC-Tgfβ1+, Arf−/− embryos at E13.5 (Fig. 1, B and D, bottom panel). Hence, the capacity for Tgfβ1 to arrest proliferation in vivo depends on Arf and illustrates the fact that Tgfβ2 lies upstream of Arf.
FIGURE 1.

Arf is required for the anti-proliferative effects of Tgfβ1 during eye development. A and B, representative photomicrographs of hematoxylin- and eosin-stained slides of E13.5 embryos showing the primary vitreous hyperplasia in Tgfβ2−/− and Arf−/− embryos (middle panels) is corrected by ectopic Tgfβ1 in AC-Tgfβ1 animals only when Arf is present (right panels). Arrows denote the cellular area of the primary vitreous. C and D, shown are a representative photomicrograph (C) and quantification (D) of BrdU incorporation in the vitreous of E13.5 mouse embryo eyes of the indicated genotypes. Original magnification: 200× (A and B); 400× (C). Quantitative data are expressed as average percent of total cells in the vitreous space. Increased proliferation in absence of Arf or Tgfβ2 is statistically significant (lane 2 versus 1; *). p < 0.002 (top) and <0.01 (bottom). Decreased proliferation by ectopic expression of Tgfβ1 was not significant in the absence of Arf (lane 3 versus 2). p < 0.001 (top, #) and p = 0.35 (bottom, @).
Arf-expressing Cells in the Eye Originate from Wnt1-expressing Neural Crest
The fact that some Arf-expressing cells co-express TβrII in the eye (13) indicates that Tgfβ2 might signal directly to cells expressing p19Arf. To further address this, we took advantage of the fact that members of the Sommer laboratory (23) previously showed that blocking Tgfβ signals in cells derived from a Wnt1-expressing lineage leads to primary vitreous hyperplasia. This was accomplished by breeding Wnt1-Cre mice into the Tgfβr2fl/fl mouse strain in which exon 4 Tgfbr2 can be conditionally deleted by Cre-mediated recombination (19). We investigated whether primary vitreous hyperplasia would similarly occur in animals in which Arf is inactivated in the same lineage.
We accomplished this by breeding Wnt1-Cre mice with animals in which with exon 1β of Arf is flanked by LoxP sites (17). These Arffl/fl animals were previously used with ArfCre/Cre and Pdgfrβfl/fl animals by Gromley et al. (17) to formally show that conditional deletion of Pdgfrβ anatomically and functionally rescues the postnatal Arf−/− eye phenotype. As a control, we confirmed that Cre promoted recombination in cells populating the primary vitreous by analyzing Wnt1-Cre, Rosa26lacZ/+ mouse embryos.4
Histological examination of eyes from postnatal day (P) 15 Wnt1-Cre, Arffl/fl mice revealed a hyperplastic retrolental mass that was not observed in Wnt1-Cre, Arffl/+ or Arffl/fl littermates (Fig. 2A; additional data not shown). Vitreous hyperplasia was also observed in Wnt1-Cre, Arffl/fl embryos at E13.5 (Fig. 2B), and this correlated with increased BrdU incorporation in these cells (Fig. 2, C and D); both of these findings reflect the phenotype of Arf−/− mice (11, 21). This finding demonstrates that the cells that are controlled by p19Arf arise in the Wnt1 expressing neural crest and further supports a model in which Tgfβ directly influences the cells expressing Arf.
FIGURE 2.

Conditional loss of Arf in Wnt1-expressing neural crest cells causes primary vitreous hyperplasia. A and B, shown are representative photomicrographs of hematoxylin- and eosin-stained sections through Arffl/fl (a), Wnt1-Cre, Arffl/+ (b), and Wnt1-Cre, Arffl/fl (c) eyes at P15 (A) and at E13.5 (B). Hyperplastic retrolental mass observed in the postnatal period (A, *) is evident as early as E13.5 (B, arrow). Arrows (A) indicate remnants of normal, regressing hyaloid vessels when functional p19Arf is present. C and D, shown are representative photomicrographs (C) and quantification (D) of BrdU incorporation in the vitreous E13.5 mouse embryo eyes of the indicated genotypes. Note the expansion of BrdU-positive, proliferating cells (C, *) in the primary vitreous between the lens (L) and the neuroretina (NR) in Wnt1-Cre, Arffl/fl mice. Quantitative data are expressed as average percent of total cells in the vitreous space. Increased BrdU+ cells in Wnt1-Cre, Arffl/fl embryos (D, lane 3) is statistically significant. p < 0.001(*) for lane 3 versus lanes 1 or 2).
Tgfβ1, -2, and -3 Induce p19Arf Expression in Cultured MEFs
Having confirmed the functional relationship between these two proteins, we sought to better define the mechanisms by which Tgfβ controls Arf. We took advantage of a cell culture system using early passage MEFs from wild type E14.5 embryos. To understand the kinetics of p19Arf induction, we treated the MEFs with Tgfβ2 for 1.5, 24, and 48 h. In this context, p19Arf protein was minimally increased at 24 h and was significantly higher at 48 h (Fig. 3A), suggesting that p19Arf induction was not an immediate Tgfβ2 response.
FIGURE 3.

Tgfβ1, -2, and -3 induce p19Arf and the Arf promoter in cultured MEFs in a manner that depends on TβrII. A–C, shown is a representative Western blot of lysates from wild type MEFs (A) and β-galactosidase (β-Gal) activity in ArflacZ/lacZ MEFs (B and C) showing the time course of Arf induction after 48 h of exposure to Tgfβ1, -2, or -3 (Τ1, T2, T3), BMP2, or vehicle (V). Induction by each Tgfβ protein was statistically significant when compared with respective vehicle (B and C, *), as was the slight repression by BMP2 (C, #) (p ≤ 0.0002 in each case). D, shown is a representative Western blot for the indicated proteins using lysates from Tgfbr2fl/fl MEFs infected with either adenovirus encoding red fluorescent protein (RFP) or Cre recombinase and exposed to Tgfβ2 or vehicle for 48 h. Note that p19Arf induction (lane 2 versus lane 1) is blunted after inactivation Tgfbr2 (lane 4 versus lane 3).
We also measured the ability of each of the closely related Tgfβ1, -2, and -3 proteins to enhance Arf expression by using MEFs derived from an ArflacZ/lacZ reporter mouse in which the first exon of Arf is replaced by lacZ cDNA (13). ArflacZ/lacZ MEFs derived are functionally Arf−/−, and β-galactosidase expression can be used as a surrogate for Arf promoter activity. Our previous work showed that the time course for β-galactosidase induction by Tgfβ2 parallels that of p19Arf protein (Fig. 3A) (13). All three of the related Tgfβ proteins induced β-galactosidase expression in the MEFs (Fig. 3B, lanes 2, 4, and 6). The relatively small increases in the Arf promoter in ArflacZ/lacZ MEFs as compared with p19Arf induction (compare Fig. 3, A, lanes 5 and 6 to B, lanes 3 and 4) indicated that increased Arf transcription might be complemented by additional, post-transcription effects such as transcript stabilization or increased translation. In contrast, BMP-2, another Tgfβ superfamily member (16, 24), failed to induce β-galactosidase (Fig. 3C). These findings suggest that Tgfβ1, -2, or -3 act through the classical pathway involving ALK-5 and TβrII rather than BMP type I receptors (ALK-2, ALK-3, and ALK-6) (16, 24).
We previously showed Arf and TβrII to be co-expressed in some pericyte-like cells in the primary vitreous in the mouse (13). To directly confirm the importance of this receptor in Arf gene activation, we used MEFs derived from the above-mentioned Tgfbr2fl/fl mouse. When the MEFs were infected with adenovirus encoding Cre recombinase, TβrII expression fell (Fig. 3D, lanes 3 and 4 versus 1 and 2). Tgfβ2 augmented p19Arf in Tgfbr2fl/fl MEFs when they were infected with control adenovirus encoding red fluorescent protein (Fig. 3D, lane 2 versus lane 1) but not in MEFs in which Cre recombinase had diminished TβrII expression (Fig. 3D, lane 4 versus lane 3).
Smads 2 and 3 Cooperatively Induce p19Arf in Response to Tgfβ2
Given the role that Smads 2 and 3 play in response to Tgfβ2, we sought to define the relative importance of these two proteins in Arf regulation. We used gene-specific siRNA to knock down their expression singly and in combination in wild type and ArflacZ/lacZ MEFs (Fig. 4, A and B, top panel; the supplemental figure). Despite the very low Smad3 expression as compared with Smad 2, knockdown of either one impaired Tgfβ2 induction of the native Arf locus and β-galactosidase in wild type and ArflacZ/lacZ MEFs, respectively; interestingly, targeting both proteins further limited Arf induction (Fig. 4, A and B, bottom panel). These findings indicate that Smad2 and -3 cooperatively control Arf induction by Tgfβ2.
FIGURE 4.

siRNA targeting Smad2 and -3 impairs Tgfβ2 induction of Arf. Shown are representative Western blots (A and B, upper panel) of lysates from wild type MEFs (A) or ArflacZ/lacZ MEFs (B) and β-galactosidase assay (β-gal, B, lower panel) in ArflacZ/lacZ MEFs treated with Tgfβ2 (T2) or vehicle (V) for 72 (A) or 60 (B) h after transfection with either siRNA control (Scram) or siRNA targeting Smad2 and/or Smad3 as indicated. Western blotting confirms partial knockdown of Smad2. (longer exposure confirming Smad3 knock-down is available as the supplemental figure). β-Galactosidase activity, normalized to expression in respective vehicle, is expressed as the average of three or more replicates from separate experiments. Knockdown of Smad2 or -3 or both significantly decreased β-galactosidase as compared with siRNA control. p < 0.007 for each case (lower panel, *).
The p38 MAPK-dependent Pathway Also Mediates Tgfβ2 Effects on p19Arf
To investigate whether Smad-independent signals also influence Tgfβ2 induction of p19Arf, we used a panel of chemical inhibitors targeting a variety of pathways recognized to be influenced by Tgfβ (Fig. 5A). As a control, we included SB431542, an inhibitor of TβrI, which blocks Smad2 phosphorylation and Tgfβ2-driven induction of p19Arf and lacZ expression in wild type and ArflacZ/lacZ MEFs, respectively (13) (Fig. 5, B, lanes 5 and 6, C, lanes 11 and 12, and D, lane 7).
FIGURE 5.

Smad-independent pathways influence Tgfβ induction of Arf. A, shown is a schematic diagram showing Smad-dependent and Smad-independent and specific inhibitors for each target: SB43, SB431542; SB20, SB203580; SP60, SP600125; U01, U0126; LY29, LY294002. B and C, shown is a representative Western blot for the indicated proteins from wild type MEFs (B) and β-galactosidase activity in ArflacZ/lacZ MEFs (C) exposed to Tgfβ2 (T2) or vehicle (V) for 48 h and either DMSO or the indicated chemical inhibitors. Quantitative analysis (C) shows that Tgfβ2 significantly changed β-galactosidase activity when compared with relevant vehicle (p < 0.013); however, the induction of β-galactosidase was significantly blunted by SB203580 and SB431542 when compared with DMSO (p = 0.0015 (#) and <0.001 (*), respectively), and it was significantly enhanced by U0126 when compared with DMSO (p = 0.047 (#)). D, a representative Western blot shows the corresponding targets for individual inhibitors in ArflacZ/lacZ MEFs exposed to Tgfβ2, and the indicated chemical inhibitor confirms that LY294002 blocks AKT phosphorylation (lane 4), U0126 blocks p42/44 phosphorylation (lane 6), and SB431542 blocks Smad2 and p38 MAPK phosphorylation (lane 7). E, representative Western blot using ArflacZ/lacZ MEFs exposed to the indicated chemical inhibitors or DMSO for 20 min shows that SB20 blunts anisomycin-stimulated, p38 MAPK-dependent phosphorylation ATF-2. F, a representative Western blot using ArflacZ/lacZ MEFs exposed SP600125 for 20 min blunts UV-stimulated phosphorylation of JNK.
Application of SB203580, a chemical inhibitor of p38 MAPK, blunted Arf expression in both models (Fig. 5, B, lanes 3 and 4, and C, lanes 3 and 4) but did not interfere with Smad 2 phosphorylation (Fig. 5D, lane 3). Consistent with a previous publication, SB203580 was not able to block p38 MAPK phosphorylation (25, 26), but it did block the phosphorylation of its downstream target ATF-2 (Fig. 5E). Of note, blocking TβrI activity with SB431542 also dampened p38 MAPK activation, consistent with existence of parallel pathways downstream from this receptor (Fig. 5D, lane 7). Although the absence of a measurable effect on Smad2 phosphorylation suggests that SB203580 may act independently of Smads, we cannot formally exclude the possibility that p38 MAPK inhibition does influence Smad-dependent activity.
Other chemical inhibitors had less robust effects on Arf induction. Although p42/p44 MAPK blockade by U0126 (Fig. 5D, lane 6) modestly augmented Tgfβ2 induction of β-galactosidase in ArflacZ/lacZ MEFs (Fig. 5C, lane 10), a similar effect on p19Arf protein was not evident in wild type MEFs (Fig. 5B, lane 10). Inhibition of PI3K/Akt by LY294002 or JNK by SP600125 had no measurable effect on Arf induction by Tgfβ2 (Fig. 5, B, lanes 12 and 8, and C, lanes 6 and 8) even though the compounds were active in MEFs (Fig. 5, D, lane 4, and F, lane 4).
Because SB203580 might have off-target effects, we used a genetic approach to confirm the importance of p38 MAPK in Tgfβ-mediated Arf regulation. Consistent with our results from chemical inhibition, even partial knockdown of p38 MAPK blocked the effects of Tgfβ2 on Arf expression (Fig. 6, A and B).
FIGURE 6.
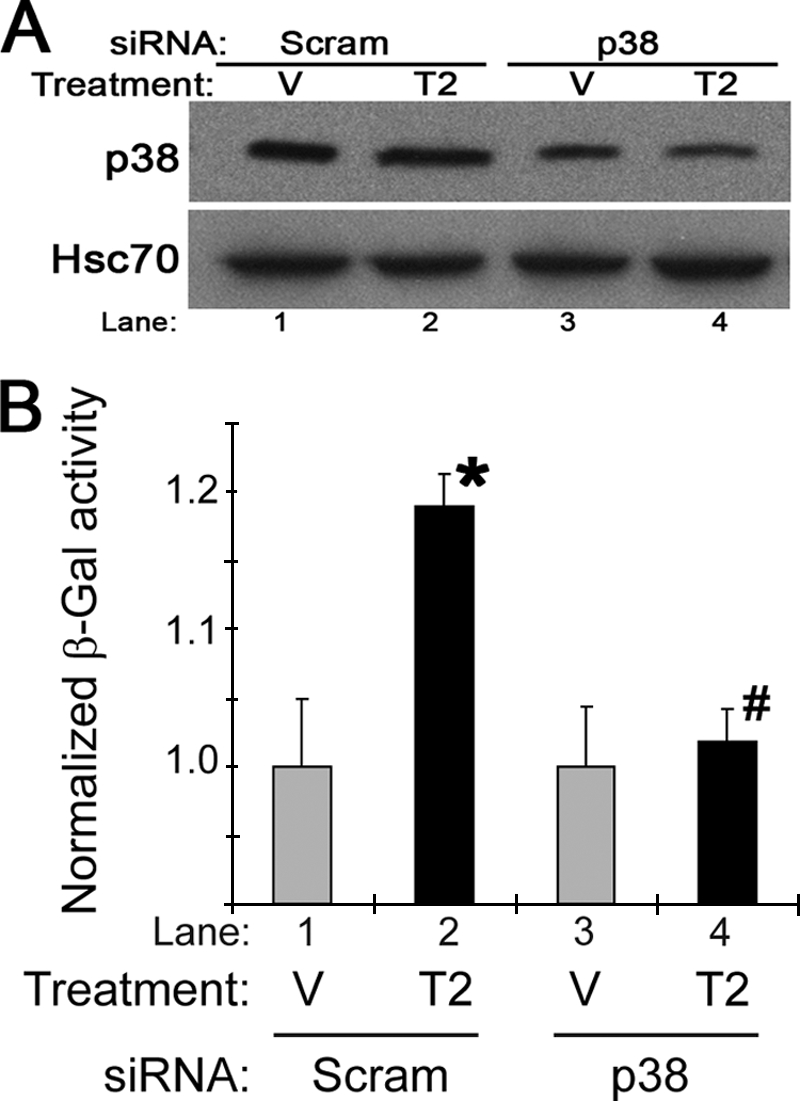
Knockdown of p38 limits Tgfβ induction of the Arf promoter. Shown is a representative Western blot (A) and β-galactosidase assay (B) of ArflacZ/lacZ MEFs transfected with either siRNA control (Scram) or siRNA targeting p38 for 24 h before exposure to vehicle (V) or Tgfβ2 (T2) for 60 h. β-Galactosidase (β-Gal) induction by Tgfβ2, normalized to vehicle, was significant when compared with vehicle in control cells (p < 0.001 (*)) but not in cells transfected with p38 siRNA (p = 0.31 (#)).
Tgfβ Fosters Chromatin Remodeling of the Arf Gene
We previously demonstrated Smad 2/3 had the capacity to bind to two regions ∼2.2 and ∼1.5 kb upstream of the Arf translation initiation codon in ArflacZ/lacZ MEFs (13); however, we did not understand how Tgfβ2 influenced the binding, the kinetics of the effect, and any other coincident changes to the Arf gene. To begin to address these issues, we first examined the binding of Smad2/3 to these regions in vehicle- and cytokine-treated MEFs. We used wild type MEFs to preclude adverse positional effects from insertion of the lacZ-Neo cassette into the Arf locus in ArflacZ/lacZ MEFs; however, more limited analysis of these MEFs showed essentially the same results (data not shown).
We observed that Smad2/3 binding at both the distal and proximal Tgfβ2 binding elements (formerly P3 and P7, respectively, in Freeman-Anderson et al. (13)) increased within 1.5 h after the addition of Tgfβ2 (Fig. 7Ba); the effects were more pronounced at the promoter proximal element. In contrast, Tgfβ2 did not significantly enhance Smad2/3 binding over base line at the other, previously identified distal site (formerly called P1 in Freeman-Anderson et al. (13)) (data not shown) or at putative Tgfβ2 binding elements in the first intron (formerly P11 in Freeman-Anderson et al. (13)) (Fig. 7, A and Ba, lanes 3–6).
FIGURE 7.

Tgfβ2 promotes Smad2/3 binding, histone H3 acetylation, and RNA Pol II localization to the Arf locus in MEFs. A, a schematic diagram shows Distal, Proximal. and Intron regions of Arf locus used in ChIP assays. B, shown is quantitative analysis of representative ChIP assays of using wild type MEFs exposed to vehicle (V) or Tgfβ2 (T2) for 1.5 h (a–c) or 24 h (c) after serum deprivation. A ChIP assay was carried out using antibodies specific to Smad2/3 (a), histone H3 acetylated at lysines 9 and 14 (H3-Ac) (b), and RNA polymerase II (RNA Pol II) (c). Immunoprecipitated DNA and input DNA were amplified with primers for genomic regions indicated in A or for the Tgfβ2 non-responsive gene Necdin. p values are as follows: a, 0.081 (*), 0.001 (#), 0.55 (@); b, 0.045 (*), 0.183 (#), 0.738 (@); (c) 0.059 (*) for Tgfβ2 versus corresponding vehicle.
Smad binding can foster histone modification and the recruitment of the RNA Polymerase II (RNA Pol II) complex at Tgfβ-responsive genes (27, 28). In our experiments, histone H3 acetylation paralleled changes in Smad2/3 binding at the distal and proximal elements 1.5 h after Tgfβ2 treatment; histone acetylation was not observed at the Necdin promoter, a locus that remains silent in MEFs (Fig. 7Bb, lanes 1–4 versus 5 and 6). In contrast to early Smad2/3 binding and histone acetylation, RNA Pol II binding at the proximal site was absent at 1.5 h but became detectable by 24 h after Tgfβ (Fig. 7Bc).
Tgfβ Treatment Increases Arf Transcription
We used quantitative, real-time RT-PCR to explore how the events at the Arf promoter correlated with increased transcription. The mature Arf mRNA transcript increased in wild type MEFs from nearly base line at 24 h through 72 h after Tgfβ2 addition to the culture media (Fig. 8, top panel) in a time course paralleling p19Arf induction (Fig. 3A). Similarly, quantitative, real-time RT-PCR of the primary, unprocessed Arf transcript increased (Fig. 8, bottom panel). The parallels between RNA Pol II localization and Arf primary transcript induction indicate that Tgfβ2 enhances Arf transcription between 24 and 72 h even though Smad2/3 binding and histone modification near exon 1β are enhanced as early as 1.5 h after exposure to this cytokine.
FIGURE 8.
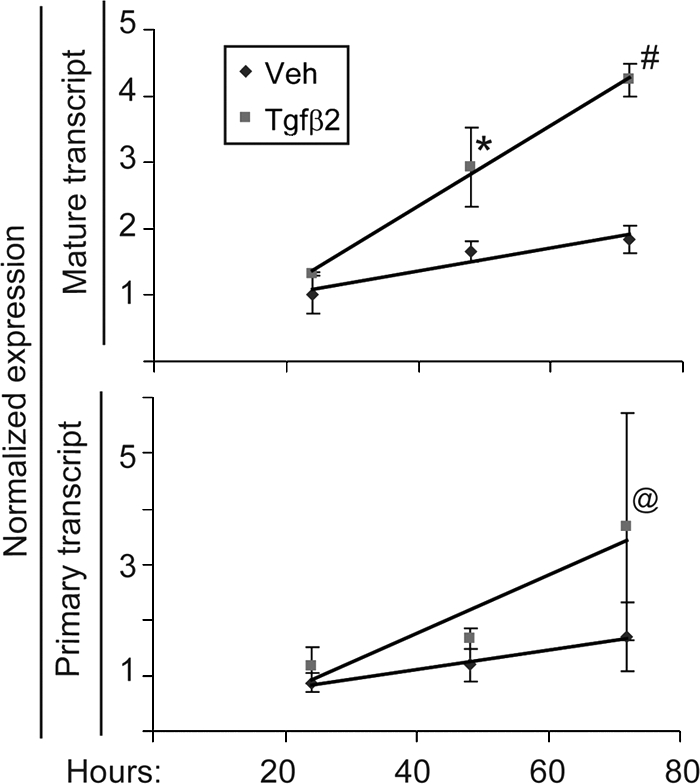
Tgfβ2 increases both the primary and mature Arf transcript. Shown is quantitative analysis of real time, RT-PCR using total RNA isolated from wild type MEFs exposed to vehicle (Veh) or Tgfβ2 for the indicated times. Differences in transcript level between Tgfβ2- and vehicle-treated MEFs are significant for the mature transcript at 48 and 72 h (p < 0.04 (*) and < 0.005 (#)) and for the primary transcript at 72 h (p = 0.039 (@)).
Oncogenic RAS Activates Arf Independently of Tgfβ Signaling to Smad2/3
Arf was initially described as an oncogene sensor that checks inappropriate or excessive cell proliferation signals. For example, ectopic expression of oncogenic H-RASV12 activates Arf via the Raf-ERK-Dmp1 pathway (29). We investigated whether the developmental signaling pathway outlined above was relevant to Arf induction by oncogenic RAS expression in MEFs.
Ectopic expression of human H-RASV12 induced p19Arf 48 h later in early passage, wild type MEFs (Fig. 9, lanes 4 versus 1). Inhibition of TβrI phosphorylation of Smad2/3 by SB431542 had no obvious effect Arf induction (Fig. 9, lane 5 versus 2), but it did slightly increase base-line p19Arf (Fig. 9, lane 2 versus 1). In contrast, inhibition of p38 MAPK using SB203580 significantly blocked p19Arf induction by H-RASV12 (Fig. 9, lane 6 versus 3). Of note, p19Arf induction by ectopically expressed cyclin D1 was not measurably affected by either chemical inhibitor.5 Thus, p38 MAPK is needed for full induction of Arf by both Tgfβ2 and oncogenic RAS in MEFs; in contrast, Smad2/3 phosphorylation is only required for Arf induction by Tgfβ2.
FIGURE 9.
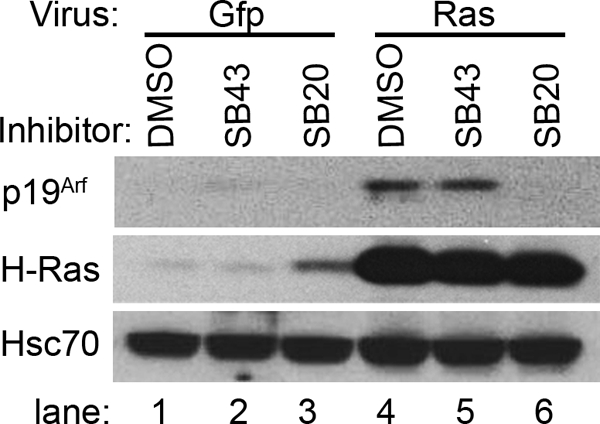
p38 MAPK signaling is needed for Arf induction by ectopically expressed RAS, but inhibition of TβrI does not. Representative Western blot for the indicated proteins using lysates from wild type MEFs, exposed to the indicated chemical inhibitors at the time of transduction using Gfp- or H-RASV12-expressing retrovirus. Lysates were collected 48 h after transduction. Fluorescence microscopy detection of Gfp from the murine stem cell virus-based vectors showed no differences in transduction efficiency across the samples.
DISCUSSION
The central role that p19Arf plays as a tumor suppressor in incipient cancer cells is well established. In contrast, relatively little is known about Arf regulation in physiological contexts. We recently demonstrated that the Tgfβ2 gene is essential for Arf induction at several sites in the developing mouse embryo (13). Here we further explore the functional relationships between Tgfβ and p19Arf in vivo, and we uncover molecular mechanisms underlying Arf induction by this signaling protein.
First, we demonstrate that the capacity for Tgfβ to arrest cell proliferation in vivo in the developing eye strictly depends on its ability to induce p19Arf. Hence, understanding of how it induces Arf becomes central to knowing how it acts in this developmental capacity. Second, we use a genetic approach to show that TβrII and p19Arf both act in cells of the same, neural crest-derived lineage to prevent primary vitreous hyperplasia; this provides in vivo evidence for cell-intrinsic signaling from TβrII to Arf. Third, the complementary use of chemical inhibitors and genetic manipulations define the signaling pathways extending from Tgfβ1, -2, or -3 binding to the TβrI/II complex to Arf gene activation. Our findings indicate that Smads 2 and 3 and p38 MAPK are necessary for full p19Arf induction. We have characterized the kinetics of Smad2/3 binding in DNA elements 5′ to Arf and subsequent changes in the chromatin architecture of the locus. Interestingly, although these events begin within a short 1.5-h timeframe after Tgfβ2 treatment, Arf transcription, measured by RNA Pol II binding and primary Arf transcript increase, are not detected until 24–48 h later. Last, we show that Arf induction by oncogenic RAS shares the p38 MAPK arm of this developmental pathway, whereas activation of Smad2/3 is dispensable.
The delay between Smad binding to the Arf gene and subsequent increases in Arf promoter and transcript levels was unexpected. Early Smad-dependent effects of Tgfβ on gene transcription are often evident within several hours of receptor activation (e.g. Gomis et al. 30)). Indeed, in cultured MEFs, early cell proliferation arrest by Tgfβ2 proceeds independently of Arf (13) and is likely mediated by earlier events like repression of Cdk4 (31) or induction of Cdk inhibitors like p21Cip1 or p15Ink4b (32, 33). We can speculate that the delayed Arf induction (although still initiated as an immediate Smad-dependent response) could have evolved to allow p19Arf to primarily contribute to the maintenance of a Tgfβ-driven proliferation arrest.
At a molecular level, the delay between Smad binding and transcriptional activation suggests the need for the recruitment of other transcription factors to Arf regulatory sequences before or coincident with RNA Pol II binding. Candidate transcription factors already implicated as direct Arf regulators (by virtue of binding to the Arf promoter) include E2f 1 and 3 (34), Dmp1 (35), Pokemon (36), and FoxO3a (37), which positively or negatively regulate mouse Arf expression. Of these, FoxO3a (which binds to intronic DNA ∼8 kb 3′ of exon 1β) (37) is particularly interesting because FoxO proteins directly interact with Smads 2/3 and 4 to induce p21Cip1 (38). Functional cooperation between FoxO and Smad proteins is also found in a cluster of genes that seem to control stress and adaptive cell signaling responses at least in cultured HaCaT cells (30). However, this work focused on immediate responses within 3 h, a time point at which Arf transcription is not observed. In our preliminary studies, Tgfβ2 did not alter the levels of FoxO3a in MEFs, nor did inhibition of Akt (a negative regulator of FoxO (39)). Additional work is required to confirm or dispel the importance FoxO proteins in Arf regulation in the eye.
That a p38-dependent signaling pathway also contributes to the regulation of Arf by Tgfβ is consistent with the growing understanding of cross-talk between Smad-dependent and -independent effectors of Tgfβ (16). p38 MAPK has previously been implicated in Ink4/Arf regulation; for example, decreased expression of Wip1 phosphatase in Ppm1d−/− MEFs increases both p16Ink4a and p19Arf in a p38 MAPK-dependent manner (40). It also contributes to other Tgfβ effects like the arrested DNA synthesis in primary mouse vascular smooth muscle cells at 48 h (26), induction of type I collagen in a cultured retinal pigment epithelial cell line at 24 h (41), and increased α-smooth muscle actin expression at 48–72 h in cultured primary human fibroblasts (42). We previously showed that some of the Arf-expressing perivascular cells also express α-smooth muscle actin in the newborn mouse eye and that ectopic p19Arf expression in 10T1/2 fibroblasts (in which Arf is deleted) can promote α-smooth muscle actin and smooth muscle myosin expression 48–96 h later (43). Conceivably, the induction of Arf with smooth muscle proteins may represent part of a Tgfβ- and p38 MAPK-dependent transcriptional routine leading to the maturation and cell cycle arrest of a subtype of vascular smooth muscle cells surrounding the hyaloid vessels.
How Smad and p38 MAPK signaling cooperates to induce Arf is not clear. Direct cooperation between the two is possible, and our preliminary studies indicate that SB203580 slightly decreases Smad2/3 occupancy at the proximal element, but the decrease is not statistically significant (negative data not shown). Instead, the positive interactions we found may lie in the ability of p38 MAPK to activate potential transcriptional co-factors, such as p300, C/EBPβ, or ATF2 (16, 44, 45). Of these, p300 is known to cooperate with Smads to modulate histone acetylation (46). Putative binding sites for C/EBPβ and ATF-2 are present in genomic DNA flanking the Arf first exon. C/EBPβ was shown to be required for RasV12-mediated senescence in MEFs but it was not needed for p19Arf induction by this oncogene (47); instead, p19Arf negatively regulates C/EBPβ (48). ATF2 has a variety of activities that could play a role in cancer biology, but there is no clear connection to Arf (49).
Given the broad role of Arf in both tumor suppression and eye development, our findings may help us understand certain human diseases. For instance, so far the molecular pathogenesis of persistent hyperplastic primary vitreous, which the Arf−/− model mimics (11, 12), is unclear. Occasional familial cases of this disease suggest an underlying genetic basis (50–52). Elucidating the complete series of components necessary for Tgfβ-mediated Arf transcription will allow us to potentially interrogate the genomic DNA extracted from either involved tissue samples or from blood of diseased patients in a more focused way. Because a persistent hyperplastic primary vitreous-like disease can also develop in somatic mosaic mice in which Arf is missing in only a subset of cells in the mouse (43), such an analysis should be accomplished in a way that can also detect mosaicism of the key gene(s).
We hope that our findings may also provide some insight into tumor suppression by Arf in incipient, oncogene-stressed cancer cells. This may involve both how Arf is induced and how Tgfβ acts as a tumor suppressor. Arf induction by oncogenic RAS, E1A, or Myc in cultured cells occurs over ∼48 h (4, 5), a time course that is similar to induction by Tgfβ (Fig. 3A). It is interesting that Tgfβs were initially described in oncogene-transformed fibroblasts (53, 54). Furthermore, using cultured mouse keratinocytes, v-RasHa and Tgfβ1 cooperatively induce p19Arf and p16Ink4a, which is also encoded at the Arf/Ink4a locus, and base-line p19Arf expression is decreased in Smad3−/− keratinocytes transduced by v-RasHa-transduced cells (55). These findings imply that Arf induction by certain oncogenes might be driven by an autocrine or paracrine loop. In our analysis of cultured MEFs, RASV12 does not require TβrI/Smad signaling, although full Arf induction by ectopic RAS depends on p38 MAPK. Because tissue-specific factors control Arf induction by oncogenic Ras (56), a more robust investigation into the relationship between Arf regulation by Tgfβ and by different oncogenes will require analysis of a variety of cell types and contexts.
The anti-tumor effects of Tgfβ have been well described (57, 58), and disruption of components of the pathway is particularly common in certain cancers, like those involving the cervix, gastrointestinal epithelium, and liver (59–61). In other contexts, Tgfβ appears to promote tumorigenesis (58, 62). One might reconcile these apparent discrepancies if the presence or absence of Arf, which is required for anti-proliferative effects of Tgfβ2 in the eye, also determines the effectiveness of Tgfβ-mediated tumor suppression.
Supplementary Material
Acknowledgments
We gratefully acknowledge T. C. He (University of Chicago) for providing adenovirus vectors, C. J. Sherr (St. Jude Children's Research Hospital) for providing Arffl/fl mice before their publication, and helpful discussions with D. Dighe, S. Volchenboum, R. Widau, and A. Zelivianskaia (all at the University of Chicago).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 EY0194368 and EY019942 (NEI, to S. X. S.).

The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
S. X. Skapek, unpublished data.
Y. Zheng and S. X. Skapek, negative data not shown.
- MEF
- mouse embryo fibroblast
- RNA Pol II
- RNA polymerase II
- TβrI and TβrII
- Tgfβ type I and type II receptors, respectively.
REFERENCES
- 1.Sherr C. J. (2006) Nat. Rev. Cancer 6, 663–673 [DOI] [PubMed] [Google Scholar]
- 2.Kamijo T., Zindy F., Roussel M. F., Quelle D. E., Downing J. R., Ashmun R. A., Grosveld G., Sherr C. J. (1997) Cell 91, 649–659 [DOI] [PubMed] [Google Scholar]
- 3.Zindy F., Eischen C. M., Randle D. H., Kamijo T., Cleveland J. L., Sherr C. J., Roussel M. F. (1998) Genes Dev. 12, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Stanchina E., McCurrach M. E., Zindy F., Shieh S. Y., Ferbeyre G., Samuelson A. V., Prives C., Roussel M. F., Sherr C. J., Lowe S. W. (1998) Genes Dev. 12, 2434–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin A. W., Lowe S. W. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5025–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmero I., Pantoja C., Serrano M. (1998) Nature 395, 125–126 [DOI] [PubMed] [Google Scholar]
- 7.Williams R. T., Roussel M. F., Sherr C. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6688–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zindy F., Quelle D. E., Roussel M. F., Sherr C. J. (1997) Oncogene 15, 203–211 [DOI] [PubMed] [Google Scholar]
- 9.Lowe S. W., Sherr C. J. (2003) Curr. Opin. Genet. Dev. 13, 77–83 [DOI] [PubMed] [Google Scholar]
- 10.Krishnamurthy J., Torrice C., Ramsey M. R., Kovalev G. I., Al-Regaiey K., Su L., Sharpless N. E. (2004) J. Clin. Invest. 114, 1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeller R. N., Fowler J. L., Cunningham J. J., Warner N., Smeyne R. J., Zindy F., Skapek S. X. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3848–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin A. C., Thornton J. D., Liu J., Wang X., Zuo J., Jablonski M. M., Chaum E., Zindy F., Skapek S. X. (2004) Invest. Ophthalmol. Vis. Sci. 45, 3387–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman-Anderson N. E., Zheng Y., McCalla-Martin A. C., Treanor L. M., Zhao Y. D., Garfin P. M., He T. C., Mary M. N., Thornton J. D., Anderson C., Gibbons M., Saab R., Baumer S. H., Cunningham J. M., Skapek S. X. (2009) Development 136, 2081–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T. (1997) Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saika S., Saika S., Liu C. Y., Azhar M., Sanford L. P., Doetschman T., Gendron R. L., Kao C. W., Kao W. W. (2001) Dev. Biol. 240, 419–432 [DOI] [PubMed] [Google Scholar]
- 16.Derynck R., Zhang Y. E. (2003) Nature 425, 577–584 [DOI] [PubMed] [Google Scholar]
- 17.Gromley A., Churchman M. L., Zindy F., Sherr C. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 6285–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zindy F., Williams R. T., Baudino T. A., Rehg J. E., Skapek S. X., Cleveland J. L., Roussel M. F., Sherr C. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15930–15935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chytil A., Magnuson M. A., Wright C. V., Moses H. L. (2002) Genesis. 32, 73–75 [DOI] [PubMed] [Google Scholar]
- 20.Srinivasan Y., Lovicu F. J., Overbeek P. A. (1998) J. Clin. Invest. 101, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silva R. L., Thornton J. D., Martin A. C., Rehg J. E., Bertwistle D., Zindy F., Skapek S. X. (2005) EMBO J. 24, 2803–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao S., Overbeek P. A. (2001) Dev. Biol. 237, 45–53 [DOI] [PubMed] [Google Scholar]
- 23.Ittner L. M., Wurdak H., Schwerdtfeger K., Kunz T., Ille F., Leveen P., Hjalt T. A., Suter U., Karlsson S., Hafezi F., Born W., Sommer L. (2005) J. Biol. 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh S., Itoh F., Goumans M. J., Ten Dijke P. (2000) Eur. J. Biochem. 267, 6954–6967 [DOI] [PubMed] [Google Scholar]
- 25.ten Hove T., van den Blink B., Pronk I., Drillenburg P., Peppelenbosch M. P., van Deventer S. J. (2002) Gut 50, 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seay U., Sedding D., Krick S., Hecker M., Seeger W., Eickelberg O. (2005) J. Pharmacol. Exp. Ther. 315, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 27.Verdone L., Agricola E., Caserta M., Di Mauro E. (2006) Brief. Funct. Genomic. Proteomic. 5, 209–221 [DOI] [PubMed] [Google Scholar]
- 28.Clayton A. L., Hazzalin C. A., Mahadevan L. C. (2006) Mol. Cell 23, 289–296 [DOI] [PubMed] [Google Scholar]
- 29.Sreeramaneni R., Chaudhry A., McMahon M., Sherr C. J., Inoue K. (2005) Mol. Cell. Biol. 25, 220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomis R. R., Alarcón C., He W., Wang Q., Seoane J., Lash A., Massagué J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 12747–12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewen M. E., Sluss H. K., Whitehouse L. L., Livingston D. M. (1993) Cell 74, 1009–1020 [DOI] [PubMed] [Google Scholar]
- 32.Cordenonsi M., Dupont S., Maretto S., Insinga A., Imbriano C., Piccolo S. (2003) Cell 113, 301–314 [DOI] [PubMed] [Google Scholar]
- 33.Hannon G. J., Beach D. (1994) Nature 371, 257–261 [DOI] [PubMed] [Google Scholar]
- 34.Aslanian A., Iaquinta P. J., Verona R., Lees J. A. (2004) Genes Dev. 18, 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoue K., Roussel M. F., Sherr C. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 3993–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeda T., Hobbs R. M., Merghoub T., Guernah I., Zelent A., Cordon-Cardo C., Teruya-Feldstein J., Pandolfi P. P. (2005) Nature 433, 278–285 [DOI] [PubMed] [Google Scholar]
- 37.Bouchard C., Lee S., Paulus-Hock V., Loddenkemper C., Eilers M., Schmitt C. A. (2007) Genes Dev. 21, 2775–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seoane J., Le H. V., Shen L., Anderson S. A., Massagué J. (2004) Cell 117, 211–223 [DOI] [PubMed] [Google Scholar]
- 39.Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 40.Bulavin D. V., Phillips C., Nannenga B., Timofeev O., Donehower L. A., Anderson C. W., Appella E., Fornace A. J., Jr. (2004) Nat. Genet. 36, 343–350 [DOI] [PubMed] [Google Scholar]
- 41.Kimoto K., Nakatsuka K., Matsuo N., Yoshioka H. (2004) Invest Ophthalmol. Vis. Sci. 45, 2431–2437 [DOI] [PubMed] [Google Scholar]
- 42.Meyer-Ter-Vehn T., Gebhardt S., Sebald W., Buttmann M., Grehn F., Schlunck G., Knaus P. (2006) Invest Ophthalmol. Vis. Sci. 47, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 43.Thornton J. D., Swanson D. J., Mary M. N., Pei D., Martin A. C., Pounds S., Goldowitz D., Skapek S. X. (2007) Invest Ophthalmol. Vis. Sci. 48, 491–499 [DOI] [PubMed] [Google Scholar]
- 44.Wagner E. F., Nebreda A. R. (2009) Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]
- 45.Euler-Taimor G., Heger J. (2006) Cardiovasc. Res. 69, 15–25 [DOI] [PubMed] [Google Scholar]
- 46.Ross S., Cheung E., Petrakis T. G., Howell M., Kraus W. L., Hill C. S. (2006) EMBO J. 25, 4490–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebastian T., Malik R., Thomas S., Sage J., Johnson P. F. (2005) EMBO J. 24, 3301–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sebastian T., Johnson P. F. (2009) Cancer Res. 69, 2588–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhoumik A., Ronai Z. (2008) Cell Cycle 7, 2341–2345 [DOI] [PubMed] [Google Scholar]
- 50.Lin A. E., Biglan A. W., Garver K. L. (1990) Ophthalmic Paediatr. Genet. 11, 121–122 [DOI] [PubMed] [Google Scholar]
- 51.Wang M. K., Phillips C. I. (1973) Acta Ophthalmol. 51, 434–437 [DOI] [PubMed] [Google Scholar]
- 52.Khaliq S., Hameed A., Ismail M., Anwar K., Leroy B., Payne A. M., Bhattacharya S. S., Mehdi S. Q. (2001) Invest Ophthalmol. Vis. Sci. 42, 2225–2228 [PubMed] [Google Scholar]
- 53.de Larco J. E., Todaro G. J. (1978) Proc. Natl. Acad. Sci. U.S.A. 75, 4001–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marquardt H., Hunkapiller M. W., Hood L. E., Twardzik D. R., De Larco J. E., Stephenson J. R., Todaro G. J. (1983) Proc. Natl. Acad. Sci. U.S.A. 80, 4684–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vijayachandra K., Higgins W., Lee J., Glick A. (2009) Mol. Carcinog. 48, 181–186 [DOI] [PubMed] [Google Scholar]
- 56.Young N. P., Jacks T. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 10184–10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexandrow M. G., Moses H. L. (1995) Cancer Res. 55, 1452–1457 [PubMed] [Google Scholar]
- 58.Derynck R., Akhurst R. J., Balmain A. (2001) Nat. Genet. 29, 117–129 [DOI] [PubMed] [Google Scholar]
- 59.Ki K. D., Tong S. Y., Huh C. Y., Lee J. M., Lee S. K., Chi S. G. (2009) J. Gynecol. Oncol. 20, 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katuri V., Tang Y., Marshall B., Rashid A., Jogunoori W., Volpe E. A., Sidawy A. N., Evans S., Blay J., Gallicano G. I., Premkumar, Reddy E., Mishra L., Mishra B. (2005) Oncogene 24, 8012–8024 [DOI] [PubMed] [Google Scholar]
- 61.Kiss A., Wang N. J., Xie J. P., Thorgeirsson S. S. (1997) Clin. Cancer Res. 3, 1059–1066 [PubMed] [Google Scholar]
- 62.Murata M., Matsuzaki K., Yoshida K., Sekimoto G., Tahashi Y., Mori S., Uemura Y., Sakaida N., Fujisawa J., Seki T., Kobayashi K., Yokote K., Koike K., Okazaki K. (2009) Hepatology 49, 1203–1217 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.