

Abstract
BACKGROUND & AIMS
Diarrhea results from reduced net fluid and salt absorption caused by an imbalance in intestinal absorption and secretion. The bulk of sodium and water absorption in the intestine is mediated by Na+/H+ exchanger 3 (NHE3), located in the luminal membrane of enterocytes. We investigated the effect of lysophosphatidic acid (LPA) on Na+/H+ exchanger activity and Na+-dependent fluid absorption in the intestine.
METHODS
We analyzed the effects of LPA on fluid absorption in intestines of wild-type mice and mice deficient in Na+/H+ exchanger regulatory factor 2 (NHERF2; Nherf2−/−)or LPA2 (Lpa2−/−). Roles of LPA5 and NHERF2 were determined by analysis of heterologous expression.
RESULTS
Under basal conditions, LPA increased fluid absorption in an NHE3-dependent manner and restored the net fluid loss in a mouse model of acute diarrhea. Expression of the LPA receptor LPA5 was necessary for LPA-induced stimulation of NHE3 activity in colonic epithelial cells. Stimulation of NHE3 by the LPA-LPA5 signaling required coexpression of NHERF2, which interacted with LPA5. LPA-mediated intestinal fluid absorption was impaired in Nherf2−/− mice, demonstrating the requirement for NHERF2 in LPA5 activity. However, fluid absorption was unaltered in Lpa2−/− mice. LPA stimulated NHE3 and fluid absorption in part by increasing NHE3 protein abundance at the brush border membrane of intestinal epithelial cells.
CONCLUSIONS
LPA is a potent stimulant of NHE3 and fluid absorption in the intestine, signaling through LPA5. Regulation by LPA5 depends on its interaction with NHERF2. LPA might be useful in the treatment of certain diarrheal diseases.
Acute diarrhea is an unpleasant digestive disorder that affects nearly everyone at one time or another, but still remains a major cause of morbidity and mortality in the elderly and the very young. Diarrhea is caused by a decrease in net salt and fluid absorption, which can result from a decrease in salt absorption or an increase in anion secretion, or both.1 The major pathway for sodium and water absorption in the intestine is thought to involve Na+/H+ exchanger type 3 (NHE3 or SLC9A3) in the brush border membrane of enterocytes, where it acts in concert with an anion exchanger to mediate electroneutral NaCl absorption.2 Consequently, genetic disruption of NHE3 expression in mice resulted in diarrhea.3
Lysophosphatidic acid (LPA) is one of the simplest phospholipids found in nature. LPA is primarily produced from preexisting lysophospholipids or phosphatidic acid by lysophopholipase D and phospholipases, respectively.4,5 LPA acts upon the cognate G-protein–coupled receptors: LPA1–LPA55-7. The growth factor-like effects of LPA in cell proliferation, cell survival, and migration have been studied broadly.4,5 In addition, LPA regulates inflammatory and innate immune responses by activating expression and secretion of cytokines and chemokines.8 Recent studies have shown that LPA inhibits the cystic fibrosis transmembrane conductance regulator (CFTR)-dependent Cl− secretion in mouse intestine,9,10 and NHE3 activity in opossum kidney (OK) cells is stimulated by LPA.11,12 However, the effect of LPA on intestinal Na+ absorption has not been reported. Here, we measured basal and LPA-induced Na+ and fluid absorption in mice and investigated the roles of LPA receptor subtypes in regulation of NHE3.
Methods
Animals
Lpa2−/− mice in C57BL/6 background were reported recently.13 Nherf2−/− mice10 were bred in C57BL/6 or FVB/N background for at least 15 generations. Nherf2−/− mice on C57BL/6 background were compared with Lpa2−/− mice, and Nherf2−/− mice on the FVB/N background were compared with mice deficient for other NHERF proteins.10 Age- and sex-matched littermates of the same respective genetic background were used as controls. It was made certain that both genetic backgrounds responded similarly to LPA (Figure 1B on C57BL/6 vs Figure 1D on FVB/N, and Figure 6A on C57BL/6 and Figure 6B on FVB/N). Genotypes were determined by polymerase chain reaction as described previously.10,13 Animals were maintained and experiments were performed under institutional guidelines of Emory University or the Hannover Medical School.
Figure 1.
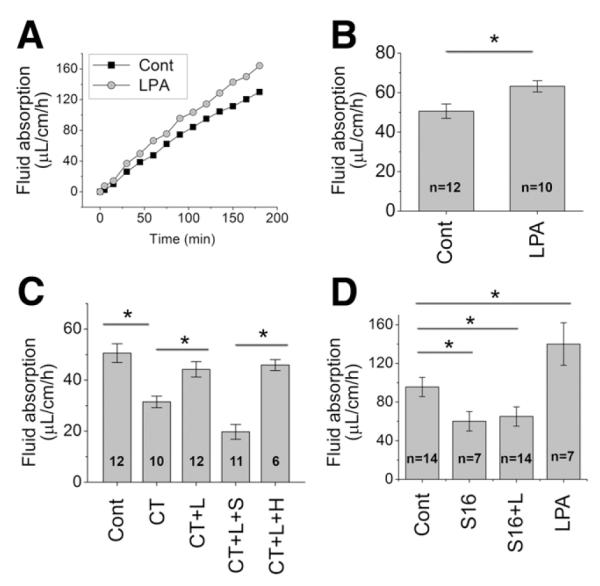
LPA stimulates fluid absorption in the mouse intestine. (A) In vivo perfusion system was used to determine absorption of fluid in the mouse intestine. Representative traces of fluid absorbed in the absence (filled square) and presence (open circle) of 20 μM LPA in the perfusion buffer are shown. (B) Rates of fluid absorption in the absence and presence of LPA are shown; *P < .001. (C) To determine the efficacy of LPA as an agonist of fluid absorption under pathological conditions, acute diarrhea was induced by CT. L: LPA, S: S3226, H; HOE694; *P < .001. (D) LPA-induced fluid absorption is determined in the presence of the NHE3 (specific inhibitor S1611 (S16). *P < .001.
Figure 6.
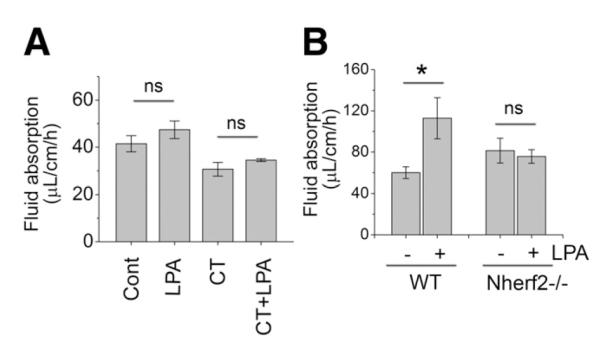
NHERF2 is necessary for stimulation of fluid absorption by LPA. (A) Effects of LPA on the rate of fluid absorption in Nherf2−/− (C57BL/6) mice under basal and CT-treated conditions are shown. Twenty micromolar LPA was added to the perfusion buffer when indicated. n = 6. ns, not significant. (B) To ascertain the lack of effect by LPA in Nherf2−/− (FVB/N) mice, 50 μM LPA was used. *P < . 05.
Plasmids and Cell Culture
Human NHE3 fused with a C-terminal VSVG tag, hNHE3V, and human LPA5, with N-terminal fusion of hemagglutinin, HA-LPA5, were cloned in pcDNA3.1 (Invitrogen, Carlsbad, CA). PS120, Caco-2, and Caco-2bbe (C2b) cells were grown as described previously.14 PS120/NHE3V, PS120/NHE3V/NHERF1, and PS120/NHE3V/NHERF2 cells have been described previously.14 1-oleoyl LPA (Avanti Polar, Alabaster, AL) was prepared in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA).8
See Supplementary Materials for real-time reverse transcription (RT) PCR, in vitro overlay assay, Na+-dependent intracellular pH recovery, and surface biotinylation.
Intestinal Water Flux Measurement
Intestinal water flux was measured by adapting the protocol reported previously.15 Seven-to-9-week-old sex-matched mouse littermates were fasted overnight and anesthetized with pentobarbital sodium. The abdomen was opened by a midline incision, and an approximately 10-cm loop of ileum was cannulated at the proximal and distal ends with 0.76-mm internal diameter polyethylene tubing. Flushing solution (mM: 118.4 NaCl, 4.7 KCl, 2.52 CaCl2, 1.18 MgSO4, 25 NaHCO3, 1.18 KH2PO4, pH 7.4) warmed to 37°C was first perfused through the ileal loop at 1 mL/min for 10 minutes using a peristaltic pump. This was followed by perfusion of prewarmed perfusion solution (mM: 118.4 NaCl, 4.7 KCl, 2.52 CaCl2, 1.18 MgSO4, 25 Na gluconate, 1.18 KH2PO4, pH 7.425 mM Na gluconate) in a recirculating manner at 1 mL/min for 2–3 hours. The abdominal cavity was covered with moistened gauze and was maintained at 37°C using a heating lamp. Water flux was measured in a real-time manner by the connected metered tube. The perfusate volume change was recorded every 15 minutes. When necessary, cholera toxin (CT; 50 μg/mL), S3226 (10 μM), S1611 (20 μM), or HOE694 (50 μM) was added to the perfusate. Twenty micrometers LPA was used unless otherwise specified.
See Supplementary Materials for immunofluorescence and confocal studies and statistical analysis.
Results
LPA Induces NHE3-Dependent Water Absorption In Vivo
We sought to test whether LPA can regulate NHE3-dependent intestinal fluid absorption, which is the hallmark of diarrhea. We adapted the in vivo perfusion system in which a small loop of intestine of an anesthetized mouse is perfused in a recirculating manner.15 Figure 1A depicts typical changes in fluid absorption over 3 hours. There was a net absorption of fluid in the ileum at the rate of 50.6 ± 3.7 μL/cm/h at basal conditions (Figure 1B). To determine the effect of LPA, 20 μM LPA was supplemented in the perfusion buffer. The presence of LPA increased the fluid absorption to 63.2 ± 2.9 μL/cm/h, a 25% increase over the basal rate, indicating a stimulatory effect of LPA in Na+ and fluid absorption (Figure 1A and B).
We next tested the efficacy of LPA as a regulator of fluid absorption under the condition that mimics a pathological situation. Infection by Vibrio cholera leads to diarrhea by altering electrolyte transport:1 increased Cl− secretion and decreased Na+ absorption as a result of the presence of CT secreted by the bacteria. Adding CT to the perfusion buffer reduced the amount of fluid absorbed by 40% (Figure 1C). More importantly, addition of LPA in the perfusion buffer reconstituted the rate of fluid absorption almost to the basal level.
To ensure the stimulatory effect of LPA on fluid absorption, we used tumor necrosis factor—α (TNF-α)to induce diarrhea. TNF-α disrupts the epithelial barrier and induces diarrhea as in the setting of inflammatory bowel disease.15 Injecting mice with TNF-α resulted in decreased fluid absorption (Supplementary Figure 1). The presence of LPA in the perfusion buffer significantly increased the amount of fluid absorbed in the intestine of the TNF-treated animals. These data clearly demonstrate that LPA activates fluid absorption in vivo.
A recent study showed that LPA inhibited Cl− secretion by the epithelial anion channel CFTR.9 To ascertain that the effect of LPA in fluid absorption is mediated through NHE3, the activity of NHE3 was blocked by the NHE3-specific inhibitor S1611.16 The presence of S1611 abrogated LPA-induced stimulation of fluid absorption, demonstrating LPA-induced fluid absorption is mediated by NHE3 (Figure 1D). Similarly, another NHE3-specific inhibitor S322614 blocked LPA-induced fluid absorption during CT treatment (Figure 1C). On the other hand, HOE694,16 which blocks NHE1 and NHE2, but not NHE3, had no effect on fluid absorption. These results suggest that NHE3, but not NHE1 and NHE2, mediates LPA-induced fluid absorption.
LPA Does Not Activate NHE3 in Intestinal Epithelial Caco-2 Cells
In order to identify an LPA receptor subtype(s) that regulates NHE3 activity, we initially tested the effect of LPA in Caco-2 cells. A polarized layer of Caco-2 cells was perfused with Na+-buffer complemented with 1 μM LPA or BSA/PBS. All Na+-dependent pH measurements were carried out in the presence of 50 μM HOE694 to block endogenous NHE1 and NHE2 activities. LPA treatment had an insignificant effect on the transport activity of NHE3 (Figure 2A). The transport activity of NHE3 in Caco-2 cells is relatively low and the low activity can lead to a significant margin of error that may obscure an effect on the transport activity by LPA. To circumvent this limitation, we ectopically expressed human NHE3 (hNHE3V) in C2b cells. However, despite the high NHE3 activity in these transfected cells, LPA did not alter NHE3 activity (Figure 2B).
Figure 2.
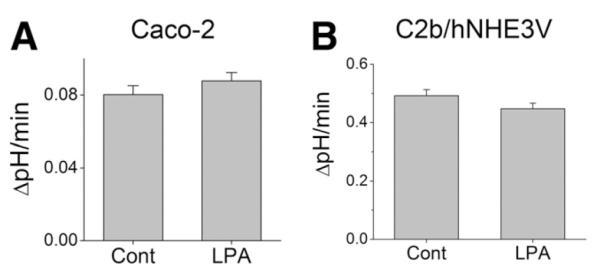
LPA does not stimulate NHE3 activity in Caco-2 cells. Na+/H+ exchange by NHE3 in (A) Caco-2 and (B) C2b/hNHE3V cells was determined in the presence of 50 μM HOE694. NHE3 activity was represented as the rate of Na+-dependent pH recovery, ΔpH/min. n = 8.
Intestinal Expression of LPA Receptors
We showed previously that the expression level of LPA2 is elevated in Caco-2 cells, and a recent study showed that LPA-induced regulation of CFTR is mediated by LPA2.8,9 Therefore, it was surprising that LPA failed to regulate NHE3 in Caco-2 cells and suggested that an LPA receptor other than LPA2 might be needed. Previously, high expression of LPA5 in the gastrointestinal track was reported,6,7 but its expression relative to other LPA receptors is not known. Real-time RT-PCR was performed to determine the relative expression levels of LPA receptors in mouse ileum and proximal colon. Figure 3A shows that LPA1 messenger RNA (mRNA) was most abundant, followed by LPA5, whereas the expression levels of LPA2, LPA3, and LPA4 were relatively low. Expression level of LPA5 mRNA was significantly higher in the intestine and colon compared with liver or kidney (Figure 3B). The abundance of LPA1 and LPA5 was confirmed in nontransformed rat intestinal IEC6 cells (Figure 3C). Interestingly, expression level of LPA5 was low in Caco-2 human colon cancer cells as opposed to abundantly expressed LPA2.
Figure 3.
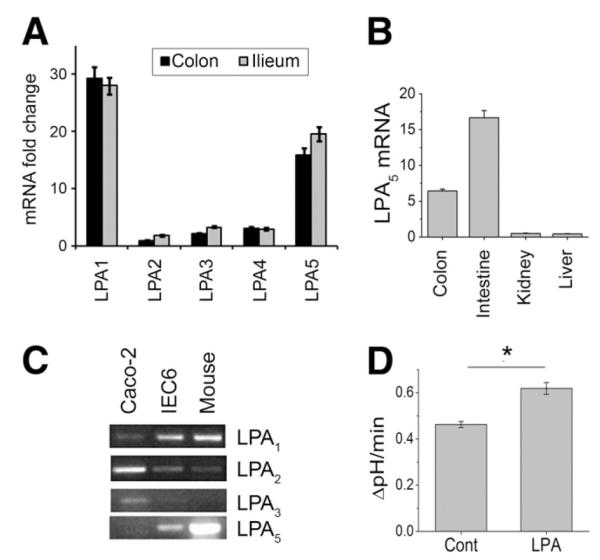
Expression of LPA receptors in rat tissues. (A) Relative levels of LPA1-LPA5 mRNA in mouse colon and ileum were determined by real-time RT-PCR. Data are expressed as fold changes relative to the expression level of LPA2 in the colon. (B) LPA5 mRNA expression levels were determined in the mouse colon, small intestine, kidney, and liver. Expression level represented as fold changes relative to the expression level in the liver. (C) RT-PCR was performed on total RNA isolated from Caco-2, IEC cells, and mouse intestine. (D) LPA significantly stimulated NHE3 activity when LPA5 was ectopically expressed in C2b/hNHE3V cells. n = 8. *P < .001 compared with the control.
Because of its abundance in the intestine,6,7 we considered the possibility that LPA5 may be needed for the regulation of NHE3. Therefore, we ectopically expressed HA-LPA5 in C2b/hNHE3V cells.7 As shown in Figure 3D, LPA treatment resulted in a significant increase in NHE3 activity when HA-LPA5 was coexpressed. These results demonstrate that the signaling by LPA5, but not by LPA2, stimulates NHE3 activity in intestinal epithelial cells.
LPA5 Binds to NHERF2
Several lines of evidence have shown that postsynaptic density 95, discs large, and zonula occludens—1 (PDZ) domains play a central role in mediating interaction of NHE3 with several signaling and structural proteins.2,10,14,16-18 LPA5 receptor has a prototypical Class I PDZ binding sequence, -DSAL, at the C-terminal end, rendering it a strong candidate for interaction with PDZ domains. To determine whether LPA5 is capable of binding to PDZ domains, we tested whether a GST-fusion protein of the carboxyl terminal 55aa of LPA5 receptor (GST-LPA5) interacts with PDZ proteins that are known to be expressed in the intestinal epithelial cells: Na+/H+ exchanger regulatory factor 1 (NHERF1; also known as EBP50 or SLC9A3R1), NHERF2 (also known as E3KARP or SLC9A3R2), and PDZ domain containing protein in kidney 1 (also known as NHERF3 or CAP70).10,16-18 Figure 4A shows that LPA5 receptor bound to NHERF2, but not to NHERF1 or PDZ domain containing protein in kidney 1 (PDZK1). No interaction with GST itself was observed. Figure 4B shows that the interaction of LPA5 with NHERF2 was mediated via the second PDZ (PDZ2) domain, but not the first PDZ (PDZ1) domain of NHERF2.
Figure 4.

The interaction of LPA5 with NHERF2. (A) GST-LPA5 or GST recombinant proteins were overlaid onto membranes containing recombinant NHERF1, NHERF2, and PDZK1. The bottom panel: Ponceau-S—stained membrane. (B) GST-LPA5 bound to the second PDZ domain, PDZ2, of NHERF2. The bottom panel: Ponceau-S. (C) Lysates from PS120/NHERF1 and PS120/NHERF2 cells transfected LPA5 or pcDNA were immunoprecipitated with either an anti-NHERF1 or anti-NHERF2 antibody. The presence of co-immunoprecipitated LPA5 was detected with an anti-HA antibody. The left 2 lanes show the expression of HA-LPA5 in transfected cells. Representative data from 3 independent experiments are shown.
Next, we confirmed the interaction between LPA5 and NHERF2 in PS120 cells by co-immunoprecipitation. HA-LPA5 was co-immunoprecipitated in PS120/NHE3V/NHERF2 (Figure 4C, lane 7), but not from PS120/NHE3V/NHERF1 cells (lane 5), confirming its interaction with NHERF2.
LPA5-Mediated Regulation of NHE3 Requires NHERF2
To determine the functional relevance of NHERF2 in LPA5-mediated NHE3 regulation, we again utilized PS120/NHE3V cells. Treatment with LPA showed no effect on NHE3 activity in PS120/NHE3V cells (Figure 5A). Ectopic expression of LPA5 did not result in LPA-induced activation of NHE3 (Figure 5B). Because LPA5 interacts with NHERF2, we co-expressed LPA5 and NHERF2 (PS120/NHE3V/LPA5/NHERF2), and observed that co-expression conferred stimulation of NHE3 by LPA (Figure 5C). On the contrary, co-expression of LPA5 and NHERF1 could not reproduce activation of NHE3 by LPA, consistent with the inability of LPA5 to bind NHERF1 (Figure 5D). Similarly, the presence of NHERF2 alone was not sufficient to reconstitute the LPA-mediated activation of NHE3 (Figure 5E). Together with previous studies showing that NHE3 interacts with NHERF2,17 these results demonstrate that NHERF2 forms a functional basis for LPA5-dependent regulation of NHE3.
Figure 5.

NHERF2 is necessary for LPA5-mediated stimulation NHE3. NHE3 activity in the absence of presence of LPA was determined. (A) PS120/NHE3V. (B) PS120/NHE3V/LPA5. (C) PS120/NHE3V/LPA5/NHERF2. (D) PS120/NHE3V/LPA5/ NHERF1. (E) PS120/NHE3V/NHERF2. n > 6. *P < .001 compared with the control.
Absence of NHERF2 Abrogates LPA-Mediated Fluid Absorption
Having demonstrated the necessity of NHERF2 in stimulation of NHE3 activity by LPA in PS120 cells, we sought to determine the importance of NHERF2 for LPA-induced fluid absorption in vivo. The rate of fluid absorption of Nherf2−/− mice was comparable to that of wild-type (WT) indicating that the basal absorptive function is unperturbed. However, the presence of LPA (20 μM) in the perfusion buffer did not alter the rate of fluid absorption in Nherf2−/− mice (Figure 6A). To affirm the above results, we performed the similar experiment on Nherf2−/− mice on the FVB/N background with increased LPA concentration (50 μM). Again no change in fluid absorption by LPA was noted in Nherf2−/− mice (Figure 6B). The deficiency in LPA-mediated fluid absorption in Nherf2−/− mice was further corroborated under the condition of CT treatment. CT inhibited fluid absorption in Nherf2−/− mice, revealing that the adenylate cyclase-PKA cascade is intact in Nherf2−/− mice (Figure 6A, right half). However, LPA again failed to alter fluid absorption in the intestine, demonstrating that NHERF2 is necessary for LPA-elicited fluid absorption.
LPA2 Is Not Necessary for LPA-Mediated Fluid Absorption
Recently, it was shown that LPA2-mediated signaling regulated the anion channel CFTR.9 In contrast, in our studies using Caco-2 or C2b cells in which LPA2 is the major LPA receptor, NHE3 activity did not respond to LPA. The role of LPA2 in Na+ absorption was re-examined by determining LPA-elicited fluid absorption in Lpa2−/− mice (Figure 7). As in WT mice, LPA induced an increase in the rate of fluid absorption in Lpa2−/− intestine both in the basal state (53.5 ± 4.6 μL/cm/h from 47.0 ± 2.4 μL/cm/h) and after CT treatment (39.0 ± 2.9 μL/cm/h from 27.8 ± 4.8 μL/cm/h). These results indicate that LPA2 is not obligatory for LPA-mediated increase in intestinal fluid absorption.
Figure 7.
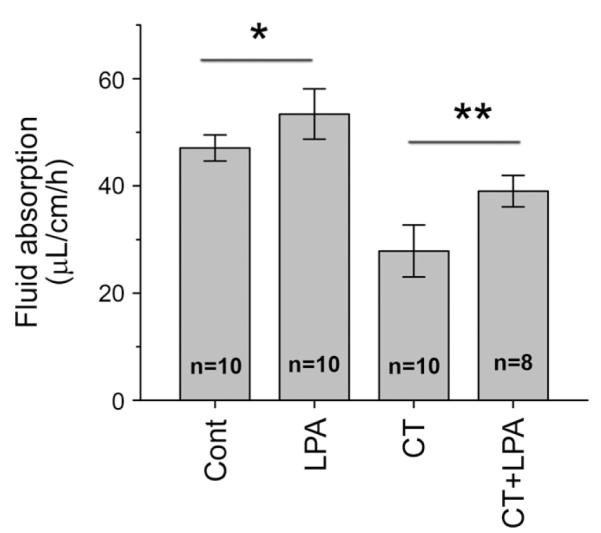
LPA2 is not necessary for stimulation of fluid absorption by LPA. Effects of LPA on the rate of fluid absorption in Lpa2−/− mice under basal or cholera toxin (CT)-treated conditions are shown. *P < .04; **P < .01.
LPA Increases the Abundance of NHE3 Protein at the Cell Surface in an NHERF2-Dependent Fashion in a Heterologous Expression System as Well as In Vivo
Regulation of NHE3 often involves trafficking of NHE3 proteins between the surface membrane and the intracellular pool. To further delineate the mechanism of NHE3 regulation by LPA, we determined whether LPA alters the surface abundance of NHE3 protein initially in PS120 cells by using surface biotinylation. Figure 8A shows that ectopic expression of LPA5 in PS120/NHE3V/NHERF2 cells resulted in a marked increase (45%) in NHE3 surface abundance, but not in control cells transfected with pcDNA. Consistent with the effect on NHE3 activity, there was no change in NHE3 surface expression in the absence of LPA5 (PS120/NHE3V/NHERF2/pcDNA) or when NHERF2 was replaced with NHERF1 despite the presence of LPA5 (PS120/NHE3V/NHERF1/ LPA5).
Figure 8.

LPA induced NHE3 trafficking within the brush border membrane (BBM) of wild-type (WT) and Nherf2−/− mice. (A) PS120NHE3 derivatives were treated with LPA, and the amount of NHE3V protein in the plasma membrane was determined by surface biotinylation. The amount of surface NHE3 (S-NHE3) was normalized to total NHE3 (T-NHE3). n = 4. *P <.01 compare with control treatment. (B) Immunofluorescence labels of NHE3 (green), F-actin (red), and nuclei (blue) in the ileum of WT and Nherf2−/− mice are shown. (C) NHE3 fluorescence intensity in the respective 1 μM microvillar (MV) and terminal web (TW) zones during control conditions (left panel) and after LPA perfusion in WT mice is shown. Confocal analysis of LPA-induced NHE3 trafficking was performed as described in Supplementary Figure 2. *P < .05. (D) Effects of LPA on the relative NHE3 expression in MV and TW of WT and Nherf2−/− mice are shown. *P < .05; **P < .01. (E)A much higher “LPA translocation quotient” in WT compared to Nherf2−/− mice is shown. For (C—E), BBMs were analyzed in approximately 10 randomly selected areas of 3 individual sections from each mouse, and 3 pairs of WT and Nherf2−/− mice were analyzed. *P <.05.
Having shown the changes in NHE3 abundance in cultured cells, we next studied LPA-mediated NHE3 trafficking and its dependency on NHERF2 expression in vivo. The terminal 3-cm ileum of anesthetized Nherf2−/− or WT mice was perfused with 50 μM LPA or saline for 20 minutes, the tissue excised and stained for NHE3 and F-actin. NHE3 immunofluorescence intensity was quantified in the most internal part (terminal web) and the most external (tip of the microvilli) 1 μm brush border membrane—cytoskeletal network, as illustrated in detail in Supplementary Figure 2A. Figure 8B shows immunofluorescence labeling of mouse small intestine villus cells. Under control conditions, a much higher percentage of NHE3 was found in the terminal web region than the microvillus tip region (Figure 8B, upper part of upper panel, and Figure 8C, left bars). After LPA perfusion, the percentage of NHE3 in the tip of the microvilli increased markedly, while the amount of NHE3 in the terminal web did not significantly decrease due to trafficking of NHE3 from the subapical vesicle pool into the terminal web region. In the Nherf2−/− mice (Figure 8B, upper part of lower panel), the ratio of NHE3 fluorescence intensity at the tip of the microvilli to the terminal web was ~2-fold greater compared with WT (Figure 8D), but after LPA perfusion, a significantly less increase of NHE3 in relation to the NHE3 in the terminal web was seen compared with WT. The actual “LPA-dependent translocation quotient” was approximately 7-fold higher in WT compared to Nherf2−/− (Figure 8E). These results indicate that NHERF2 is required for LPA-induced NHE3 trafficking.
We further corroborated the changes in NHE3 expression by immunohistological analysis of intestinal tissue sections from the same intestines used in the perfusion study (Supplementary Figure 3) to correlate the changes in fluid absorption and NHE3 expression. LPA increased NHE3 abundance in WT and Lpa2−/− mice compared with controls, but no significant change was observed in Nherf2−/− mice. Similarly, LPA treatment resulted in a significant increase in NHE3 intensity in CT-treated WT and Lpa2−/− intestine, but not in Nherf2−/− intestine (Supplementary Figure 3). These data show that LPA stimulates NHE3 activity and fluid absorption in part by modulating NHE3 expression at the microvilli.
Discussion
In the present study, we investigated the effect of LPA on NHE3 activity and NHE3-mediated fluid absorption in the intestine. We found that LPA is a potent stimulator of Na+ and fluid absorption. The presence of LPA is able to overcome diarrhea induced by CT or TNF-α, suggesting its potential therapeutic usage. LPA specifically acted on NHE3, but not on NHE1 or NHE2. Moreover, this regulation was dependent on the presence of NHERF2 as demonstrated both in vitro and in vivo. Our study identifies LPA5 as the major LPA receptor modulating NHE3 activity in the intestine based on at least 3 observations. First, upon ectopic expression of LPA5, LPA stimulated NHE3 activity in both PS120 and Caco-2 cells. Second, the lack of NHE3 regulation in C2b and Caco-2 cells that express high levels of LPA2 and the preservation of fluid absorption in the intestine of LPA2−/− mice ruled out a role for LPA2. Third, NHERF2 interacted with LPA5 and the presence of NHERF2 was necessary for the stimulation of NHE3 by LPA. To our knowledge, this is the first study demonstrating a physiological function of LPA5.
LPA5 is an atypical LPA receptor with ~22% identity to the classical LPA receptors, LPA1–3, but displays ~35% identity with LPA4.7 Expression of LPA5 is high in the small intestine and colon, but low in most other tissues, including the brain, liver, lung, skin, spleen, stomach, and thymus.6,7 By real-time RT-PCR we confirmed the high expression of LPA5 in mouse small intestine and IEC6 cells. Interestingly, we found that the level of LPA5 was low in colon cancer cells, such as Caco-2 (Figure 3), HCT1116, and SW480 cells (Zhang and Yun, unpublished data). Although most of G-protein—coupled receptors respond to a single endogenous ligand, LPA5 has been shown to be activated by a number of lipid-derived molecules, including LPA, farnesyl pyrophosphate, N-arachidonylglycine, and peptone, which mimics protein digest in the luminal chime.6,19 In addition to the synthesis via hydrolysis of phospholipids, LPA can be derived from foods, such as soybean and hen egg.4,20-23 Previous studies have shown that a meal stimulates the Na+/H+ exchanger NHE3, and that Na+/H+ exchange plays a major role in the postprandial increase in water and electrolyte absorption in the ileum.24 In view of the activation of LPA5 by peptone, it remains an intriguing possibility that LPA5 functions as the sensory mechanism on the surface of enterocytes, which responds to dietary intake and regulates NHE3 as part of normal physiological response to a meal.
The effect of LPA on NHE3 was shown previously using OK cells.11 However, there is a notable difference between this report and our study. In the aforementioned study, OK cells were treated with a high concentration of LPA (100 μM), whereas we saw the stimulatory effect using 1 μM within 5 minutes. Although high concentrations of LPA have been reported, such occurrences are limited to patients with epithelial ovarian cancer.25 In addition, OK cells show predominant expression of LPA1 (Zhang and Yun, unpublished data) and, hence, the specific pathways elicited by LPA in OK cells remain to be elucidated.
The physiological and cellular functions of LPA2 have gained increasing attention in recent years. We reported recently that the absence of LPA2 in the mouse gut protected the animals from chemically induced colonic dysplasia.13 In addition, LPA was shown to inhibit CFTR-dependent Cl− and fluid secretion in WT mouse intestine, but not in Lpa2−/− intestine.9 In the present work, LPA did not affect NHE3 activity in Caco-2 or C2b/hNHE3V cells, which have elevated expression of LPA2.8 The lack of NHE3 regulation in Caco-2 cells implies that LPA2 does not significantly contribute to NHE3 regulation. Moreover, the absence of LPA2 did not abrogate the NHE3-dependent stimulatory effect of LPA on fluid absorption, suggesting that LPA receptor(s) other than LPA2 must mediate NHE3-dependent fluid absorption. Although these findings appear to contradict the previous study on the effect of LPA on CFTR,9 the disparity in regulation of CFTR and NHE3 can be attributed to the regional difference in the expression of these proteins. There is a division of absorptive and secretory functions along the villus-crypt axis in the intestine.1 Although this division is not absolute, NHE3 expression is predominant in the villi, whereas CFTR is most abundant in the crypts. It was shown previously that LPA2 and CFTR mRNA expression is enriched in the crypt region.10 On the other hand, Choi et al6 found LPA5 mRNA level to be highest in the villus tip of the brush border where NHE3 is expressed. Therefore, the lack of effect by LPA on NHE3-mediated fluid absorption in Lpa2−/− mice can partly be attributed to the difference in the expression of NHE3/LPA5 and CFTR/LPA2 along the villus-crypt axis.
In the intestine, 4 members of the NHERF family protein are present.2,26 The mechanisms by which the NHERF proteins affect the functions of NHE3 and other transporters are primarily based on heterologous expression studies, but the recent generation of mice lacking 1 or more of these proteins have revealed subtle and yet significant differences in the functions of these proteins in vivo.10,16,27,28 In the current study, LPA5 specifically interacted with NHERF2, but not with NHERF1 or PDZK1. The importance of NHERF2 interaction was demonstrated using heterologous expression in which LPA5 stimulates NHE3 only in the presence of NHERF2 (Figure 5). Recently, we reported that the absence of NHERF2 abrogates LPA-mediated inhibition of anion secretion by CFTR in mouse intestine.10 Similarly, LPA cannot modulate NHE3 activity in Nherf2−/− mice, suggesting an obligatory role of NHERF2. As discussed earlier, we infer that the LPA-elicited modulation of Cl− secretion by CFTR and Na+ absorption by NHE3 is mediated by LPA2 and LPA5, respectively, both of which bind to NHERF2. Although an animal deficient in LPA5 expression is necessary for a definitive proof of LPA5-mediated fluid absorption in the intestine, the absence of an effect of LPA in Nherf2−/− mice provides credible evidence that LPA5 is the major LPA receptor regulating NHE3. NHERF2 interaction is limited to LPA2 and LPA5, but not LPA1 or LPA3.8,29 The absence of an effect in Nherf2−/− mice precludes LPA1 and LPA3 in LPA-mediated fluid absorption based on their inability to bind NHERF2. Finally, LPA had normal effects on fluid absorption in the Lpa2−/− mice (Figure 7), and LPA activation of NHE3 in Caco-2 cells required the presence of LPA5 but not LPA2. We do not know the reason for the insufficiency of LPA2 to regulate NHE3 despite its ability to bind NHERF2. It seems unlikely that simple binding to NHERF2 is a determinant for the specific effect of LPA5 vs LPA2. Hence, we suggest that the presence of NHERF2 and distinct elements of LPA5 permits the assembly of signaling proteins that enables regulation of NHE3 activity. Further investigation is needed to decipher the underlying molecular mechanisms.
The activity of NHE3 at the apical membrane of intestinal epithelial cells is modulated by a number of mechanisms, including protein—protein interaction, trafficking, phosphorylation, and membrane locale.2 Our data show that LPA stimulates NHE3 activity in part via altering its membrane locale and trafficking. The surface NHE3 abundance and NHE3 transport activity were concomitantly elevated in PS120 cells only when both LPA5 and NHERF2 were coexpressed. In the intact ileal tissues, a shift in NHE3 protein location was observed from the terminal web at the base of the microvilli to the tips of microvilli in response to LPA. Surprisingly, LPA induced trafficking of NHE3 protein to the tips of microvilli to some extent in Nherf2−/− mice, although the shift was much less distinct and diffuse compared with WT. This change in the Nherf2−/− ileum is clearly disengaged from the lack of changes in NHE3-dependent fluid absorption. The reason behind this discrepancy is not clear, but it suggests that trafficking of NHE3 protein to the microvilli is not the only step in the functional activation of NHE3 transport activity by LPA.
One important finding of the current study is that LPA stimulates fluid absorption in the mouse bowel exposed to CT or TNF-α, implicating the utility of LPA as a potential antidiarrheal therapy and its use in an oral rehydration solution. However, there is a practical concern over the stability of LPA in the lumen of the gut. LPA can be dephosphorylated by phosphatase or deacylated by phospholipases.30 However, at least 2 studies showed that orally administered LPA is effective in mediating its biological effects in the small intestine and colon.9,13 In addition, there is a continuing effort to synthesize metabolically inert LPA analogues.31 Because of the plethora of biological effects mediated by LPA, one has to be concerned with undesirable effects of LPA, such as the effects on cell survival and tumor promoting properties. In a recent study, we orally administered LPA for 4 weeks to WT and mice heterozygous for the adenomatous polypopsis coli (Apc) allele (ApcMin/+); LPA promoted tumor progression in Apcmin/+ mice, but not in WT mice.13 This study suggests that LPA is not sufficient to cause cancer by itself in the absence of a genetic predisposition to develop cancer. Moreover, use of LPA in an oral rehydration solution will be relatively short, lasting only several days. Therefore, together with the previous report that LPA inhibits CFTR,9 our study suggests that there is a clinical potential for LPA against diarrheal syndromes.
Supplementary Material
Acknowledgments
Funding This work was funded by National Institutes of Health grants DK061416 (CCY), MH51699 and HD50685 (JC) and by DFG SFB621-C9 (US). SL was supported by the Crohn’s and Colitis Foundation of America. PH was supported by the American Heart Association. AS and MC were supported by postdoctoral stipends of the Hannover biomedical Research School that were funded by the DAAD and Abbot GmbH via the “matching fund” program. We thank the Emory Digestive Disease Research Development Center (supported by DK064399) for the use of light microscope and the Zeiss 510 confocal microscope and the Volkswagen Stiftung for funding of the Leica LSM.
Abbreviations used in this paper
- Apc
adenomatous polypopsis coli
- BSA
bovine serum albumin
- CT
cholera toxin
- CFTR
cystic fibrosis transmembrane conductance regulator
- LPA
lysophosphatidic acid
- mRNA
messenger RNA
- NHE3
Na+/H+ exchanger type 3
- NHERF
Na+/H+ exchanger regulatory factor
- OK
opossum kidney
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PDZ
postsynaptic density 95, discs large, and zonula occludens–1
- PDZK1
PDZ domain containing protein in kidney 1
- RT
reverse transcription
- TNF-α
tumor necrosis factor—α
- WT
wild-type.
Footnotes
Supplementary Materials Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2009.09.055.
Conflicts of interest The authors disclose no conflicts.
References
- 1.Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931–943. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchanger. Ann Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 3.Schultheis PJ, Clarke LL, Meneton P, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 4.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 5.Noguchi K, Herr D, Mutoh T, et al. Lysophosphatidic acid (LPA) and its receptors. Curr Opin Pharmacol. 2009;9:15–23. doi: 10.1016/j.coph.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Choi S, Lee M, Shiu AL, et al. Identification of a protein hydrolysate responsive G protein-coupled receptor in enterocytes. Am J Physiol. 2007;292:G98–G112. doi: 10.1152/ajpgi.00295.2006. [DOI] [PubMed] [Google Scholar]
- 7.Lee C-W, Rivera R, Gardell S, et al. GPR92 as a New G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J Biol Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 8.Yun CC, Sun H, Wang D, et al. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol. 2005;289:C2–C11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Dandridge KS, Di A, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh AK, Riederer B, Krabbenhoft A, et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J Clin Invest. 2009;119:540–550. doi: 10.1172/JCI35541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee-Kwon W, Kawano K, Choi JW, et al. Lysophosphatidic acid stimulates brush border Na+/H+ exchanger 3 (NHE3) activity by increasing its exocytosis by an NHE3 kinase A regulatory protein-dependent mechanism. J Biol Chem. 2003;278:16494–16501. doi: 10.1074/jbc.M300580200. [DOI] [PubMed] [Google Scholar]
- 12.Choi JW, Lee-Kwon W, Jeon ES, et al. Lysophosphatidic acid induces exocytic trafficking of Na+/H+ exchanger 3 by E3KARP-dependent activation of phospholipase C. Biochim Biophys Acta. 2004;1683:59–68. doi: 10.1016/j.bbalip.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Lin S, Wang D, Iyer S, et al. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology. 2009;136:1711–1720. doi: 10.1053/j.gastro.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yun CC, Chen Y, Lang F. Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J Biol Chem. 2002;277:7676–7683. doi: 10.1074/jbc.M107768200. [DOI] [PubMed] [Google Scholar]
- 15.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cinar A, Chen MM, Riederer B, et al. Nhe3 inhibition by cAMP and Ca2+ is abolished in PDZ-domain protein PDZK1-deficient murin enterocytes. J Physiol. 2007;581:1235–1246. doi: 10.1113/jphysiol.2007.131722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yun CC, Oh S, Zizak M, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci U S A. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gisler SM, Pribanic S, Bacic D, et al. PDZK1: I. A major scaffolder in brush borders of proximal tubular cells1. Kidney Int. 2003;64:1733–1745. doi: 10.1046/j.1523-1755.2003.00266.x. [DOI] [PubMed] [Google Scholar]
- 19.Oh DY, Yoon JM, Moon MJ, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054–21064. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyazawa D, Ikemoto A, Fujii Y, et al. Dietary alpha-linolenic acid suppresses the formation of lysophosphatidic acid, a lipid mediator, in rat platelets compared with linoleic acid. Life Sci. 2003;73:2083–2090. doi: 10.1016/s0024-3205(03)00564-2. [DOI] [PubMed] [Google Scholar]
- 21.Nakane S, Tokumura A, Waku K, et al. Hen egg yolk and white contain high amounts of lysophosphatidic acids, growth factor-like lipids: distinct molecular species compositions. Lipids. 2001;36:413–419. doi: 10.1007/s11745-001-0737-1. [DOI] [PubMed] [Google Scholar]
- 22.Tokumura A, Kanaya Y, Kitahara M, et al. Increased formation of lysophosphatidic acids by lysophospholipase D in serum of hypercholesterolemic rabbits. J Lipid Res. 2002;43:307–315. [PubMed] [Google Scholar]
- 23.Aoki J, Taira A, Takanezawa Y, et al. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 24.Maher MM, Gontarek JD, Jimenez RE, et al. Role of brush border Na+/H+ exchange in canine ileal absorption. Dig Dis Sci. 1996;41:651–659. doi: 10.1007/BF02213119. [DOI] [PubMed] [Google Scholar]
- 25.Mills GB, May C, McGill M, et al. A putative new growth factor in ascitic fluid from ovarian cancer patients: identification, characterization, and mechanism of action. Cancer Res. 1988;48:1066–1071. [PubMed] [Google Scholar]
- 26.Lamprecht G, Seidler U. The emerging role of PDZ adapter proteins for regulation of intestinal ion transport. Am J Physiol. 2006;291:G766–G777. doi: 10.1152/ajpgi.00135.2006. [DOI] [PubMed] [Google Scholar]
- 27.Shenolikar S, Voltz JW, Minkoff CM, et al. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci U S A. 2002;8:8. doi: 10.1073/pnas.162232699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broere N, Chen M, Cinar A, et al. Defective jejunal and colonic salt absorption and altered Na+/H+ exchanger 3 (NHE3) activity in NHE regulatory factor 1 (NHERF1) adaptor protein-deficient mice. Pflugers Arch. 2009;457:1079–1091. doi: 10.1007/s00424-008-0579-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh YS, Jo NW, Choi JW, et al. NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-beta3 activation. Mol Cell Biol. 2004;24:5069–5079. doi: 10.1128/MCB.24.11.5069-5079.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiroyama M, Takenawa T. Isolation of a cDNA encoding human lysophosphatidic acid phosphatase that is involved in the regulation of mitochondrial lipid biosynthesis. J Biol Chem. 1999;274:29172–29180. doi: 10.1074/jbc.274.41.29172. [DOI] [PubMed] [Google Scholar]
- 31.Deng W, Shuyu E, Tsukahara R, et al. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology. 2007;132:1834–1851. doi: 10.1053/j.gastro.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.