

Abstract
In drug development, phase 1 first-in-human studies represent a major milestone as the drug moves from preclinical discovery to clinical development activities. The safety of human subjects is paramount to the conduct of these studies and regulatory considerations guide activities. Forces of evolution on the pharmaceutical industry are re-shaping the first-in-human dose selection strategy. Namely, high attrition rates in part due to lack of efficacy have led to the re-organization of research and development organizations around the umbrella of translational research. Translational research strives to bring basic research advances into the clinic and support the reverse transfer of information to enhance compound selection strategies. Pharmacokinetic/pharmacodynamic (PK/PD) modeling holds a unique position in translational research by attempting to integrate diverse sets of information. PK/PD modeling has demonstrated utility in dose selection and trial design for later stages of drug development and is now being employed with greater prevalence in the translational research setting to manage risk (i.e., oncology and inflammation/immunology). Moving from empirical Emax models to more mechanistic representations of the biological system, a higher fidelity of human predictions is expected. Strategies that have proven useful for PK predictions are being applied to PK/PD predictions. This review article examines examples of the application of PK/PD modeling in establishing target concentrations for supporting first-in-human study design.
Keywords: biomarker, drug development, pharmacodynamics, pharmacokinetics, PK/PD, translation
INTRODUCTION
Phase 1 studies involving the first-in-human dose administration represent the transition from preclinical studies to the clinic. The studies are designed to capture fundamental properties of a new chemical entity or biologic such as safety, tolerability, pharmacokinetics (PK), and in the case of biologics, immunogenicity while minimizing the potential risk to subjects. A renewed emphasis of phase 1 study design is being recognized in part due to the need to reduce attrition in later phases of development, decreasing number of innovative new medicines, and impending patent expiration for major products (1–3). Higher upfront investment to de-risk projects prior to advancement to later, more costly phases will be important to improve R&D productivity (2). Inclusion of pharmacodynamic measures (both biomarkers and exploratory endpoints) in early clinical studies has become critical for building confidence, defining mechanism of action, and supporting earlier decisions for projects which merit further investment (4,5). In the model-based drug development paradigm, this information can be leveraged by using mathematical models to inform the study design for future studies (6).
The significance of transition from preclinical to clinical studies has been recognized leading to the re-organization of research units into translational groups bringing involved disciplines closer together. The intent is to leverage basic science to better support the translation of in vitro and in vivo preclinical data into clinical decisions (7,8). In addition, the reverse transfer is encouraged such as using clinical information from lead molecules to inform discovery strategies. The challenges of animal to human predictions with existing disease models and shift away from precedented mechanisms to novel therapeutic approaches has increased the demand for biomarker development (9). Biomarkers have been broadly characterized into target, mechanism, and outcome categories (5,10). With a focus on strategies to increase confidence in the molecule or the approach early in clinical development, it is envisioned that better use of resources will follow improving the delivery of novel therapeutics to patients.
Several work streams converge providing important inputs in the consideration of first-in-human study design. Work streams include research pharmacology, preclinical safety evaluation, risk assessment, and biomarkers (Fig. 1). Translational research pharmacology groups define the mechanism of action of a new therapeutic through in vitro and in vivo studies. In addition, key differences across species are defined in target expression and affinity of the drug for the target. Preclinical safety studies establish the patterns of toxicity arising from drugs after the intended route of administration in animal species. In addition, doses and exposures associated with toxicity are defined, serving as upper limits to the concentration range to be explored in clinical studies involving healthy volunteers. Risk assessment involves gathering available information about the pharmacological target and related mechanisms, and the available compound safety database to define the level of caution around first-in-human dose selection. Molecular scientists help to define the strategy with respect to biomarker selection and validation, building confidence that a robust signal of drug effect can be defined. The inputs from the above work streams sets the foundation upon which a dose selection rationale can be proposed. The PK/PD model serves as a tool to integrate a wide range of data and facilitate the exchange of information.
Fig. 1.

Work streams contributing to first-in-human study design
Application of the target concentration approach in drug development has been described and is a fundamental concept in translational PK/PD efforts (11). Key steps in the target concentration approach include selection of a target effect, defining the target concentration associated with target effect, and use of predicted PK and PK/PD parameters to calculate a dosing regimen to maintain the target concentration for a chronic dosing regimen. Recent publications demonstrate the use of the target concentration approach in the prediction of human dose regimens (12,13). Continued advancement of computational tools and translational focus in drug discovery have increased opportunities for physiological or mechanism-based PK/PD approaches to be applied much earlier in the lead optimization phase in discovery (14–16). These activities help to build confidence in the mechanism of action, demonstrate the robustness of the pharmacological measurement, and enable in vitro to in vivo potency comparisons which guide PK/PD optimization.
The integration of successful translational research efforts is embodied in the prediction of human target concentrations. Early selection of the target effect is important to facilitate compound selection. Predictions of human PK need to be merged with PK/PD relationships to predict a dose having the greatest likelihood of achieving the target effect. In the context of reducing attrition, this may lead to more efficient drug development by rationally selecting compounds with the greatest chance of success, and discarding compounds which are unable to produce desired concentrations due to PK or toxicity profiles. The relationship between concentration and effect is derived from responses measured in animal models. Where possible, mechanism-based PK/PD models are developed to describe the system and the impact of drug treatment on underlying physiology. These models have shown utility in helping to bridge preclinical experience to effective human doses and concentrations to be explored in early clinical studies. The objective of this review is to provide examples of the application of preclinical PK/PD modeling highlighting translational efforts in drug development as a rational basis for dose selection in first-in-human studies.
Regulatory Considerations
Given the importance of safe guarding human subjects, regulatory guidance documents have been issued by the Food and Drug Administration (FDA) and the Committee for Medicinal Products of the European Medicines Agency (EMEA). The intent of these guidance documents related to first-in-human starting dose is to provide a robust scientific rationale for the dose selection to avoid toxicity in the first dose cohort. An overview of the starting dose recommendation based on FDA and EMEA regulatory guidance documents is shown in Fig. 2. It is recognized that the appropriate starting dose will balance the need to limit risk for the subject population, but also dose escalate to therapeutic concentrations in an efficient manner (17). An algorithm to estimate the maximum recommended starting dose (MRSD) for first-in-human studies in healthy volunteers was proposed for new drugs and biologics (18). The document provides a basis for the consistent use of terminology, scaling factors across species, and overall strategy. Of note, two parallel approaches are envisioned to the selection of starting dose. The first involves scaling of doses to calculate the MRSD, while the other is based on animal exposures and modeling. The major elements of the MRSD process include the determination of the no observed adverse effect level (NOAEL) in animal species, conversion of NOAEL to the human equivalent dose (HED) through the application of scaling factors, selection of the most appropriate species, and use of a safety factor. The safety factor is to provide an additional margin of safety for humans given the possibility that humans could be more sensitive to the toxic effects of a drug.
Fig. 2.

Overview of starting dose recommendation based on FDA and EMEA regulatory guidance documents
An alternative to scaling the NOAEL dose involves the use of the pharmacologically active dose (PAD). This approach may be particularly useful when the effects in humans may arise from exaggerated pharmacology (i.e., anticoagulants, vasodilators, and biologics). Scaling the PAD to a HED may give rise to an estimate that is lower than the MRSD derived from the NOAEL. In particular, demonstration of a robust dose–response relationship is needed and consideration of the translation of the animal response to humans is integral to the use of PAD.
In the aftermath of the TGN1412 case, the EMEA issued a guidance document on first in human dose selection which emphasized identification of factors influencing risk (19). In situations where the translation of toxicological data from preclinical species to human is uncertain additional considerations are needed. Knowledge of mode of action, nature of the pharmacological target, and relevance of animal models are additional considerations in assessing the potential for severe adverse reactions. The concept of the minimum anticipated biological effect level (MABEL) approach was introduced for medicinal products which fall into the high risk category. The MABEL approach integrates pharmacology and toxicology information into the selection of the first-in-human starting dose. In particular, use of PK/PD models is suggested to integrate available in vitro and in vivo information. The specific types of information to be integrated included (a) in vitro studies to characterize binding and affinity to the pharmacological target, (b) in vitro concentration–response evaluation in test species and human cellular systems, and (c) concentration–response in animal models.
In the discovery setting, PK/PD models are being developed and used to integrate in vitro and in vivo data to predict efficacious plasma concentrations as part of candidate selection process during lead optimization (20). This additional effort, while not always a requirement, may help to prioritize lead molecules with respect to PK/PD properties, give early information about dosing requirements prior to or in parallel with the collection of safety data, and support the selection of optimal dosing schedules.
Small Molecule PK Predictions
The development and refinement of PK prediction methods have continued to advance, in part leading to a reduction in compound failure rates during early development due to unacceptable PK properties over the last decade (1). PK predictions define the dose–concentration relationship necessary for guiding dose selection and are based on in vitro and in vivo data. The key parameters to be defined by PK predictions include clearance, bioavailability and volume of distribution. For drugs which are intended to be given orally, clearance and bioavailability are the determinants of the resulting area under the concentration-time curve for a given dose. Many small molecule drugs (MW < 1,000 Da) are cleared by hepatic metabolism. For the prediction of hepatic clearance, in vitro methods have been proposed including the use of hepatic subcellular fractions such as microsomes, hepatocytes, and expressed enzymes (21). Fundamental differences in protein binding, blood flow, and organ size can be included in the well-stirred model of hepatic clearance to predict hepatic clearance. The validity of in vitro predictions can be assessed within each species to build confidence in the prediction method.
In vivo methods utilize PK data collected in one or more species to scale-up human predicted PK parameters based on allometric principles (22,23). The underlying principle is that physiological functions can be scaled across a wide range of body sizes. The power law function that is typically applied to allometric scaling is shown in Eq. 1:
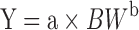 |
1 |
where Y is the predicted parameter, a is the intercept, BW is body weight, and b is the exponent. The value of the exponent for clearance and volume of distribution scaling tend to be 0.75 and 1, respectively. It is also possible to estimate the exponent for a particular parameter when data from several species are available. Whole-body physiologically based PK (PBPK) models combine in vitro and in vivo data. PBPK models reflect major tissues and compartments in the body on the basis of flow models. Model parameters based on PK data in preclinical species is replaced with human values with assumptions around tissue distribution. PBPK approaches have been applied to both small molecules and monoclonal antibodies (24,25).
PK prediction methods attempt to predict human clearance or oral clearance within a factor of 2-fold. In a large retrospective analysis including 50 development compounds for which prediction methods were employed using a standardized single-species scaling approach, the human oral clearance predictions were within twofold of predicted for approximately 32–46% of the cases depending on the species chosen (26).
Large Molecule PK Predictions
For monoclonal antibodies, physical size (∼150 kDa) dictates many disposition properties simplifying PK predictions (27). Often, extravascular distribution of antibodies is limited, and volume of distribution is found to be similar to plasma volume (40 mL/kg) (27). Elimination is in part controlled by catabolism by the reticuloendothelial system and recycling after binding to the FcRn receptor. Recent examples from the literature suggest a high success rate in the prediction of clearance for antibodies with linear elimination properties using monkey pharmacokinetic data and fixed exponent values (28,29).
Other factors influencing the disposition of antibodies involve the specific interaction with the pharmacological target (soluble ligand or receptor). The binding of the antibody with the pharmacological target can modulate the apparent volume of distribution and removal of the complex can enhance clearance in a dose- and concentration-dependent manner and has been referred to as target-mediated drug disposition (30). In cases where pharmacodynamic data on the target interaction can be obtained, the target-mediated drug disposition model (31,32) may serve as a basis for interspecies PK predictions enabling the inclusion of known species differences in target expression and affinity to be explicitly recognized in the model. Alternatively, for cases where pharmacodynamic information on the interaction with the target cannot be gathered, allometric scaling of available PK data may be utilized. Immunogenicity is another factor which can confound the interpretation of preclinical PK data, and may lead to enhanced elimination of the antibody in some cases (33,34). Generally, antibody-positive animals are excluded from the prediction of human clearance because immunogenicity in animals is a poor predictor of immunogenicity in humans.
PK/PD Predictions
In addition to the need for successful PK predictions, a new emphasis on successful pharmacodynamic predictions is emerging. Driven by the activities of the translational medicine initiatives, the development of biomarkers to detect drug responses early in development have lead to more opportunities to integrate PK and pharmacodynamic understanding into formal PK/PD models of drug effects. The value of PK/PD modeling to support dose selection in later phases of drug development is well-recognized (35) with examples of the PK/PD modeling applications in discovery are being reported with greater frequency. For the product of PK predictions to be fully leveraged, the relationship between concentration and effect must be understood (36). Knowledge of the concentration–effect relationship enables the time-course of effect to be defined. While the prediction of human PK parameters involves similar parameters for each new compound (i.e., clearance, volume of distribution, and bioavailability), the nature of PK/PD predictions will depend on the pharmacological target and biomarker upon which the prediction is based. In general, the major components of PK/PD predictions can be classified into two areas, drug-specific and system-specific parameters (37). The drug-specific parameters refer to the biological interaction between the drug and its pharmacological target. The system-specific parameters describe the physiology of the biological system which has been perturbed by drug administration. A wide range of mechanism-based PK/PD models exists for application to in vivo models (38). Examples of these models have been applied to a range of efficacy models across therapeutic areas as highlighted in Table I. A third area where predictions are needed involves the study of the interrelationship between biomarkers and outcome. This information may come from clinical or preclinical studies, and is critical when informing dose selection using a biomarker.
Table I.
Examples of commonly used animal models used for pharmacodynamic evaluation of new chemical entities or biologicals (adapted with permission from (57))
| Therapeutic Area | Examples of in vivo models | Biological endpoint | Therapeutic agents evaluated | References |
|---|---|---|---|---|
| Oncology | Xenograft models | Tumor volume | Cytotoxics, and receptor tyrosine kinase inhibitiors | (48) |
| Angiogenesis | Vascular permeability | VEGF receptor inhibitors | (58) | |
| Hematology | Normal animals | Reticulotcytes, RBC, WBC, hemoglobin, and lymphocytes | Erythropoiesis-stimulating agents, and granulocyte colony stimulating factor | (50) |
| Chemotherapy-induced neutropenia/anemia | WBC, RBC and hemoglobin | |||
| Diabetes | Glucose tolerance test performed in normal animals | Glucose, insulin, and other biomarkers | DPP-4 inhibitors, thiazolidinediones, metformin | (59) |
| Obese-hyperglycemic mice (ob/ob mice) | ||||
| Zucker fatty rats | ||||
| Central Nervous System | 5-HTP potentiation | 5-HTP potentiated behavior score | Anti-depressants | (60) |
| Conditioned avoidance response | Rat behaviors | Anti-psychotics | (61) | |
| Infectious Disease | Murine infection models of the thigh and lung | Colony forming units of a bacterial strain | Penicillins, cephalosporins, | (62) |
The time course of pharmacodynamic effects can be viewed as either direct or indirect. For direct PK/PD relationships, concentrations are correlated with effects in a reversible manner with the peak pharmacodynamic effect observed at the same time as peak drug concentrations. The sigmoid Emax model (Hill equation) is based on the receptor occupancy theory and used to describe the nonlinear concentration–effect relationship as shown below in Eq. 2.
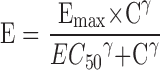 |
2 |
where the effect of the drug (E) can be described by some maximal effect (Emax) and the concentration associated with half of the maximal effect (EC50). The Hill slope coefficient (γ) increases or decreases the steepness of the concentration–effect relationship depending on whether the value of γ is greater or less than 1, respectively. Alternatively, for drugs that act as an antagonist, an inhibitory model can be selected with an Imax, IC50, and γ parameters in the form of Eq. 2 above. The EC50 value reflects potency of the molecule and can be compared back to in vitro potency estimates. Major considerations for translation of Emax and EC50 values from in vitro to in vivo and across species include protein binding, pharmacological target affinity and expression, tissue distribution, and active metabolites. For small molecules which may be subject to extensive plasma protein binding, it is necessary to correct for the fraction unbound which may differ substantially across species. It is assumed that the free concentration in plasma is in equilibrium with free concentration in tissues, but interaction with efflux and uptake transporters may confound the relationship. In addition, for small molecules and biologics, species differences in the interaction between the drug and pharmacological target should also be considered. During the drug discovery and lead optimization process, there may be an opportunity to confirm that a consistent relationship exists between in vitro and in vivo potency parameters providing greater confidence in the measurements. In cases where the in vitro potency is predictive of in vivo potency, correcting for species differences in potency is warranted as described by Chien et al. (12). Distribution of the drug to its target site of action can impact the pharmacodynamics of a drug (39). Therefore, where possible, measurement of tissue drug concentrations that represent the site of action should be included to define the time-course of drug delivery to the target. This additional information can guide help to distinguish between potency and distribution properties which may be import when selecting from several potential candidates. Distinguishing between active free drug and drug bound to specific and/or non-specific binding sites remains challenging. An understanding of the metabolism of the drug is also crucial because active metabolites can contribute to the drug effects and may confound the concentration–effect relationship. Modeling approaches to include the effect of AR-HO47108 and its active metabolite AR-HO47116 on gastric acid inhibition in dogs have been described (40).
Optimal delivery of the drug to its site of action or biophase must also be considered as part of PK/PD predictions. However, depending on the site of action, tools to assess biophase concentrations may or may not exist. If available, compound permeability and distribution to the site of action may be evaluated in conjunction with in vivo measures of activity to define distributional properties. Drug distribution to target tissue is frequently investigated in disease models of the central nervous system, oncology, and infectious disease. The hypothetical effect compartment model was proposed to describe temporal delays in effect where drug distribution is rate-limiting as shown in Eq. 3 (41):
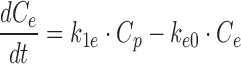 |
3 |
where the rate of change of concentration in the effect compartment is described by k1e is the first order rate constant describing movement of drug out of the central compartment, Cp is the plasma concentration of drug, ke0 is the first-order rate constant describing drug elimination from the effect compartment, and Ce is the effect compartment concentration. It is assumed that the effect compartment is sufficiently small so as not to influence the disposition of the drug in the central compartment. As demonstrated by Kalvass et al., accounting for differences in the brain distribution of μ-opioid receptor agonists enabled a consistent in vitro to in vivo potency correlation across seven compounds (42).
For therapeutic monoclonal antibodies, proteins, and peptides, the highly specific affinity for the human target may limit their use in other animal species in characterizing pharmacological activity. The use of surrogate antibodies is often employed to demonstrate in vivo activity in disease models. Correction for in vitro potency differences between the surrogate antibody and the clinical candidate was proposed by Mordenti (43). In addition, studies which investigate antibody PK after changes in pharmacological target expression may help to better predict dose requirements in disease versus healthy populations as demonstrated by Vugmeyster et al. (44).
System-specific parameters are defined by the underlying physiology of the biological system that is being studied. As an example, the indirect response model has been used extensively to characterize drug effects for drugs which act on turnover processes (45,46). The general equation for the indirect response model is shown below in Eq. 4.
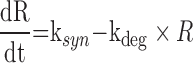 |
4 |
where R is the response, ksyn is the zero-order synthesis rate, and kdeg is the first-order degradation rate. A family of four indirect effect models has been applied. Drug effects can include (1) inhibition of input, (2) inhibition of output, (3) stimulation of input, and (4) stimulation of output, where model selection is based on an understanding of the mechanism of drug action. For chemotherapeutic agents such as anti-cancer and anti-bacterial agents, irreversible effect models are used to describe the delay between concentration and effect (47). System parameters for the growth and death of cells are used to describe the time course of cell proliferation. Drug effects can be incorporated to impair growth or stimulate cell death depending on the mechanism of action. Further modifications to account for time delays due to signal transduction events (48) or time-dependent changes in cellular resistance may be required. Additionally, a wide range of other mechanism-based models exist, but are beyond the scope of this review (38).
Approaches to the PK/PD predictions involving system parameters from preclinical species to humans have involved the use of allometric scaling techniques or utilized available data from humans directly. Two recent examples illustrate the successful application of allometric scaling of PK/PD parameters, and involved in 5-Ht1A receptor mediated responses in rats and red blood cell responses to recombinant human erythropoietin (rhuEpo) (49,50). Allometric scaling principles were applied to mechanism based PK/PD models to predict the time-course of response in humans. The allometric coefficient for parameters that are related to physiologic time was 0.25. When human data exist for system parameters, it is possible to incorporate them directly into the model avoiding the need for allometric scaling (36) and discussed in more detail below. Phase zero studies may play a role in the future to define system parameters where this information is critical.
Leveraging PK/PD Predictions in First-in-Human Study Design
Several areas of the first-in-human study design can be informed by PK/PD modeling. Effective integration of the product of PK/PD predictions is an evolving area. An iterative process is involved for defining key parameters for the first in human study which considers the products of parallel work streams defined in Fig. 1. Areas of focus are considered in more detail below:
Select Safe Starting Dose
As defined by regulatory guidance, a safe starting dose should minimize risk to healthy human subjects by avoiding concentrations that may produce profound pharmacological effects and toxicity. A robust scientific rationale should effectively integrate available information. Alternatively, in settings involving patient-centered first-in-human studies, PK/PD modeling can help to avoid starting at unnecessarily low doses which would not be expected to offer any benefit.
Define Appropriate Concentration Range
PK/PD modeling can help to define the most appropriate concentration range to study drug response(s), inform the choice of dose escalation scheme through translation of preclinical dose-concentration-response, define doses associated with concentrations that may give rise to secondary pharmacology signals (such as QTc or blood pressure), and avoid doses that may exceed NOAEL exposure margins for healthy volunteer studies.
Inform Study Design Elements
Several study design elements can be supported with PK/PD model predictions. These elements include the selection of sample collection times for PK observations, pharmacodynamic observations, appropriate washout times, and duration of study. Duration of study is important particularly for some monoclonal antibodies may have an extended duration of exposure after a single administration.
Preclinical PK/PD Modeling Examples
During compound selection, PK/PD models help to define the relationship between in vitro potency measurements and in vivo potency, support the quantitative understanding of the mechanism of action linking biomarkers to downstream effects, and support human predictions of effect accounting for species differences. To date, a comprehensive analysis is not available to chart the increasing application of PK/PD predictions. Several recent publications illustrating the concepts involved in preclinical PK/PD modeling in the oncology and inflammation/immunology therapeutic areas are described in more detail below.
Oncology
Several translational efforts in oncology have been successfully applied and presented in the literature. The value of translational PK/PD modeling is in supporting the quantitative selection of doses that will produce effective concentrations and avoid severe toxicity, while lessening the chance of including additional cohorts at suboptimal doses with little hope of providing a benefit to patients enrolled in the study. In addition, it is recognized that animal models of disease can be misleading in terms of their prediction of clinical response. Some of the challenges involving PK/PD predictions for anti-cancer drugs involve the selection of the appropriate xenograft model in the face of a range of drug sensitivity across cell lines, differences in the dynamics of tumor growth between xenograft models and human tumors, and defining drug distribution to the tumor. The following examples demonstrate the integration of in vitro and in vivo data with PK/PD models to support human dose selection.
The utility of target concentration predictions arising for xenograft studies was evaluated by Rochetti et al. (51). Tumor growth inhibition and PK of ten marketed anti-cancer agents which produce chemotherapeutic effects through cytotoxicity were evaluated in a common xenograft model, nude mice bearing human A2780 ovarian carcinoma implanted subcutaneously. A PK/PD model of tumor growth inhibition was used to estimate the anti-tumor potency of each agent (48). A threshold concentration was derived from the tumor growth inhibition model to guide dose selection. In the PK/PD model, drug concentrations were linked to the rate constant describing the rate of cell death using the potency term k2. An inverse linear relationship (r = −0.92) was observed between k2 and human systemic exposures observed in the clinic over the course of a typical 3-week cycle such that greater potency translated into a lower cumulative exposure requirement. For the development of future cytotoxic agents, the experimental approach and PK/PD model may be useful to help optimize dose selection for the clinic.
For emerging targeted therapeutics, PK/PD modeling plays an important part of target validation and supports translational efforts. Unlike cytotoxics, a window is expected between the dose that produces efficacy and the dose that produces toxicity. There does not appear to be a consensus as to whether dosing should be based on concentrations producing a specific level of effect in a sensitive xenograft model or a target biomarker response measured in the tumor or clinically accessible surrogate site (i.e., blood).
Preclinical PK/PD relationships for the small molecule cMet receptor tyrosine kinase inhibitor, PF02341066, were characterized in GTL16 gastric carcinoma xenografts after daily oral administration to support candidate selection and first-in-human dose selection (52). This example of a mechanism-based PK/PD analysis demonstrates the combined assessment of drug to biomarker, and biomarker to outcome relationships. A one compartment model was used to describe the steady-state PK of PF02341066 after doses that ranged from 6.25 to 50 mg/kg. The authors applied a hypothetical effect compartment (link) model to account for the observed time delay between peak concentrations of PF02341066 in plasma and reductions in tumor cMet phosphorylation (biomarker). In addition, the relationship between plasma concentration of PF02341066 and tumor growth inhibition (outcome) were characterized with a first-order tumor growth model where the drug inhibited tumor growth with Emax and EC50 values. The PK/PD analysis indicated that the EC50 for cMet phoshorylation was 18.5 ng/mL with a ke0 of 0.135 h−1. The EC50 value for GTL16 tumor growth inhibition was comparable to the EC90 for cMet phosphorylation (213 vs. 167 ng/mL, respectively) suggesting that near complete inhibition of cMet was required for tumor growth inhibition by PF02341066.
A second example involving the small molecule B-Raf kinase inhibitor, GDC-0879, integrated PK, biomarker, and tumor growth inhibition into a PK/PD model to support first-in-human dose selection (53). The PK of GDC-0879 was characterized by a one-compartment model with first order absorption after daily oral administration of 15–200 mg/kg to mice. An indirect response model with the drug inhibiting the zero order synthesis rate of the biomarker, pMEK1, was used to characterize the pharmacodynamics of GDC-0879 in nu/nu mice bearing A375 melanoma xenografts. To characterize the biomarker to outcome relationship, the inhibition of pMEK1 (%I) acted on the first-order growth rate constant of tumor volume with a sigmoidal Emax model. Findings from these studies indicated that approximately 60% inhibition of biomarker (pMEK1) was required to achieve stasis in the A375 melanoma xenograft at GDC-0879 plasma concentrations of 4.5 μM. Interestingly, the hill slope coefficient was 8 indicating a very steep biomarker–response relationship. This example demonstrates a mechanism-based PK/PD approach to define target plasma concentrations and important insights into the mechanism of action.
A recent publication utilized preclinical PK/PD modeling to support selection of dose and regimen for the mammalian target of rapamycin (mTOR) inhibitor everolimus in cancer patients (54). The PK of everolimus in blood and tissues were characterized in rats after oral administration, and scaled to humans using a physiologically based PK model. In addition, the pharmacodynamics of a marker of the mTOR pathway, inactivation of S6K1, was characterized in xenografts of CA20948 pancreatic tumors and peripheral blood mononuclear cell (PBMC) extracts. A direct effect model linked unbound everolimus concentrations to S6K1 activity. Simulations were performed using the predicted human PK parameters and the PK/PD model developed in rats under the assumption that an effective human dosage regimen would require a degree and duration of S6K1 inhibition in PBMC and tumor comparable to rats bearing everolimus sensitive tumors. The model developed from rats accurately predicted the S6K1 inhibition profile in human PBMC. Simulations suggested that 20–30 mg weekly doses of oral everolimus were required to provide comparable S6K1 inhibition to that observed in rats and providing >98% inhibition over the dosing interval. Higher doses of 50 and 70 mg weekly everolimus were not predicted to offer any additional benefit with respect to the degree or duration of S6K1 inhibition. An expansion study successfully demonstrated intratumoral mTOR pathway inhibition at the predicted doses (55).
For monoclonal antibodies, the key steps in translational PK/PD modeling to support first-in-human dose selection were presented by Mordenti et al. (43). The exposure–response relationship for a murine anti-vascular endothelial growth factor (VEGF) monoclonal antibody (muMAb) was described with a process to perform human dose predictions. Beige nude mice were implanted with A673 human rhadomyosarcoma cells and received twice weekly intraperitoneal doses of 0.05–5 mg/kg muMAb for 4 weeks. Steady-state plasma concentrations were linked to the inhibition of tumor growth in model using a modified version of the sigmoidal Emax equation at week 4 or at the time of euthanasia. The target plasma concentration of the muMAb was estimated for >80% inhibition of tumor growth (based on tumor volume measurements). Correction factors were applied to convert the muMAb target concentration to a recombinant human monoclonal antibody (rhuMAb) target concentration based on comparisons of in vitro potency and binding affinity which were reported to be similar for muMAb and rhuMAb. Human PK predictions were based on previous observations for another antibody, rhuMAb-HER2, under the assumption that the two antibodies would have similar PK. Simulations were conducted to estimate the human dosing regimen that would achieve predicted target plasma concentration for rhuMAb. The authors reported that concentrations >10 μg/mL were required for full efficacy in the xenograft model, and exposures from breast cancer patients in a Phase II confirmed the predictions. This study demonstrates the application of PK and PD in oncology to guide dose regimen selection for a monoclonal antibody targeting VEGF.
Inflammation/Immunology
For the sphingosine-1-phosphate receptor agonist FTY720, a physiologically based PK/PD model of lymphocyte trafficking was applied to predict target concentrations (36). Lymphocyte counts were determined over time after a single IV administration in rats and monkeys. An indirect response model was used to characterize the time-course of drug effects where the drug inhibited the appearance of lymphocytes in blood in a concentration dependent manner characterized by an Imax and IC50. Rats and monkeys had distinct IC50 estimates of 90 and 407 pg/mL, respectively, with similar Imax values of 1 and 0.87, respectively. A physiologically based approach was used to predict human PK parameters which were based on in vivo and in vitro data. Consistently high protein binding was reported across species ranging from 99.74% to 99.87%. The authors did not correct for differences in the affinity of FTY720 for S1P1 across species, if any.
Human PK/PD predictions were based on rat and monkey IC50 values, and literature values for the system-specific parameters of human basal lymphocyte counts and turnover were used in place of the rats or monkey values. Simulations of the predicted human time-course of lymphocyte depletion were performed at proposed first-in-human doses for comparison with observed values. PK predictions were in close agreement with observed concentration time profiles though a slight over-prediction of Cmax was noted by the authors. Comparison of the observed and predicted lymphocyte time-course indicated that the physiologically based PK/PD model described the dose-dependent effects of FTY720 on lymphocytes. The IC50 obtained from the monkey more closely approximated the human; however the actual human IC50 value was not given for a quantitative comparison. Application of PK/PD models to the prediction of the time course of effect can help to design first-in-human studies.
The application of preclinical PK/PD modeling to support dose selection for a second generation anti-IgE monoclonal antibody was recently presented (56). The model included data collected as part of the pharmacological characterization of HAE1 and extensive clinical data derived from the lead molecule, omalizumab (Xolair®). The backup molecule was selected on the basis of it’s the apparent equilibrium dissociation constant (Kd) of approximately 23-fold lower than omalizumab. A mechanism-based PK/PD model was used to integrate information on the PK of HAE1, binding to IgE, and turnover of IgE using a physiologic receptor binding model. Key assumptions included in the analysis were as follows: (a) omalizumab/HAE1 in vitro and in vivo KD ratios were constant; (b) similar PK/PD parameters with the exception of Kd which was drug-specific; (c) similar PK/PD covariates such as baseline IgE and body weight. Based on the relationship between a biomarker response (free IgE) and exacerbations of asthma symptoms after administration of omalizumab, 10 IU/mL was selected as the target biomarker suppression for the backup molecule HAE1. The resulting single dose, dose escalation study design encompassed three cohorts of subjects with allergic rhinitis with or without atopic dermatitis administered either 30/90, 180, or 360 mg SC HAE1. In the initial cohort, a minimum biologic dose of 30 mg was selected to ensure toleration before escalating to higher doses. Rapid analysis of PK/PD data from subjects revealed that 30 mg SC produced greater than expected lowering of free IgE below the target free IgE level of 10 IU/mL. A protocol amendment enabled the addition of an additional cohort of 7.5 mg SC to fully characterize the dose–response relationship. This example demonstrates an example of incorporating data from a lead molecule into the quantitative dose selection rationale for a backup molecule.
CONCLUSIONS
Advancements in translational research have increased opportunities to apply information and knowledge gained during drug discovery into the clinical development program. The strategic application of biomarkers to inform scientists about the successful interaction between drug and pharmacological target, or physiological responses arising from receptor occupancy helps to efficiently guide drug development. At the transition from preclinical to clinical stages, PK/PD modeling provides a conceptual framework to integrate existing information from a variety of in vitro and in vivo sources. While originally seen as an opportunity to mitigate risk in dose selection for high-risk biologics, preclinical PK/PD modeling is being applied in other therapeutic areas, particularly in oncology. Gains in the science of PK/PD predictions will help to deliver new medicines to patients.
References
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Paul S, Mytelka D, Dunwiddie C, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 3.Kaitin K. Deconstructing the drug development process: the new face of innovation. Clin Pharmacol Ther. 2010;87:356–361. doi: 10.1038/clpt.2009.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Severino ME, Dubose RF, Patterson SD. A strategic view on the use of pharmacodynamic biomarkers in early clinical drug development. IDrugs. 2006;9:849–853. [PubMed] [Google Scholar]
- 5.Sultana SR, Marshall S, Davis J, Littman BH. (2007) Experiences with dose finding in patients in early drug development: the use of biomarkers in early decision making. Ernst Schering Research Foundation Workshop. 65–79. [DOI] [PubMed]
- 6.Lalonde RL, Kowalski KG, Hutmacher MM, et al. Model-based drug development. Clin Pharmacol Ther. 2007;82:21–32. doi: 10.1038/sj.clpt.6100235. [DOI] [PubMed] [Google Scholar]
- 7.Horig H, Marincola E, Marincola FM. Obstacles and opportunities in translational research. Nat Med. 2005;11:705–708. doi: 10.1038/nm0705-705. [DOI] [PubMed] [Google Scholar]
- 8.Littman BH, Di Mario L, Plebani M, Marincola FM. What’s next in translational medicine? Clin Sci. 2007;112:217–227. doi: 10.1042/CS20060108. [DOI] [PubMed] [Google Scholar]
- 9.Kelland LR. Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer. 2004;40:827–836. doi: 10.1016/j.ejca.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 10.Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G. Mechanism-based pharmacokinetic-pharmacodynamic modeling—a new classification of biomarkers. Pharm Res. 2005;22:1432–1437. doi: 10.1007/s11095-005-5882-3. [DOI] [PubMed] [Google Scholar]
- 11.Holford NH. The target concentration approach to clinical drug development. Clin Pharmacokinet. 1995;29:287–291. doi: 10.2165/00003088-199529050-00001. [DOI] [PubMed] [Google Scholar]
- 12.Chien JY, Friedrich S, Heathman MA, de Alwis DP, Sinha V. Pharmacokinetics/pharmacodynamics and the stages of drug development: role of modeling and simulation. AAPS J. 2005;7:E544–E559. doi: 10.1208/aapsj070355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowe PJ, Hijazi Y, Luttringer O, et al. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica. 2007;37:1331–1354. doi: 10.1080/00498250701648008. [DOI] [PubMed] [Google Scholar]
- 14.Agoram BM, Martin SW, van der Graaf PH. The role of mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modelling in translational research of biologics. Drug Discov Today. 2007;12:1018–1024. doi: 10.1016/j.drudis.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Gabrielsson J, Dolgos H, Gillberg PG, et al. Early integration of pharmacokinetic and dynamic reasoning is essential for optimal development of lead compounds: strategic considerations. Drug Discov Today. 2009;14:358–372. doi: 10.1016/j.drudis.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 16.Gabrielsson J, Green AR, Van der Graaf PH. Optimising in vivo pharmacology studies--Practical PKPD considerations. Journal of Pharmacological & Toxicological Methods 61:146–156. [DOI] [PubMed]
- 17.Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotechnol. 2009;20:722–729. doi: 10.1016/j.copbio.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 18.FDA . Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Washington, DC: Guidance for Industry; 2005. [Google Scholar]
- 19.EMEA/CHMP. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal agents. 2007. [DOI] [PMC free article] [PubMed]
- 20.Gabrielsson J, Green AR. Quantitative pharmacology or pharmacokinetic pharmacodynamic integration should be a vital component in integrative pharmacology. J Pharmacol Exp Ther. 2009;331:767–774. doi: 10.1124/jpet.109.157172. [DOI] [PubMed] [Google Scholar]
- 21.Obach RS, Baxter JG, Liston TE, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 22.Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm. 1982;10:201–227. doi: 10.1007/BF01062336. [DOI] [PubMed] [Google Scholar]
- 23.Boxenbaum H. Interspecies pharmacokinetic scaling and the evolutionary–comparative paradigm. Drug Metab Rev. 1984;15:1071–1121. doi: 10.3109/03602538409033558. [DOI] [PubMed] [Google Scholar]
- 24.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34:687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 25.Kawai R, Mathew D, Tanaka C, Rowland M. Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther. 1998;287:457–468. [PubMed] [Google Scholar]
- 26.Hosea NA, Collard WT, Cole S, et al. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol. 2009;49:513–533. doi: 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 28.Ling J, Zhou H, Jiao Q, DAvis H. Interspecies scaling of therapeutic monoclonal antibodies: initial look. Journal of Clinical Pharmacology. 2010. [DOI] [PubMed]
- 29.Dong J, Salinger D, Endres C, et al.(2010) Quantitative prediction of human pharmacokinetics for monoclonal antibodies: analysis of monkey as a single species for first-in-human prediction. Clinical Pharmacokinetics (in press). [DOI] [PubMed]
- 30.Levy G. Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56:248–252. doi: 10.1038/clpt.1994.134. [DOI] [PubMed] [Google Scholar]
- 31.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–532. doi: 10.1023/A:1014414520282. [DOI] [PubMed] [Google Scholar]
- 32.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72:1–10. doi: 10.1016/j.bcp.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 33.Pendley C, Schantz A, Wagner C. Immunogenicity of therapeutic monoclonal antibodies. Curr Opin Mol Ther. 2003;5:172–179. [PubMed] [Google Scholar]
- 34.Ponce R, Abad L, Amaravadi L, et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul Toxicol Pharmacol. 2009;54:164–182. doi: 10.1016/j.yrtph.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 35.Dingemanse J, Appel-Dingemanse S. Integrated pharmacokinetics and pharmacodynamics in drug development. Clin Pharmacokinet. 2007;46:713–737. doi: 10.2165/00003088-200746090-00001. [DOI] [PubMed] [Google Scholar]
- 36.Meno-Tetang GM, Lowe PJ. On the prediction of the human response: a recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin Pharmacol Toxicol. 2005;96:182–192. doi: 10.1111/j.1742-7843.2005.pto960307.x. [DOI] [PubMed] [Google Scholar]
- 37.Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29:186–191. doi: 10.1016/j.tips.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31:510–518. doi: 10.1124/dmd.31.5.510. [DOI] [PubMed] [Google Scholar]
- 39.Lin JH. Tissue distribution and pharmacodynamics: a complicated relationship. Curr Drug Metab. 2006;7:39–65. doi: 10.2174/138920006774832578. [DOI] [PubMed] [Google Scholar]
- 40.Abelo A, Andersson M, Holmberg AA, Karlsson MO. Application of a combined effect compartment and binding model for gastric acid inhibition of AR-HO47108: a potassium competitive acid blocker, and its active metabolite AR-HO47116 in the dog. Eur J Pharm Sci. 2006;29:91–101. doi: 10.1016/j.ejps.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 41.Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham J. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther. 1979;25:358–371. doi: 10.1002/cpt1979253358. [DOI] [PubMed] [Google Scholar]
- 42.Kalvass JC, Olson ER, Cassidy MP, Selley DE, Pollack GM. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50, u and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther. 2007;323:346–355. doi: 10.1124/jpet.107.119560. [DOI] [PubMed] [Google Scholar]
- 43.Mordenti J, Thomsen K, Licko V, et al. Efficacy and concentration-response of murine anti-VEGF monoclonal antibody in tumor-bearing mice and extrapolation to humans. Toxicol Pathol. 1999;27:14–21. doi: 10.1177/019262339902700104. [DOI] [PubMed] [Google Scholar]
- 44.Vugmeyster Y, Tian X, Szklut P, Kasaian M, Xu X. Pharmacokinetic and pharmacodynamic modeling of a humanized anti-IL-13 antibody in naive and Ascaris-challenged cynomolgus monkeys. Pharm Res. 2009;26:306–315. doi: 10.1007/s11095-008-9739-4. [DOI] [PubMed] [Google Scholar]
- 45.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–478. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma A, Jusko WJ. Characterization of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1996;24:611–635. doi: 10.1007/BF02353483. [DOI] [PubMed] [Google Scholar]
- 47.Jusko WJ. Pharmacodynamics of chemotherapeutic effects: dose-time-response relationships for phase-nonspecific agents. J Pharm Sci. 1971;60:892–895. doi: 10.1002/jps.2600600618. [DOI] [PubMed] [Google Scholar]
- 48.Simeoni M, Magni P, Cammia C, et al. Predictive pharmacokinetic-pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004;64:1094–1101. doi: 10.1158/0008-5472.CAN-03-2524. [DOI] [PubMed] [Google Scholar]
- 49.Zuideveld KP, Van der Graaf PH, Peletier LA, Danhof M. Allometric scaling of pharmacodynamic responses: application to 5-Ht1A receptor mediated responses from rat to man. Pharm Res. 2007;24:2031–2039. doi: 10.1007/s11095-007-9336-y. [DOI] [PubMed] [Google Scholar]
- 50.Woo S, Jusko WJ. Interspecies comparisons of pharmacokinetics and pharmacodynamics of recombinant human erythropoietin. Drug Metab Dispos. 2007;35:1672–1678. doi: 10.1124/dmd.107.015248. [DOI] [PubMed] [Google Scholar]
- 51.Rocchetti M, Simeoni M, Pesenti E, De Nicolao G, Poggesi I. Predicting the active doses in humans from animal studies: a novel approach in oncology. Eur J Cancer. 2007;43:1862–1868. doi: 10.1016/j.ejca.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 52.Yamazaki S, Skaptason J, Romero D, et al. Pharmacokinetic-pharmacodynamic modeling of biomarker response and tumor growth inhibition to an orally available cMet kinase inhibitor in human tumor xenograft mouse models. Drug Metab Dispos. 2008;36:1267–1274. doi: 10.1124/dmd.107.019711. [DOI] [PubMed] [Google Scholar]
- 53.Wong H, Belvin M, Herter S, et al. Pharmacodynamics of 2-[4-[(1E)-1-(hydroxyimino)-2, 3-dihydro-1 H-inden-5-yl]-3-(pyridine-4-yl)-1 H-pyrazol-1-yl]ethan-1-ol (GDC-0879), a potent and selective B-Raf kinase inhibitor: understanding relationships between systemic concentrations, phosphorylated mitogen-activated protein kinase 1 inhibition, and efficacy. J Pharmacol Exp Ther. 2009;329:360–367. doi: 10.1124/jpet.108.148189. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka C, O’Reilly T, Kovarik JM, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008;26:1596–1602. doi: 10.1200/JCO.2007.14.1127. [DOI] [PubMed] [Google Scholar]
- 55.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26:1603–1610. doi: 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 56.Putnam WS, Li J, Haggstrom J, et al. Use of quantitative pharmacology in the development of HAE1, a high-affinity anti-IgE monoclonal antibody. AAPS J. 2008;10:425–430. doi: 10.1208/s12248-008-9045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amore B, Gibbs J, Emery M. Application of in vivo animal models to characterize the pharmacokinetic and pharmacodynamic properties of drug candidates in discovery settings. Comb Chem High Throughput Screen. 2010;13:207–218. doi: 10.2174/138620710790596808. [DOI] [PubMed] [Google Scholar]
- 58.Lee L, Sharma S, Morgan B, et al. Biomarkers for assessment of pharmacologic activity for a vascular endothelial growth factor (VEGF) receptor inhibitor, PTK787/ZK 222584 (PTK/ZK): translation of biological activity in a mouse melanoma metastasis model to phase I studies in patients with advanced colorectal cancer with liver metastases. Cancer Chemother Pharmacol. 2006;57:761–771. doi: 10.1007/s00280-005-0120-6. [DOI] [PubMed] [Google Scholar]
- 59.Kim D, Wang L, Beconi M, et al. (2R)-4-oxo-4-[3-(trifluoromethyl)-5, 6-dihydro[1, 2, 4]triazolo[4, 3-a]pyrazin-7(8 H)-yl]-1-(2, 4, 5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 60.Kreilgaard M, Smith DG, Brennum LT, Sanchez C. Prediction of clinical response based on pharmacokinetic/pharmacodynamic models of 5-hydroxytryptamine reuptake inhibitors in mice. Br J Pharmacol. 2008;155:276–284. doi: 10.1038/bjp.2008.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olsen CK, Brennum LT, Kreilgaard M. Using pharmacokinetic-pharmacodynamic modelling as a tool for prediction of therapeutic effective plasma levels of antipsychotics. Eur J Pharmacol. 2008;584:318–327. doi: 10.1016/j.ejphar.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 62.Katsube T, Yano Y, Yamano Y, et al. Pharmacokinetic-pharmacodynamic modeling and simulation for bactericidal effect in an in vitro dynamic model. J Pharm Sci. 2008;97:4108–4117. doi: 10.1002/jps.21265. [DOI] [PubMed] [Google Scholar]