

Abstract
The purpose of this research was the preparation of four formulations containing hydrocortisone acetate (HCA) for topical application, including two aqueous systems (hydrophilic microemulsion and aqueous gel) and two systems with dominant hydrophobicity (hydrophobic microemulsion and ointment). The formulations were tested for the release and permeation of HCA across an animal membrane. The release of HCA was found comparable for the four systems. The two microemulsions promote permeation across an ex-vivo membrane, examined by means of a Franz cell. Hydrophobic microemulsion guarantees the highest solubility (2,370 μg/ml) and flux (133 μg/cm2.h) of the drug, since it contains almost 40% Transcutol, a permeation enhancer. Gel and ointment provide lower solubility and flux, being the values, related to the ointment, the lowest ones (562 μg/ml and 0.4 μg/cm2.h). Experimental results allow the conclusion that gel and ointment can be suitable when it is desirable to minimize absorption of topically applied HCA as to keep the drug restricted to the diseased area and prevent side effects of the systemic presence of HCA.
Key words: gel and ointment, hydrocortisone acetate, microemulsions, transdermal permeability
INTRODUCTION
Treatment of inflammatory skin diseases frequently requires a combination of systemic and topical agents, but topical substances alone are very often adequate (1–5).
As an example, systemic administration of corticosteroids is suggested only in the case of acute dermatological disorders or of chronic diseases, since they are not lacking in side effects; while topical administration offers the advantage of avoiding potential problems associated with systemic exposure of corticosteroids and is, as a consequence, widely used. In order to minimize absorption, so as to keep the drug restricted to the diseased area, the action of the drug must be limited to the surface of the skin and therefore does not need transdermal permeation: this goal can be achieved by an appropriate choice both of the pharmaceutical form and/or excipients present in the formulation (6).
Hydrocortisone acetate (HCA) is a synthetic corticosteroid efficient against cutaneous disorders, such as inflammatory dermatitis. Its parent compound hydrocortisone was the first drug of this class employed in topical formulations for this purpose: the introduction of the acetate moiety in the 21 position of the steroid ring system improved the topical activity fivefold. The drug is indicated for the relief of inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses. Commercial vehicles for systemic administration of HCA include rectal suppositories, while intra-articular injections are used for temporary relief under certain conditions. Several commercial products containing HCA are available for topical use on skin (7): formulations for topical application must allow adequate release of the active compound, be non-allergenic, non-irritating, and cosmetically acceptable and, more important, avoid transdermal permeation of the drug (8). Topical formulations of corticosteroids are usually administered in the pharmaceutical form of ointments, creams, lotions, gels, aerosol sprays, powders and foams (7). The type of delivery vehicle or formulation of excipients can markedly affect the penetrating properties and absorption of the active agent, and thereby affect drug potency or effectiveness, but also the associated side effects. In fact, despite belonging to different categories, vehicles are often structurally similar and affect the diffusion of the active agent through the matrix and can contain chemical enhancers promoting transdermal permeation.
Following these suggestions, the purpose of the research was to test four different formulations for topical administration of HCA, mainly to evaluate their properties to promote or not its flux across an animal membrane. The formulations examined in this paper range from an aqueous gel to an ointment, and from an aqueous to a non-aqueous microemulsion.
MATERIALS
Hydrocortisone acetate was a commercial sample of pharmaceutical purity (Sigma, Milan, Italy): white powder (melting point 223 °C, with decomposition), very poorly soluble in water.
Propylene glycol, 2-(2-ethoxy-ethoxy) ethanol (Transcutol®), caprylo-capryloyl macrogol-8-glyceride (Labrasol®), oleoyl macrogol-6-glyceride (Labrafil® M 1944CS), propylene glycol monolaurate (lauroglycol R90®) and polyglyceryl-6-di-oleate (Plurol oleique®) were gifts from Gattefossé and were used as received. Liquid paraffin, vaseline oil, Polyacrylamide, C13–14 isoparaffin, Laureth-7 (Sepigel 305 Farcosgel®) and cellulose acetate dialysis membrane (cutoff 14,000) were purchased from Polichimica (Bologna, Italy).
Equipments
High performance liquid chromatography (HPLC) Water Mod. 700 Satellite WISP was employed for analysis, equipped with: chromatographic column reversed phase 18, 3.9 × 300 mm Waters; automatic sampling injector Waters WISP 700; pump Mod. 600 E ultraviolet–visible detector Mod. 484; data recorder and processor PC-NEC Power Mate.
METHODS
Preparation of the formulations
The quali–quantitative composition of each vehicle is shown in Table I.
Hydrophilic microemulsion (A)—Prefixed volumes of three emulsifiers (Transcutol, Labrafil and Labrasol) were mixed, under magnetic stirring, together with Plurol oleique that represents the oil phase in this system. The mixture is added, very slowly, to water. The microemulsion thus formed, after 1 h rest, was finally added to the drug powder and stirred for 24 h, in a dark room.
Aqueous gel (B)—The gel-forming polymer (Sepigel 305) was dispersed in demineralised water under stirring; after the system has become homogeneous, the drug is slowly added up to complete dispersion.
Lipophilic microemulsion (C)—Transcutol, liquid paraffin and propylene glycol were added to the emulsifier and stirred. After 1 h rest, the microemulsion thus formed is added to the drug under stirring for 24 h, in the absence of light.
Anhydrous ointment (D)—Vaseline oil and jelly were mixed, added to the drug and stirred up to homogeneous appearance of the formulation.
Table I.
Weight Percent Composition of the Four Vehicles Examined (HCA: 0.5% w/w)
| % (w/w) | |
|---|---|
| A) Hydrophilic microemulsion | |
| Transcutol | 7.97 |
| Labrafil M 1944 CS | 4.68 |
| Plurol oleique | 6.98 |
| Labrasol | 39.02 |
| Water | 40.85 |
| B) Aqueous gel | |
| Sepigel 305 | 3.50 |
| Water | 96.50 |
| C) Hydrophobic microemulsion | |
| Transcutol | 41.54 |
| Lauroglycol 90 | 45.08 |
| Propylene glycol | 5.50 |
| Vaseline oil | 7.88 |
| D) Anhydrous ointment | |
| Vaseline oil | 11.51 |
| Vaseline jelly | 88.49 |
Solubility determination
HCA equilibrium solubility in the different vehicles was measured at 25 °C. Excess drug was added to the different vehicles, which were stirred for 24 h and then forced through a 0.20 μm nylon filter to remove the undissolved drug. After filtration an accurately weighed portion (0.5 g) of each pharmaceutical formulation was transferred into a 50 ml centrifuge tube and supplemented with 20 ml acetonitrile solution to extract HCA from each vehicle: the solvent contained dexamethasone, as internal standard (10 mg/ml). The mixture was placed in the ultrasonic bath for 10 min and then centrifuged at 3,000 rpm for 15 min. 10 μl supernatant was assayed for the content in HCA by HPLC (9).
Release test
The apparatus consists of two compartments. The donor one is a cylinder (9 mm diameter) and contains the releasing system (2 ml of each formulation); the drug reaches the receiving phase (40 ml aqueous ethanol 40/60 v/v) through a semi-permeable cellulose acetate membrane, previously wetted for 10 min. The apparatus is thermostated at 37 °C. At pre-selected times (1, 2, 3, 4, 5, 6, 7, 8 h) the receptor compartment is renewed with the same solution and the drug content is analyzed by HPLC.
Permeation experiments
Permeation of HCA from the four formulations was determined using Franz diffusion cells fitted with excised pig ear that represents the diffusional membrane. This consists of the internal portion of the pig ear, appropriately prepared by elimination of fat tissues. This study agrees with the protocol approved by the Ethics Committee at the Faculty of Pharmacy of Bologna University; this research also adheres to the Principles of Laboratory Animal Care. (NIH publication #85-23, revised in 1985). The Franz cell (Crown Glass Company Inc. New Jersey, USA) contains a donor compartment (2.5 ml), where the releasing system is positioned, separated from the receiving cell (5.0 ml) by the animal diffusional membrane (64 mm2). Membranes are obtained as 2 × 2 cm samples and after preparation are kept at −20 °C until used. The membrane is positioned between the donor and the receptor compartment of the cell, with the epidermis surface towards the upper compartment. The formulation (2 ml) to be tested fills the donor part, while the receiving volume (5 ml) contains phosphate buffer solution (pH 7.4) to simulate physiological conditions. Two ml aliquot of the solution of the receptor compartment is withdrawn at a preset time (1, 2, 3, 4, 5, 6, 7, 8 h) and replaced with the same volume of the buffer at 37 °C. Analysis of the permeated drug was carried out by means of HPLC, after filtration (0.20 μm) of the sample. All experiments were carried out in triplicate. The cumulative amount permeated (μg) divided by the surface area of the membrane (cm2) was plotted versus time in hours. J, the steady-state flux (μg/cm2.h), was obtained as the slope of the linear portion of the plot. The permeability coefficient, K (cm/h) was obtained dividing the flux by the concentration (μg/cm3) of the dissolved drug in each formulation in the donor compartment. Permeation parameters are shown in Tables II and III.
Table II.
HCA Solubility (±Standard Deviation) in the Four Systems and Release of HCA from the Four Formulations
| Formulation | Solubility (μg/ml) | Released after 8 h (μg) (released/dissolved ratio) | Diffusional exponent (n) and constant k(102) |
|---|---|---|---|
| A | 2,110 ± 20 | 188 ± 33 (0.09) | 0.56/2.3 |
| B | 1,774 ± 15 | 246 ± 11 (0.14) | 0.56/1.9 |
| C | 2,370 ± 22 | 265 ± 22 (0.11) | 0.50/3.2 |
| D | 562 ± 6 | 163 ± 20 (0.29) | 0.51/8.3 |
Table III.
HCA Permeation Parameters from the Four Formulations
| Formulation | Solubility (S, μg/ml) | Flux (J, μg/cm2h) | Permeability coefficient Kp.104 (J/S, cm/h) |
|---|---|---|---|
| A | 2,110 ± 20 | 130 ± 10 | 630 ± 58 |
| B | 1,774 ± 15 | 2 ± 1 | 11 ± 6 |
| C | 2,370 ± 20 | 133 ± 15 | 560 ± 60 |
| D | 562 ± 6 | 0.4 ± 0.2 | 8 ± 4 |
HPLC method
The mobile phase was a mixture of methanol, acetonitrile and water (15:27:58, v/v/v), degassed before application. Experimental conditions were as follows: injection volume 10 μl, the mobile phase isocratically pumped at a flow rate 0.8 ml/min at room temperature, the detection wavelength 238 nm. Linearity was tested at four concentrations (0.2, 0.5, 1 and 2 mg/ml): the limit of quantification was found to be 0.05 mg/ml (9–10).
RESULTS AND DISCUSSION
Nature of the formulations
Since the rate of transdermal permeation of a topical drug is dependent on the properties of the compound and also the physico-chemical characteristics of the vehicle, in which the compound is dissolved or dispersed, vehicles for topical HCA considered here include different formulations: two aqueous systems (hydrophilic emulsion, A and aqueous gel, B); and two systems where hydrophobicity is dominant (hydrophobic emulsion, C and ointment, D).
Microemulsions are emulsions producing a transparent product that has a very small droplet size and does not have tendency to coalesce. A large amount of drug can in fact be incorporated in these formulations, due to the high solubilizing capacity of hydrophobic drugs.
Formulation A is a hydrophilic microemulsion containing different components (Table I). Labrasol, originally developed for solubilization of hydrophobic drugs (HLB 14), is a non-ionic emulsifier, useful also in oral formulations for its high tolerance and low toxicity in animals; it also displays a strong absorption enhancing effect (11). Labrasol can be easily dissolved in water; because it displays only a low interfacial tension with water, the amount of co-surfactants can be kept low. Labrafil, is a non-ionic amphiphilic excipient, used as a co-surfactant in microemulsions. Although Labrafil is a polyethylene glycol derivative, the presence of fatty acids (mainly oleic acid) in this non-ionic amphiphilic compound granted hydrophobic properties to the molecule. This compound, coupled with polyglyceryl di-oleate (Plurol oleique), builds up the necessary surfactant and co-surfactant system for the preparation of the microemulsion.
Formulation C represents an example of a self-microemulsifying drug delivery systems and is an isotropic mixture of oil (vaseline oil), surfactant/co-surfactant and drug: a fine o/w microemulsion is rapidly formed when the components are introduced into water.
Generally, gels contain organic macromolecules forming a tridimensional skeleton with a high solvation degree: the present gel (Formulation B) is made from the unique water-soluble component, Sepigel 305, that is a blend of three ingredients (Polyacrylamide, C13–14 Isoparaffin and Laureth-7) providing increased viscosity, stability and shine to the formulation. Sepigel 305 is non-ionic and is stable over a wide range of pH values; after addition, it rapidly disperses under agitation assuming a pleasant, creamy, gel-like consistency: the viscosity increase is almost instantaneous.
The ointment (Formulation D) is a viscous semisolid preparation, hydrophobic in nature, where vaseline, a non-drying oil, disperses soft petroleum jelly or petrolatum, a semi-solid mixture of hydrocarbons; systems of this type spread easily, are protective, hydrating and lubricating.
Solubility of hydrocortisone acetate
Knowledge of the drug’s solubility for each vehicle composition is necessary to avoid preparation supersaturation: when the drug concentration is increased above its equilibrium solubility, the system becomes thermodynamically unstable and crystallization and precipitation of excess drug occurs over time. The drug solubility in the vehicle is also an important factor for drug penetration across the skin or artificial membranes: the solubility of the drug in the vehicle will influence both the drug concentration gradient in the solution and partition coefficient between the vehicle and the membrane.
HCA has a low solubility in water, but it is more soluble in propylene glycol, where the saturated solubility is 115 mg/100 ml: its poor solubility originates because the steroid ring of the parent compound hydrocortisone contains only two hydrophilic contributions on the steroid rings and in HCA the hydroxy in the 21 position is masked by the ester linkage with acetate moiety. A low solubility in water and a high partition coefficient value is therefore not surprising. LogP value is 1.61 for hydrocortisone and increases up to 2.30 for HCA, as a consequence of the masking of a hydroxy group (12). The solubility of HCA in water measured at 37 °C is 10 μg/ml, much lower than that reported in Table II for the different systems, suggesting a solubilization ability of all components used to prepare present formulations: this is particularly interesting in view of the formation of a concentration gradient, necessary for the release. HCA solubility is higher in formulations A and C, due to the presence of co-solvents or to surfactant compounds able to drive micelle solubilization. Solubility in ointment D is the lowest value found. All the values of the released HCA after 8 h are well below the starting amount of the dissolved drug in each formulation: these systems also potentially represent reservoir systems, since they also contain undissolved drug.
Release from the formulations
Each formulation was tested for the release of the drug through a dialysis membrane, which should not offer resistance to the crossing of the drug: the receptor phase contained ethanol to improve solubility of the drug and avoid back passage. Table II shows the results of the release for the different formulations as a function of time: the test demonstrated that all the formulations have the ability to release HCA to a comparable extent, despite differences in solubility. The release profiles are almost parallel for all the formulations (Fig. 1): the amount released increases with a characteristic profile during a first period of 8 h; and then reaches the 24 h value very slowly: in the second period of 16 h the amount released represents only about 15% of the total (not shown in Figure). The released amount after 8 h is comparable for the different formulations, ranging from 163 μg from the ointment (D) to 265 μg for microemulsion systems. The comparison between solubility in the vehicle and medium ability to release the drug suggests that the main driving force for HCA release is the concentration gradient of the dissolved drug. A rapid and consistent release from a topical formulation first involves the dissolved drug. Since present formulations are saturated systems, dissolution must not represent an obstacle to the supply of the active agent, when this is present in a topical formulation for transdermal absorption. When the dissolved/released amount ratio is considered, the highest value is found for the ointment, suggesting that in this vehicle the solute displays high mobility. A linear relationship can be found between the dissolved and released amount after 8 h as shown in Table II (Fig. 2): the amount released is high when solubility is high, indicating that the dissolved fraction drives the release. Formulation A does not adhere to this rule, since its release value appears lower than that expected. This fact suggests that HCA in the aqueous microemulsion attains a particular stability (also due to micelle solubilization, see below) that delays its release. On the contrary, the behaviour of non-aqueous microemulsion is more linear, since high release of the drug corresponds to high solubility.
Fig. 1.

HCA release profiles from the four formulations: unfilled triangle hydrophilic microemulsion; unfilled square aqueous gel; filled diamond lipophilic microemulsion; filled square ointment
Fig. 2.

Relationship between HCA amount released after 24 h from the four formulations and solubility
When experimental data are plotted against time in terms of the simplified equation:
 |
(where: Mt = mass released at time t; 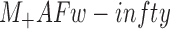 starting mass at t = 0; k = diffusional constant that depends on the characteristics of the formulations; n = diffusional exponent), it could be possible to obtain further information on the release process. n values proved similar for all the formulations in the range 0.50–0.56, very close to the reference values for a Fick type release kinetics, where the release rate decreases with time. Diffusional constant k values, as obtained by the graphical solution of the equation, suggest that the release rate is of the same order of magnitude for the four formulations and that the release of the drug is affected to a limited extent by technological forms employed for the release.
starting mass at t = 0; k = diffusional constant that depends on the characteristics of the formulations; n = diffusional exponent), it could be possible to obtain further information on the release process. n values proved similar for all the formulations in the range 0.50–0.56, very close to the reference values for a Fick type release kinetics, where the release rate decreases with time. Diffusional constant k values, as obtained by the graphical solution of the equation, suggest that the release rate is of the same order of magnitude for the four formulations and that the release of the drug is affected to a limited extent by technological forms employed for the release.
The diffusional constant k suggests that ointment (D) allows better diffusion of the drug from the bulk toward the interfaces formulation/dialysis membrane; while aqueous gel (B) slows down diffusion. In this case it must be taken into account diffusional difficulties in gelified systems of semisolid consistency and also the scarce affinity of the hydrophobic solute towards the aqueous medium.
Effect of the vehicles on HCA permeation
Table III shows that formulations A and C allow a notable permeation of the drug after 8 h. Formulations B and D on the contrary display low permeated amount and low flux, the value related to the ointment D being the lowest one. Improved flux and permeation can result from the microemulsion components (surfactants and co-surfactants) (13,14); moreover also Transcutol is present in both types of microemulsions and it is well known that it significantly increases the percutaneous penetration of various active substances, particularly if used in combination with suitable cosolvents (15,16). This association is present in formulation C, where propylene glycol acts with a possible synergistic enhancer effect and this accounts first for the satisfactory solvent power and second for the skin penetration enhancing properties of the hydrophobic microemulsion (1) (Fig. 3).
Fig. 3.

HCA Permeation profiles from: unfilled triangle hydrophilic microemulsion (A); filled diamond lipophilic microemulsion (C)
This is not the only mechanism that operates in the presence of microemulsions. Microemulsions in fact have a very low interfacial tension that improves the contact between vehicle and membrane surface; moreover the high loading capacity of microemulsions ensures a concentration gradient suitable to improve permeation of the drug (17); also the occurrence of supersaturation cannot be excluded (18), which should further increase the gradient.
The high solubility of the drug in systems A and C provides the necessary concentration gradient for high permeation: however, though higher with respect to formulations B, and especially D, it does not appear as high as expected, despite the presence of Transcutol in the formulation. Formulation C in fact contains almost 40% Transcutol, which, beside its notable transdermal permeation enhancement, also behaves as a solubilizer. It cannot be excluded that in this aqueous microemulsioned system (C), due to the high concentration of the surfactant Labrasol, the drug can be distributed over three different phases: the dispersed phase, the continuous phase and the surfactant micelles. The presence of solubilized drug tends to lower the drug concentration available for permeation, altering the drug partition process between the system and the membrane.
Formulations B and D can be suggested when it is desirable to minimize HCA absorption, so as to keep the drug restricted to the diseased area (Fig. 4): in this case topical administration offers the advantage of avoiding potential side effects associated with systemic exposure. Skin permeation can be inhibited as the affinity with the vehicle becomes greater than that the affinity with the membrane and this creates a poor transfer from the vehicle to the skin, as in the case of the ointment. Difficulty in releasing the drug was already demonstrated by this formulation also towards the dialysis membrane, therefore a low permeation promotion was expected from formulation D: in fact ointment is a hydrophobic system where the hydrophobic drug can accommodate and display little tendency to diffuse outwards. The lack of any type of enhancer can explain the low drug permeation in these last cases. In addition, due to the complete aqueous nature of gel, its application can be found to be cosmetically more attractive, compared with the ‘‘greasy’’ sensation often experienced after the use of ointments. Gel is less dense and therefore generally easier to apply to and spread on the skin surface. This may have important clinical implications, especially if the diseased area is inflamed and sensitive to mechanical shearing forces.
Fig. 4.

Permeation profiles of HCA from: unfilled square aqueous gel (B); filled square ointment (D)
As a result of the flux obtained, these formulations can be suggested for maintaining HCA on the skin surface or for its promotion across the horny layer to the systemic circulation. In this respect formulations B and D prove useful for, e.g., topical anti-micotic action on the skin surface or against psoriasis or dermal affections related to an increased cell turnover; or to promote anti-inflammatory activity below the site of application with the reduction of secretion of chemotactic and vasoactive factors. Formulations A and C appear optimal vehicles exerting a high capacity for incorporating HCA and promote its skin permeation and can be suggested when HCA absorption is required or, in more general terms, for drugs that need to be absorbed to exert their therapeutical activity, even though an important disadvantage of aqueous microemulsions is that they require a high content of surfactants, which in turn induce significant alterations to the skin barrier function and can lead to possible allergic reactions.
CONCLUSIONS
Experimental results allow the conclusion that gel and ointment represent suitable formulations to minimize absorption of topically applied HCA.
Hydrophilic and hydrophobic microemulsions promote HCA absorption, due to the presence in both formulations of Transcutol, as solubilizer and permeation enhancer.
Acknowledgments
This research was financially supported by MIUR funds. The English language and style of the text was corrected by an English translator. The Authors want to thank the reviewers for their effort in improving the text.
References
- 1.Davis A. F., Gyurik R. J., Hadgraft J., Pellet M. A., Walters K. A. Formulation strategies for modulating skin permeation. In: Walter K. A., editor. Dermatological and transdermal formulations. New York: Dekker; 2002. pp. 271–317. [Google Scholar]
- 2.Devos S. A., Van Der Valk P. G. Relevance and reproducibility of patch-test reactions to corticosteroids. Contact Dermatitis. 2001;44:362–365. doi: 10.1034/j.1600-0536.2001.044006362.x. [DOI] [PubMed] [Google Scholar]
- 3.Huang X., Tanojo H., Lenn J., Deng C. H., Krochmal L. A novel foam vehicle for delivery of topical corticosteroids. J. Am. Acad. Dermatol. 2005;53:S26–S38. doi: 10.1016/j.jaad.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 4.Panconesi E., Lotti T. Steroids versus nonsteroids in the treatment of cutaneous inflammation: therapeutic modalities for office use. Arch. Dermatol. Res. 1992;284:S37–S41. doi: 10.1007/BF00638239. [DOI] [PubMed] [Google Scholar]
- 5.Orienti I., Zecchi V., Ceschel G. C., Fini A. Controlled release of hydrocortisone acetate from dermal bases. Eur. J. Drug Metab. Pharmacokinet. 1991;3:466–472. [PubMed] [Google Scholar]
- 6.Neubert R. H., Potzsch A. Liberation of hydrocortisone acetate from different commercial formulations. Pharmazie. 2004;59:472–474. [PubMed] [Google Scholar]
- 7.Refai H., Müller-Goymann C. C. The influence of dilution of topical semisolid preparations on hydrocortisone permeation through excised human stratum corneum. Eur. J. Pharm. Biopharm. 2002;54:143–150. doi: 10.1016/S0939-6411(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 8.Ceschel G. C., Maffei P., Lombardi Borgia S., Ronchi C. Design and evaluation of buccal adhesive hydrocortisone acetate (HCA) tablets. Drug Deliv. 2001;8:161–171. doi: 10.1080/107175401316906937. [DOI] [PubMed] [Google Scholar]
- 9.Hajkova R., Solich P., Dvorak J., Sicha J. Simultaneous determination of methylparaben, propylparaben, HCA and its degradation products in a topical cream by RP-HPLC. J. Pharm. Biomed. Anal. 2003;32:921–927. doi: 10.1016/S0731-7085(03)00193-6. [DOI] [PubMed] [Google Scholar]
- 10.Grippa E., Santini L., Castellano G., Gatto M. T., Leone M. G., Saso L. Simultaneous determination of hydrocortisone, dexamethasone, indomethacin, phenylbutazone and oxyphenbutazone in equine serum by high-performance liquid chromatography. J. Chromatogr. B. Biomed. Sci. Appl. 2000;738:17–25. doi: 10.1016/S0378-4347(99)00478-8. [DOI] [PubMed] [Google Scholar]
- 11.Hu Z., Prasad R., Tawa R., Konishi T., Ishida M., Shibata N., Takada K. Diethyl ether fraction of Labrasol having a strong enhancing effect of gentamicin than Labrasol itself. Int. J. Pharm. 2002;234:223–235. doi: 10.1016/S0378-5173(01)00967-X. [DOI] [PubMed] [Google Scholar]
- 12.Kanazawa H., Sunamoto T., Ayano E., Matsushima Y., Kikuchi A., Okano T. Temperature responsive chromatography using poly-(N-isopropylacrylamide) hydrogel-modified silica. Anal. Sci. 2000;18:45–48. doi: 10.2116/analsci.18.45. [DOI] [PubMed] [Google Scholar]
- 13.Kreilgaard M. Influence of microemulsions on cutaneous drug delivery. Adv. Drug Deliv. Rev. 2002;54(Suppl. 1):S77–S98. doi: 10.1016/S0169-409X(02)00116-3. [DOI] [PubMed] [Google Scholar]
- 14.Kogan A., Garti N. Microemulsions as transdermal drug delivery vehicles. Adv. Coll. Interface Sci. 2006;123–126:369–385. doi: 10.1016/j.cis.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann L., Keipert S., Gloor M. Effects of microemulsions on the stratum corneum and hydrocortisone penetration. Eur. J. Pharm. Biopharm. 2001;52:129–136. doi: 10.1016/S0939-6411(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 16.Refai H., Muller-Goymann C. C. The influence of dilution of topical semisolid preparations on hydrocortisone permeation through excised human stratum corneum. Eur. J. Pharm. Biopharm. 2002;54:143–150. doi: 10.1016/S0939-6411(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 17.Kreilgaard M., Pedersen E. J., Jaroszewski J. W. NMR characterisation and transdermal drug delivery potential of microemulsion systems. J. Control Rel. 2000;69:421–433. doi: 10.1016/S0168-3659(00)00325-4. [DOI] [PubMed] [Google Scholar]
- 18.Kemken J., Ziegler A., Müller B. W. Influence of supersaturation on the pharmacodynamic effect of bupranolol after dermal administration using microemulsions as vehicle. Pharm. Res. 1992;9:554–558. doi: 10.1023/A:1015856800653. [DOI] [PubMed] [Google Scholar]