


Abstract
BodyMap is a human and mouse gene expression database that is based on site-directed 3′-expressed sequence tags generated at Osaka University. To date, it contains more than 300 000 tag sequences from 64 human and 39 mouse tissues. For the recent release, the precise anatomical expression patterns for more than half of the human gene entries were generated by introduced amplified fragment length polymorphism (iAFLP), which is a PCR-based high-throughput expression profiling method. The iAFLP data incorporated into BodyMap describe the relative contents of more than 12 000 transcripts across 30 tissue RNAs. In addition, a newly developed gene ranking system helps users obtain lists of genes that have desired expression patterns according to their significance. BodyMap supports complete transfer of unique data sets and provides analysis that is accessible through the WWW at http://bodymap.ims.u-tokyo.ac.jp.
INTRODUCTION
The function of a gene is determined not only by the coding information that defines the activity of a protein but also by regulation of its expression, which influences the consequences of the protein’s actions. Accordingly, the importance of systematic generation and collection of gene expression data in parallel with the genome sequencing effort has been emphasized (1,2). BodyMap is a collection of site-directed 3′-expressed sequence tags (ESTs) (gene signatures, GSs) structured as an anatomical database of human and mouse gene expression (3,4). Construction of BodyMap was started in 1991 by random sequencing of cDNA clones from libraries that preserved gene expression information contained in tissue RNAs. By compiling frequency data for the tagged sequences, the transcription patterns of genes can be reported as a matrix (genes × tissues) of isolation frequencies (1). One drawback inherent to such a study of populations by random sampling is that the sensitivity and accuracy of transcript detection are dependent on the sampling size and prone to errors originating from fluctuation. In a recent revision of the database, we addressed these problems by two different means.
EXPRESSION PROFILING BY INTRODUCED AMPLIFIED FRAGMENT LENGTH POLYMORPHISM (iAFLP)
As of August 2000, BodyMap contained 18 998 human and 16 772 mouse independent GS clusters based on 3′ sequences of more than 300 000 clones generated at Osaka University from 64 human and 39 mouse tissues. The frequencies of the cognate sequence tags were summarized as a genes × tissues matrix in which anatomical distributions of the transcripts were represented. In this matrix, however, more than half the genes were identified only once, providing little information about regulation of their expression. To provide expression data for these rare transcripts, we incorporated a set of data generated by a recently developed PCR-based expression profiling method, iAFLP (5). In iAFLP, a pool of differentially adapted 3′-ends of cDNAs from multiple tissues is used as a universal template for expression profiling. Amplification with one GS primer and one fluorescent adapter primer yields a mixture of specific products with small size differences that originate from differences in the adapter length, indicating the ratio of target molecules among tissues. In the present protocol, the relative concentrations of a transcript in six different tissues were determined by single PCR followed by fragment analysis on an autosequencer. iAFLP results for a panel of 30 representative tissue RNAs are currently being generated for every human GS cluster (Fig. 1). Thus, with our revised ‘GS card’, which contains detailed information for each GS cluster (6), users can see the absolute level of gene expression as the frequency of occurrence of a sequence tag across the libraries and the precise anatomical distribution pattern of transcript as iAFLP data. The raw electropherogram of each iAFLP–PCR product can be viewed on a web page that shows non-specific PCR products in addition to products with expected sizes, thereby allowing users to validate the iAFLP results. As of August 2000, iAFLP data were available for 12 644 human GSs. BodyMap is based on locally produced data, but users can obtain further information about each GS entry from other public data sets via links to UniGene, RefSeq and GenBank accession numbers provided at NCBI (http://www.ncbi.nlm.nih.gov).
Figure 1.

Production process for BodyMap data sets. Based on the distribution of cognate sequence tags (GSs) across cDNA libraries, expression patterns of genes were approximated as gene × tissue matrix. In the latest revision, precise anatomical distribution of the transcript represented by each GS cluster was observed by iAFLP and incorporated.
STATISTICAL EVALUATION OF DIFFERENTIAL EXPRESSION BY THE GENE RANKING SYSTEM
The frequency distributions of the three distinct GSs presented in Figure 2 illustrate the rationale behind Gene Ranking for evaluating the significance of anatomically regulated expression of genes. For example, intuitive analysis of the three distributions shown in Figure 2 would lead us to conclude that GS02047 is most specific to liver. However, this cannot be confirmed by simple comparison of the frequencies of the three genes in liver, because the frequency of GS02047 is less than the frequencies of the other two. Statistical analyses are therefore essential for rigorous measurement of the tissue specificity of genes.
Figure 2.

Distribution patterns of GSs across libraries in BodyMap. Frequencies are shown as occurrences of the cognate sequence tags.
The higher the specificity of a given cDNA to a particular tissue, the larger the discrepancy between the probability of observing the cDNA in the particular tissue versus all other tissues. Larger differences are characterized by lower values of the expected probability of y occurrences of the cDNA in N2 clones from livers when x in N1 from all other tissues are observed. Because in BodyMap cognate sequence tags are collected at random from tissues, the expected probability is derived from the following mathematical formula (7):
[(N2/N2)y] 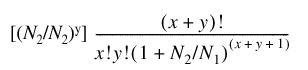
For instance, the expected probabilities of GS02047, GS02155 and GS00273 in Figure 2 are 6.58 × 10–5, 8.21 × 10–4 and 2.12 × 10–1, respectively. The ascending order obtained from expected probabilities of the three GSs agrees with the intuitive order according to specificity of expression in liver.
‘Select genes by expression patterns’, which is a query at the BodyMap web site, allows users to query expression patterns with respect to the presence or absence of tag in each library. On the web site, selection of tissues that are expected to be up-regulated, yields a list of GSs preferential to the selected tissues that is sorted by expected probabilities.
FUTURE DEVELOPMENT
The current version of Gene Ranking supports queries for GS frequencies. The expected probability is accurate for abundant GSs that are observed with sufficient frequencies, whereas the measure may be erroneous for GSs that occur infrequently. Use of iAFLP data overcomes this difficulty because it yields accurate expression patterns even for rare transcripts. In the near future, we will release the Gene Ranking system based on iAFLP data. Additionally, genes not listed in BodyMap will be included in iAFLP profiling.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas, Genome Science, Medical Genome Science, and Genome Information Science from the Ministry of Education, Science and Culture, and by Research for the Future of Japan (Society for the Promotion of Science, Japan).
References
- 1.Okubo K. and Matsubara,K. (1997) Complementary DNA sequence (EST) collections and the expression information of the human genome. FEBS Lett., 403, 225–229. [DOI] [PubMed] [Google Scholar]
- 2.Bortoluzzi S. and Danieli,G.A. (1999) Towards an in silico analysis of transcription patterns. Trends Genet., 15, 118–119. [DOI] [PubMed] [Google Scholar]
- 3.Okubo K., Hori,N., Matoba,R., Niiyama,T., Fukushima,A., Kojima,Y. and Matsubara,K. (1992) Large scale cDNA sequencing for analysis of quantitative and qualitative aspects of gene expression. Nature Genet., 2, 173–179. [DOI] [PubMed] [Google Scholar]
- 4.Kawamoto S., Matsumoto,Y., Mizuno,K., Okubo,K. and Matsubara,K. (1996) Expression profiles of active genes in human and mouse livers. Gene, 174, 151–158. [DOI] [PubMed] [Google Scholar]
- 5.Kawamoto S., Ohnishi,T., Kita,H., Chisaka,O. and Okubo,K. (1999) Expression profiling by iAFLP: A PCR-based method for genome-wide gene expression profiling. Nucleic Acids Res., 9, 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hishiki T., Kawamoto,S., Morishita,S. and Okubo,K.(2000) BodyMap: a human and mouse gene expression database. Nucleic Acids Res., 28, 136–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Audic S. and Claverie,J.M. (1997) The significance of digital gene expression profiles. Genome Res., 7, 986–995. [DOI] [PubMed] [Google Scholar]